


Evaluating the Binding Potential and Stability of Drug-like Compounds with the Monkeypox Virus VP39 Protein Using Molecular Dynamics Simulations and Free Energy Analysis

 , , , , , ,
, , , , , ,  and
and 
Abstract
1. Introduction
2. Result
2.1. Structural-Based Virtual Screening
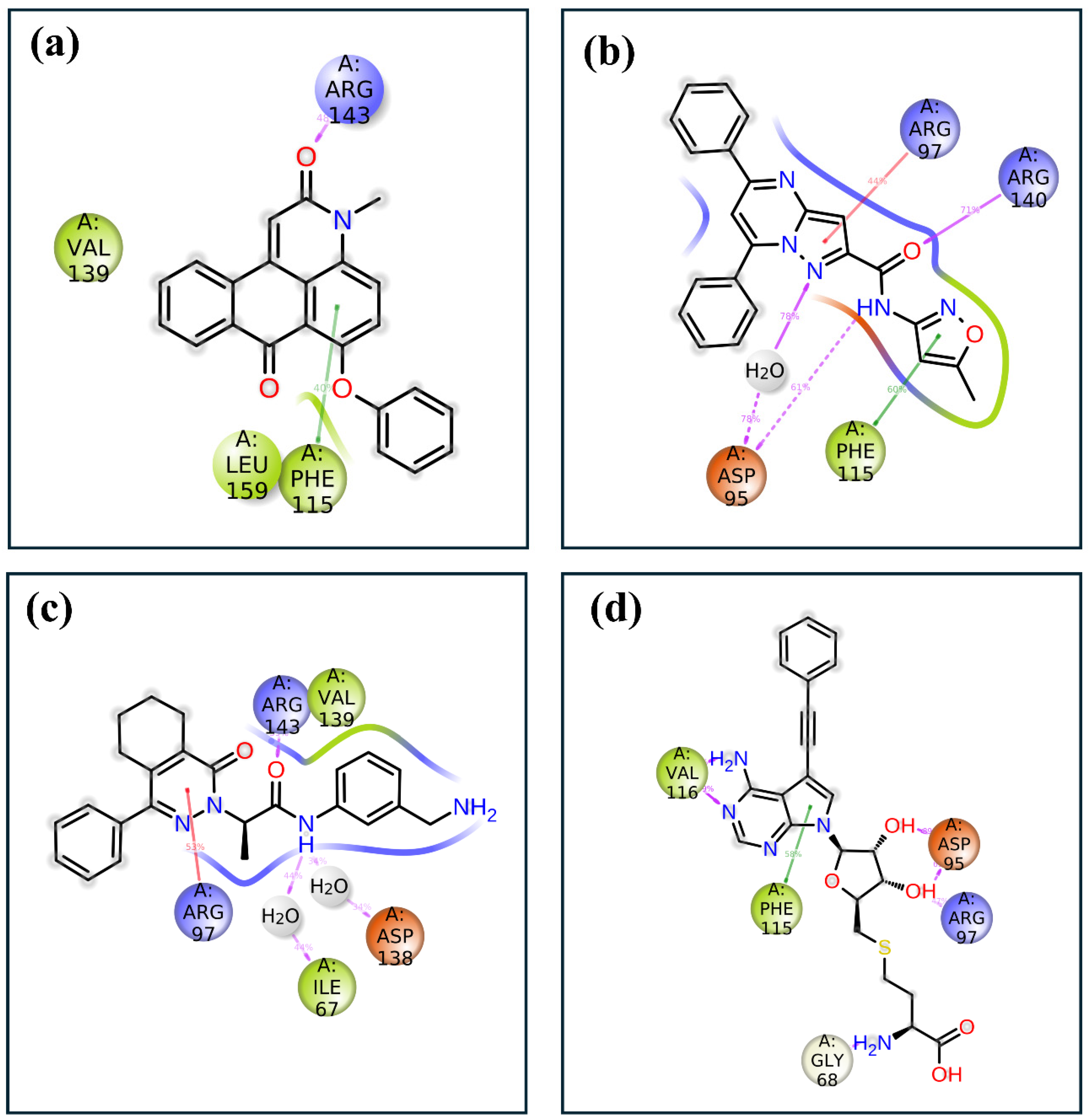
2.2. Redocking and Intermolecular Analysis and ADME Analysis
2.3. Specificity Analysis
2.4. Dynamical Analysis
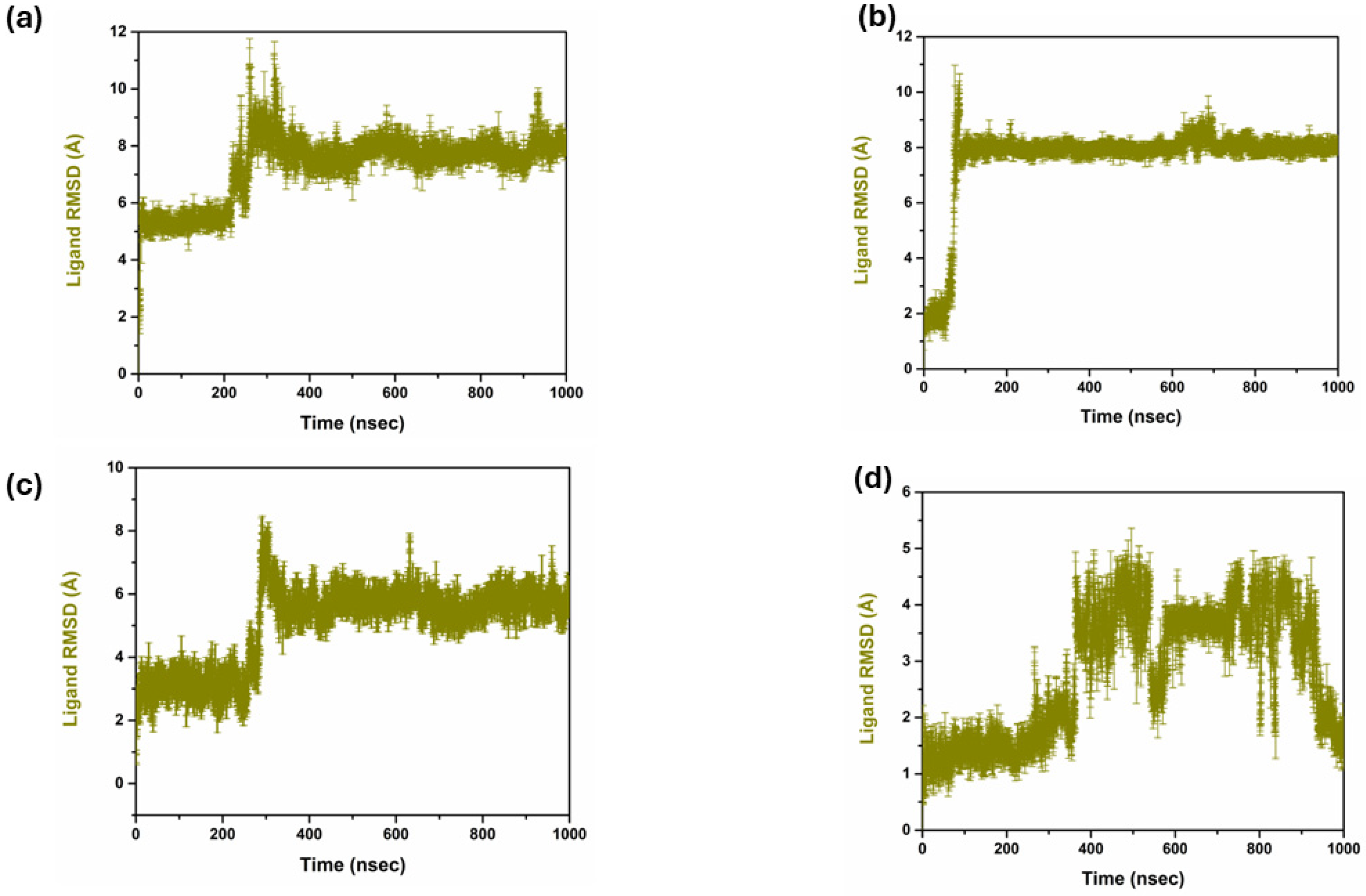
2.4.1. RMSD Analysis
2.4.2. Protein RMSF Analysis
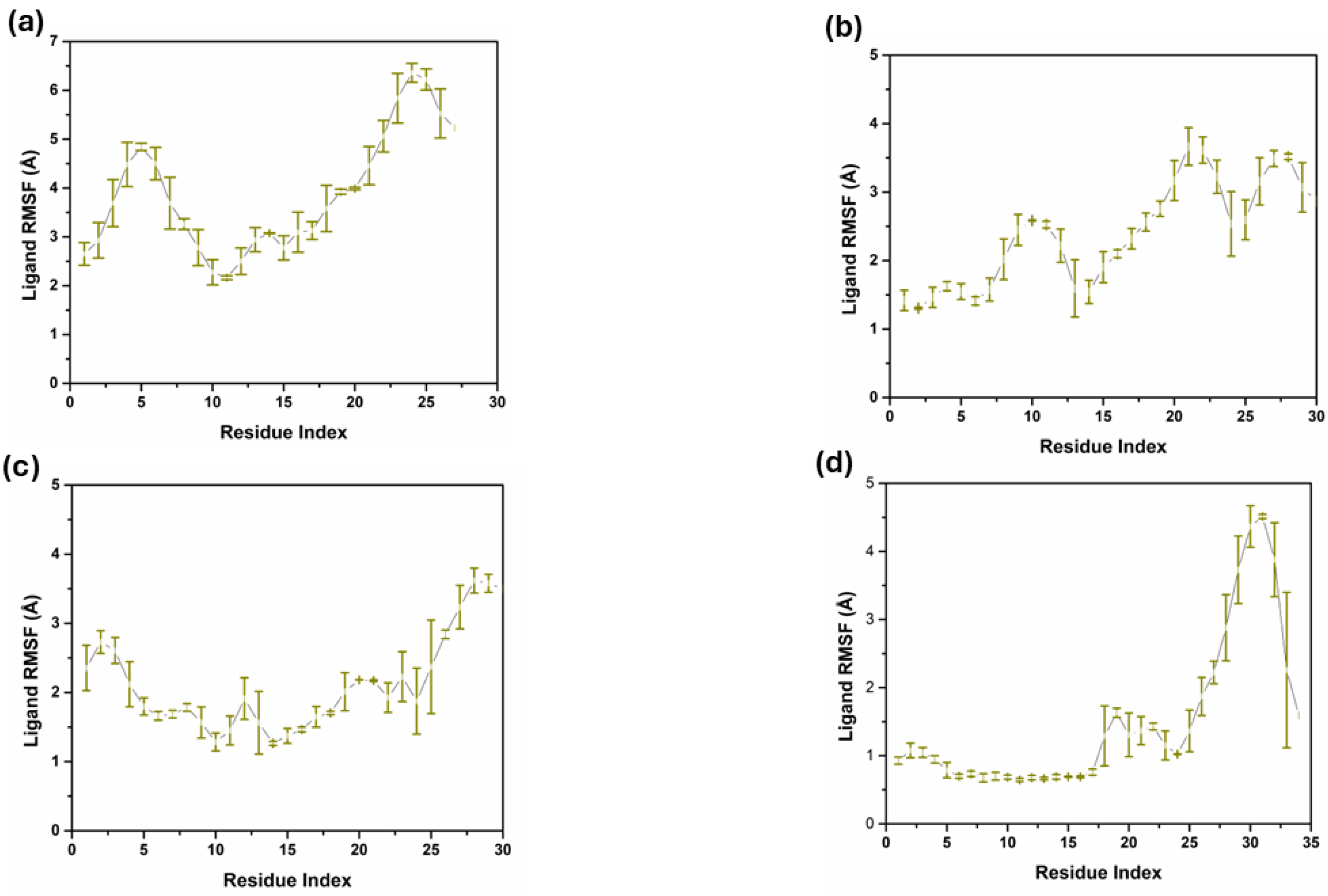
2.4.3. Ligand RMSF Analysis
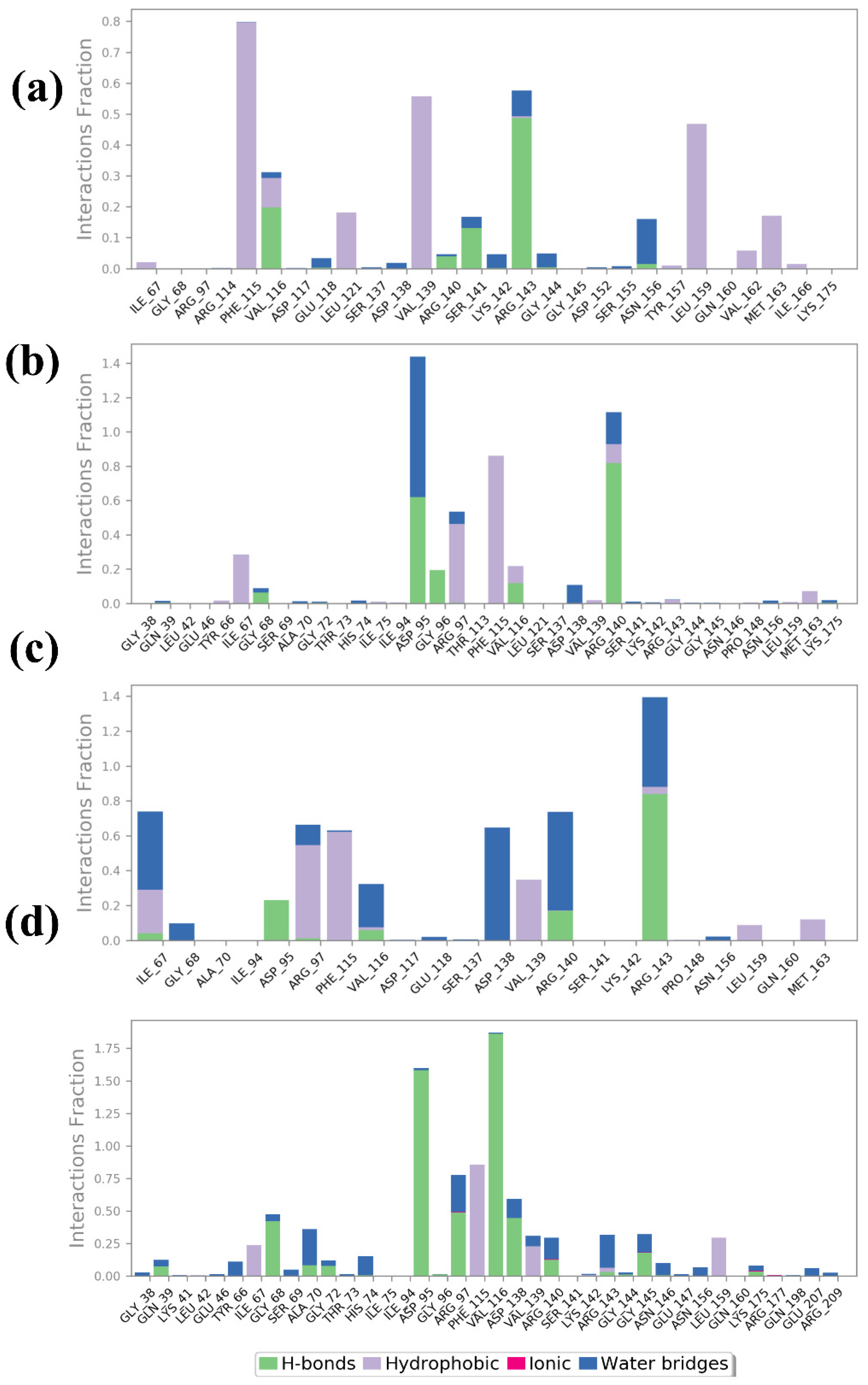
2.4.4. Protein–Ligand Profiling
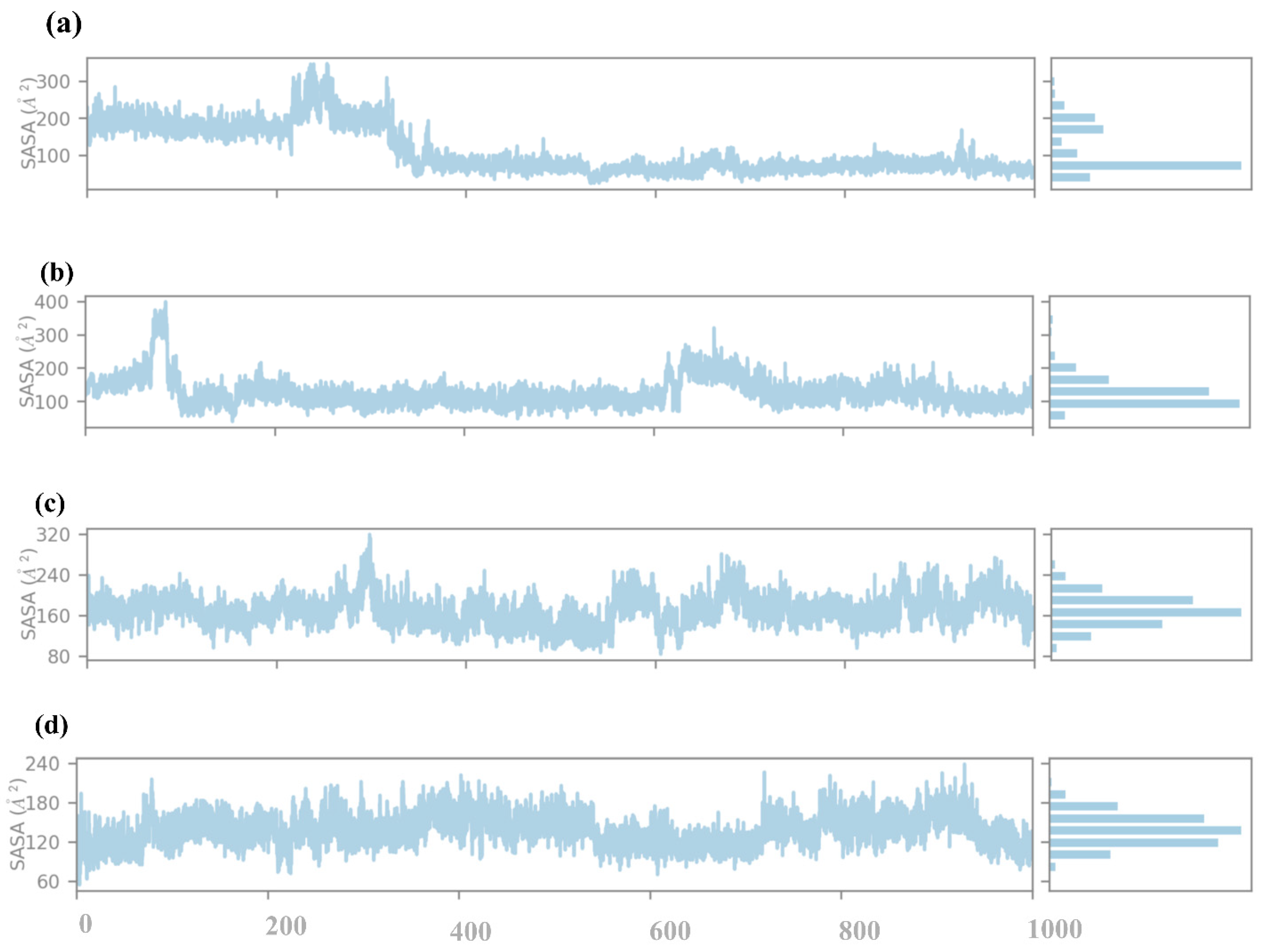
2.4.5. SASA Analysis
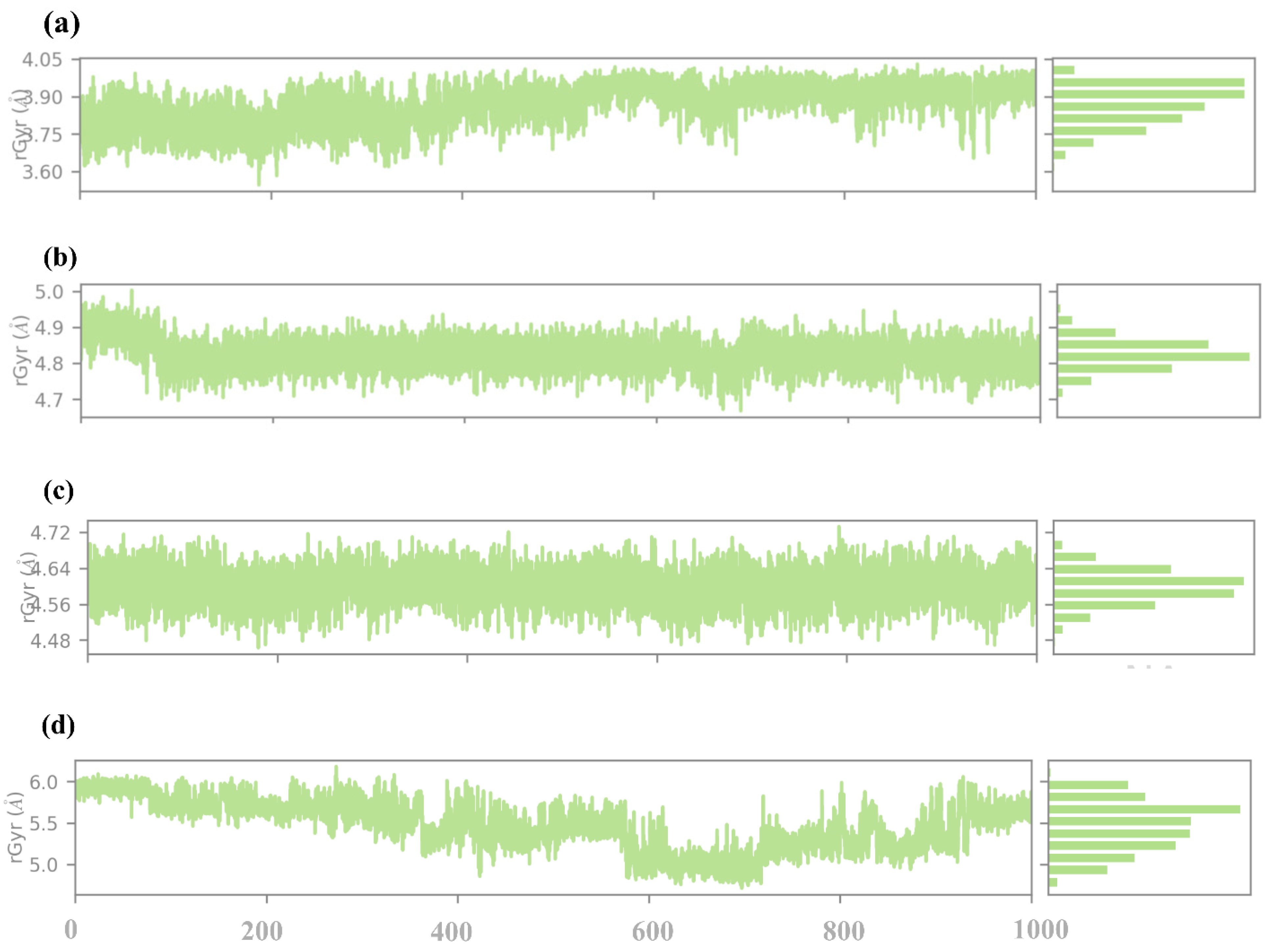
2.4.6. RG Analysis
2.5. Free Binding Energy Analysis
2.6. Negative Control Analysis
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Virtual Screening and Molecular Docking
4.3. Dynamical Analysis
4.4. Binding Free Energy
- ΔEMM is the molecular mechanics energy, which includes electrostatic and van der Waals interactions.
- ΔGsolvation is the solvation free energy, typically comprising polar and non-polar contributions.
- TΔS is the entropic contribution, often approximated or derived from normal mode analysis in certain cases.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moore, M.J.; Rathish, B.; Zahra, F. Mpox (Monkeypox). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Monkeypox—An Overview. ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/monkeypox (accessed on 3 September 2024).
- Rabaan, A.A.; Alasiri, N.A.; Aljeldah, M.; Alshukairiis, A.N.; AlMusa, Z.; Alfouzan, W.A.; Abuzaid, A.A.; Alamri, A.A.; Al-Afghani, H.M.; Al-baghli, N.; et al. An Updated Review on Monkeypox Viral Disease: Emphasis on Genomic Diversity. Biomedicines 2023, 11, 1832. [Google Scholar] [CrossRef] [PubMed]
- Adnan, N.; Haq, Z.U.; Malik, A.; Mehmood, A.; Ishaq, U.; Faraz, M.; Malik, J.; Mehmoodi, A. Human Monkeypox Virus: An Updated Review. Medicine 2022, 101, e30406. [Google Scholar] [CrossRef] [PubMed]
- Mpox (Monkeypox). Available online: https://www.who.int/news-room/fact-sheets/detail/monkeypox (accessed on 10 December 2023).
- Alakunle, E.; Moens, U.; Nchinda, G.; Okeke, M.I. Monkeypox Virus in Nigeria: Infection Biology, Epidemiology, and Evolution. Viruses 2020, 12, 1257. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Wu, Z.; Jiang, S.; Lu, L.; Liu, H. The Current Emergence of Monkeypox: The Recurrence of Another Smallpox? Biosaf. Health 2022, 4, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Gigante, C.M.; Korber, B.; Seabolt, M.H.; Wilkins, K.; Davidson, W.; Rao, A.K.; Zhao, H.; Smith, T.G.; Hughes, C.M.; Minhaj, F.; et al. Multiple Lineages of Monkeypox Virus Detected in the United States, 2021–2022. Science 2022, 378, 560–565. [Google Scholar] [CrossRef]
- Karagoz, A.; Tombuloglu, H.; Alsaeed, M.; Tombuloglu, G.; AlRubaish, A.A.; Mahmoud, A.; Smajlović, S.; Ćordić, S.; Rabaan, A.A.; Alsuhaimi, E. Monkeypox (Mpox) Virus: Classification, Origin, Transmission, Genome Organization, Antiviral Drugs, and Molecular Diagnosis. J. Infect. Public Health 2023, 16, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunova, G.A.; Shchelkunov, S.N. Smallpox, Monkeypox and Other Human Orthopoxvirus Infections. Viruses 2022, 15, 103. [Google Scholar] [CrossRef]
- Alakunle, E.; Kolawole, D.; Diaz-Cánova, D.; Alele, F.; Adegboye, O.; Moens, U.; Okeke, M.I. A Comprehensive Review of Monkeypox Virus and Mpox Characteristics. Front. Cell. Infect. Microbiol. 2024, 14, 1360586. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, Y.; Igarashi, M.; Kato, H. Targeting Cap1 RNA Methyltransferases as an Antiviral Strategy. Cell Chem. Biol. 2024, 31, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Otava, T.; Silhan, J.; Nencka, R.; Weber, J.; Boura, E. Inhibitors of Mpox VP39 2’-O Methyltransferase Efficiently Inhibit the Monkeypox Virus. Antivir. Res. 2023, 218, 105714. [Google Scholar] [CrossRef]
- Ezike, T.C.; Okpala, U.S.; Onoja, U.L.; Nwike, C.P.; Ezeako, E.C.; Okpara, O.J.; Okoroafor, C.C.; Eze, S.C.; Kalu, O.L.; Odoh, E.C.; et al. Advances in Drug Delivery Systems, Challenges and Future Directions. Heliyon 2023, 9, e17488. [Google Scholar] [CrossRef] [PubMed]
- Silhan, J.; Klima, M.; Otava, T.; Skvara, P.; Chalupska, D.; Chalupsky, K.; Kozic, J.; Nencka, R.; Boura, E. Discovery and Structural Characterization of Monkeypox Virus Methyltransferase VP39 Inhibitors Reveal Similarities to SARS-CoV-2 Nsp14 Methyltransferase. Nat. Commun. 2023, 14, 2259. [Google Scholar] [CrossRef] [PubMed]
- Flores-Padilla, E.A.; Juárez-Mercado, K.E.; Naveja, J.J.; Kim, T.D.; Alain Miranda-Quintana, R.; Medina-Franco, J.L. Chemoinformatic Characterization of Synthetic Screening Libraries Focused on Epigenetic Targets. Mol. Inform. 2022, 41, 2100285. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.M.; Rey, J.; Lagorce, D.; Vavruša, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tufféry, P.; Miteva, M.A. MTiOpenScreen: A Web Server for Structure-Based Virtual Screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Biovia, D.S. Discovery Studio Visualizer; Accelrys Software Inc: San Diego, CA, USA, 2019. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Minasov, G.; Inniss, N.L.; Shuvalova, L.; Anderson, W.F.; Satchell, K.J. Structure of the Monkeypox Virus Profilin-like Protein A42R Reveals Potential Functional Differences from Cellular Profilins. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2022, 78, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xue, H.; Hu, N.; Liu, Y.; Sun, H.; Yu, D.; Qin, L.; Shi, G.; Wang, F.; Xin, L. Discovery of the Clinical Candidate Sonrotoclax (BGB-11417), a Highly Potent and Selective Inhibitor for Both WT and G101V Mutant Bcl-2. J. Med. Chem. 2024, 67, 7836–7858. [Google Scholar] [CrossRef]
- Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. Poxviral Strategies to Overcome Host Cell Apoptosis. Pathogens 2020, 10, 6. [Google Scholar] [CrossRef]
- Hatok, J.; Racay, P. Bcl-2 Family Proteins: Master Regulators of Cell Survival. Biomol. Concepts 2016, 7, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Alcamí, A. Poxvirus Immune Evasion. Annu. Rev. Immunol. 2024, 42, 551–584. [Google Scholar] [CrossRef] [PubMed]
- Knapp, B.; Ospina, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of Protein Structure Comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Kumar Bhardwaj, V.; Singh, R.; Rajendran, V.; Purohit, R.; Kumar, S. An In-Silico Evaluation of Different Bioactive Molecules of Tea for Their Inhibition Potency against Non Structural Protein-15 of SARS-CoV-2. Food Chem. 2021, 346, 128933. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Alexey, R.; Dariya, S.; Liudmyla, I.; Lilia, V.; Valeriy, M.; Dmytro, L.; Oleksandr, B.; Svitlana, S.; Sergii, O.; Elijah, B. Structure-based Virtual Screening and Biological Evaluation of Novel Inhibitors of Mycobacterium Z-ring Formation. J. Cell. Biochem. 2022, 123, 852–862. [Google Scholar] [CrossRef]
- Venkatraman, V.; Gaiser, J.; Demekas, D.; Roy, A.; Xiong, R.; Wheeler, T.J. Do Molecular Fingerprints Identify Diverse Active Drugs in Large-Scale Virtual Screening? (No). Pharmaceuticals 2024, 17, 992. [Google Scholar] [CrossRef]
- Veljkovic, N.; Glisic, S.; Perovic, V.; Veljkovic, V. The Role of Long-Range Intermolecular Interactions in Discovery of New Drugs. Expert Opin. Drug Discov. 2011, 6, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Olaoye, F.; Potter, K. Protein-Ligand Interactions and Drug Design. Med. Chem. 2024. [Google Scholar] [CrossRef]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossváry, I.; Moraes, M.; Sacerdoti, F.; et al. Molecular Dynamics—Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC2006 Conference on High Performance Networking and Computing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Bagewadi, Z.K.; Yunus Khan, T.M.; Gangadharappa, B.; Kamalapurkar, A.; Mohamed Shamsudeen, S.; Yaraguppi, D.A. Molecular Dynamics and Simulation Analysis against Superoxide Dismutase (SOD) Target of Micrococcus Luteus with Secondary Metabolites from Bacillus Licheniformis Recognized by Genome Mining Approach. Saudi J. Biol. Sci. 2023, 30, 103753. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of Gyration as an Indicator of Protein Structure Compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Yang, W.; Zhao, L.; Hu, G. Conformations of KRAS4B Affected by Its Partner Binding and G12C Mutation: Insights from GaMD Trajectory-Image Transformation-Based Deep Learning. J. Chem. Inf. Model. 2024, 64, 6880–6898. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Pang, L.; Zhang, J.Z.H.; Zhu, T. Effect of Mutations on Binding of Ligands to Guanine Riboswitch Probed by Free Energy Perturbation and Molecular Dynamics Simulations. Nucleic Acids Res. 2019, 47, 6618–6631. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T. SWISS-MODEL: An Automated Protein Homology-Modeling Server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Barostats in Molecular Dynamics. Available online: https://www.compchems.com/barostats-in-molecular-dynamics/ (accessed on 18 April 2024).
- Petersen, H.G. Accuracy and Efficiency of the Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Schrödinger Release 2023-2: Prime; Schrödinger, LLC: New York, NY, USA, 2023.
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. no. | Complex | H-Bond | Van der Waals | π-π Stacking/ π-π Cation | Structure |
|---|---|---|---|---|---|
| 1 | 8CEQ_17444176 (PubChem SID 17444176) | -- | Lys142, Arg97, Gly68, Gly96, Asp95, Ser141, Phe115, Ile94, Ile67, Val116 Leu159 | -- |  |
| 2 | 8CEQ_17450998 (PubChem SID 17450998) | Gly68, | Val116, Leu159, Asn156, Ser141, Asp138, Ser69, Gln39, Ile75, Pro71, Gly72, Ala70, Lys142, Gly145 | Phe115 |  |
| 3 | 8CEQ_24392109 (PubChem SID 24392109) | Arg97, Asp95, Arg140, | Leu42, Asn146, Lys175, Lys142, Asp138, Gly96, Ile94, Ile67, Val116, Leu159, Arg143, Ala70, Ser69, Gly145 | Phe115 |  |
| 4 | 8CEQ_Control | Val116, Asp95, Arg97, Asp138, Gly68, His74, Gly72, Gln39 | Asn156, Ser141, Ser155, Ser69, Tyr66, Arg140, Arg114, Ile94, Ile67, Leu42, Ile75 | -- | 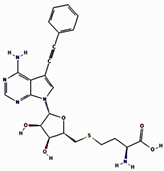 |
| 17444176 | 17450998 | 24392109 | Control | |
|---|---|---|---|---|
| ΔGBind | −87.84 ± 6.45 | −69.06 ± 4.84 | −59.55 ± 3.99 | −76.46 ± 4.89 |
| ΔGBind Coulomb | −36.93 ± 6.42 | −11.27 ± 2.76 | −14.64 ± 4.47 | −10.49 ± 3.53 |
| ΔGBind Covalent | 1.79 ± 1.41 | 4.85 ± 1.99 | −0.18 ± 1.04 | −0.16 ± 0.68 |
| ΔGBind Hbond | −4.19 ± 0.81 | −1.12 ± 0.37 | −1.21 ± 0.39 | −025 ± 0.24 |
| ΔGBind Lipo | −19.88 ± 1.16 | −15.31 ± 1.50 | −19.70 ± 1.45 | −27.97 ± 1.65 |
| ΔGBind Packing | −1.22 ± 0.43 | −7.06 ± 0.72 | −3.60 ± 0.60 | −4.72 ± 0.89 |
| ΔGBind Solv GB | 37.36 ± 4.79 | 20.92 ± 1.84 | 27.49 ± 3.17 | 19.70 ± 1.70 |
| ΔGBind vdW | −64.77 ± 3.40 | −60.04 ± 3.27 | −47.70 ± 3.08 | −52.55 ± 2.34 |
| Ligand Strain Energy | 5.45 ± 2.34 | 4.41 ± 1.57 | 5.54 ± 1.02 | 1.61 ± 0.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, A.M.; Gattan, H.S.; Faizo, A.A.; Alruhaili, M.H.; Alharbi, A.S.; Bajrai, L.H.; AL-Zahrani, I.A.; Dwivedi, V.D.; Azhar, E.I. Evaluating the Binding Potential and Stability of Drug-like Compounds with the Monkeypox Virus VP39 Protein Using Molecular Dynamics Simulations and Free Energy Analysis. Pharmaceuticals 2024, 17, 1617. https://doi.org/10.3390/ph17121617
Hassan AM, Gattan HS, Faizo AA, Alruhaili MH, Alharbi AS, Bajrai LH, AL-Zahrani IA, Dwivedi VD, Azhar EI. Evaluating the Binding Potential and Stability of Drug-like Compounds with the Monkeypox Virus VP39 Protein Using Molecular Dynamics Simulations and Free Energy Analysis. Pharmaceuticals. 2024; 17(12):1617. https://doi.org/10.3390/ph17121617
Chicago/Turabian StyleHassan, Ahmed M., Hattan S. Gattan, Arwa A. Faizo, Mohammed H. Alruhaili, Azzah S. Alharbi, Leena H. Bajrai, Ibrahim A. AL-Zahrani, Vivek Dhar Dwivedi, and Esam I. Azhar. 2024. "Evaluating the Binding Potential and Stability of Drug-like Compounds with the Monkeypox Virus VP39 Protein Using Molecular Dynamics Simulations and Free Energy Analysis" Pharmaceuticals 17, no. 12: 1617. https://doi.org/10.3390/ph17121617
APA StyleHassan, A. M., Gattan, H. S., Faizo, A. A., Alruhaili, M. H., Alharbi, A. S., Bajrai, L. H., AL-Zahrani, I. A., Dwivedi, V. D., & Azhar, E. I. (2024). Evaluating the Binding Potential and Stability of Drug-like Compounds with the Monkeypox Virus VP39 Protein Using Molecular Dynamics Simulations and Free Energy Analysis. Pharmaceuticals, 17(12), 1617. https://doi.org/10.3390/ph17121617