
Pharmaceutical Equivalence of Film-Coated and Chewable Tablets: A Comparative Dissolution Study Using Pulverized Chewable Tablets


Abstract
1. Introduction
2. Results
2.1. Preparation and Characterization of Tablets
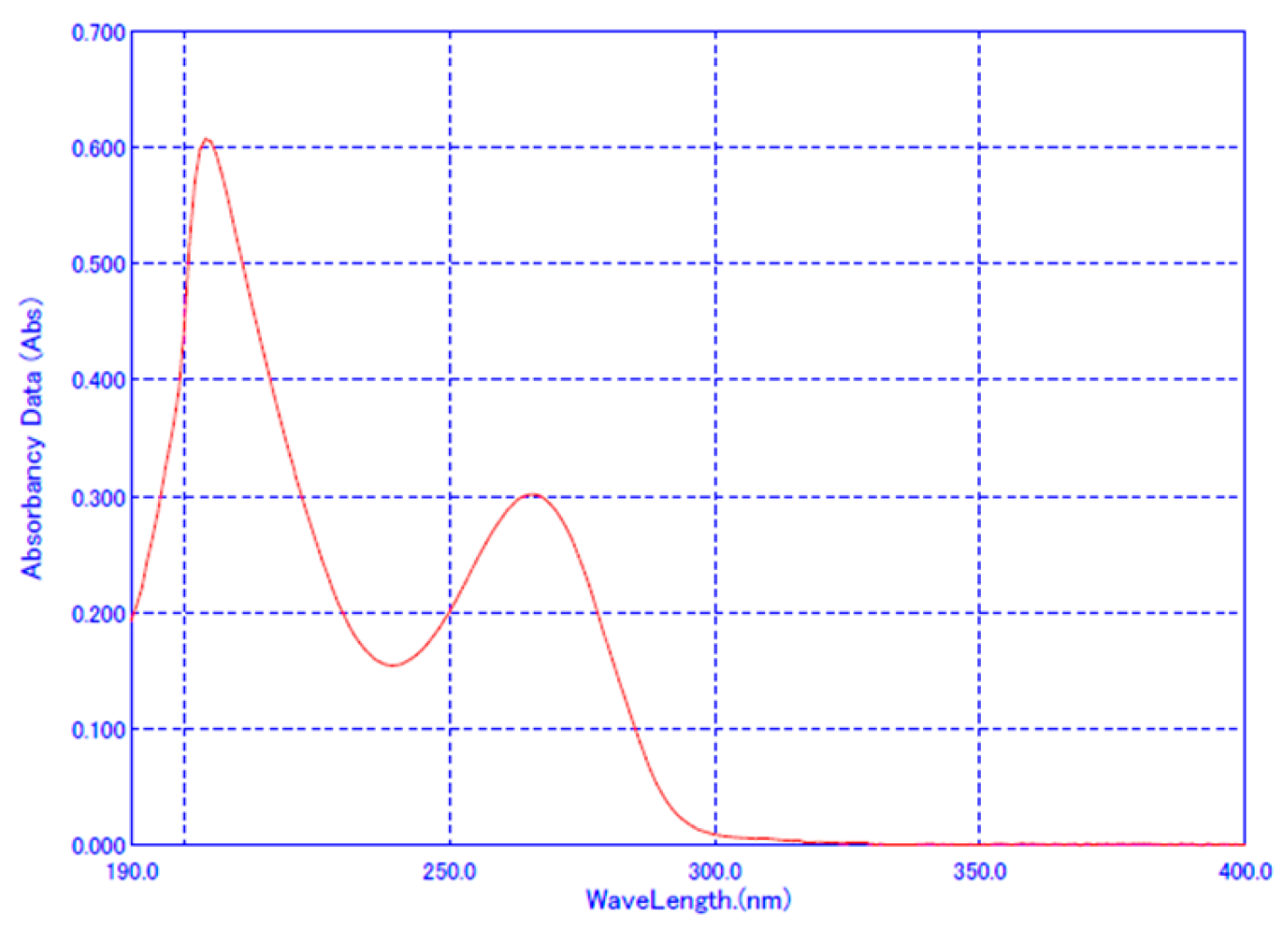
2.2. Method Validation for Dissolution Test
2.3. Dissolution Profiles
2.4. Difference Factor (f1) and Similarity Factor (f2)
3. Discussion
3.1. Selecting an Appropriate Form as the Reference Drug for the Pharmaceutical Equivalence
3.2. Determining Dissolution Similarity
4. Materials and Methods
4.1. Materials
4.2. Preparation of Famotidine Fixed-Dose Tablets (FT) with Antacids
4.3. Quality Control of the Tablets
- a: Consumed amount of 0.1 mol/L sodium hydroxide VS (mL).
- b: Consumed amount of 0.1 mol/L sodium hydroxide VS in blank test (mL).
- f: Normality factor of 0.1 mol/L sodium hydroxide VS.
- t: Weight of a tablet (mg).
- s: Sample quantity (mg).
4.4. Method Validation for the UV Analysis of Dissolution
4.5. Dissolution Comparison with Difference Factor (f1) and Similarity Factor (f2)
- Rt = the dissolution value of the reference drug at time t.
- Tt = the dissolution value of the test drug at time t.
- n = the number of time points.
- t = time point.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korean Statistics 2022. Available online: https://www.index.go.kr/unity/potal/main/EachDtlPageDetail.do?idx_cd=2770 (accessed on 15 October 2024).
- Usmani, M.T.; Shoaib, M.H.; Siddiqui, F.; Ahmed, F.R.; Yousuf, R.I.; Saleem, M.T. Formulation development, in vivo bioequivalence and pediatric PBPK modeling studies of taste-masked ciprofloxacin chewable tablets. Sci. Rep. 2023, 13, 16070. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shah, R.B.; Yu, L.X.; Khan, M.A. In vitro bioequivalence approach for a locally acting gastrointestinal drug: Lanthanum carbonate. Mol. Pharm. 2013, 10, 544–550. [Google Scholar] [CrossRef] [PubMed]
- El-Gazayerly, O.N.; Rakkanka, V.; Ayres, J.W. Novel chewable sustained-release tablet containing verapamil hydrochloride. Pharm. Dev. Technol. 2004, 9, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Food and Drug Safety. Standard on Pharmaceutical Equivalence Test; Ministry of Food and Drug Safety: Cheongju, Republic of Korea, 2021.
- Wesche, D.; Lutz, S.; Barnish, G. Comparative study of chewable pyrantel pamoate: Should standards for chewable tablets be revised? Clin. Trials PNG Med. J. 1994, 37, 12–14. [Google Scholar]
- Krogstad, P.; Samson, P.; Acosta, E.P.; Moye, J.; Townley, E.; Bradford, S.; Brown, E.; Denson, K.; Graham, B.; Hovind, L.; et al. Pharmacokinetics and Safety of a Raltegravir-Containing Regimen in Children Aged 4 Weeks to 2 Years Living With Human Immunodeficiency Virus and Receiving Rifampin for Tuberculosis. J. Pediatr. Infect. Dis. Soc. 2021, 10, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Sawatdee, S.; Atipairin, A.; Sae Yoon, A.; Srichana, T.; Changsan, N.; Suwandecha, T. Formulation Development of Albendazole-Loaded Self-Microemulsifying Chewable Tablets to Enhance Dissolution and Bioavailability. Pharmaceutics 2019, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Cánovas, M.; Arcabell, M.; Martínez, G.; Canals, M.; Cabré, F. Bioequivalence studies of film-coated tablet and chewable tablet generic formulations of montelukast in healthy volunteers. Arzneimittelforschung 2011, 61, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, L.; Han, S.; Peng, H.; Tong, M.; Gu, X.; Hu, H.; Wang, Y.; Lv, Z.; He, X. Pharmacokinetics and bioequivalence study of montelukast sodium oral soluble film and chewable tablet in healthy Chinese volunteers: Randomized trials under fasting and fed conditions. Ann. Transl. Med. 2023, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Zaid, A.N.; Abualhasan, M.N.; Watson, D.G.; Mousa, A.; Ghazal, N.; Bustami, R. Investigation of the bioequivalence of montelukast chewable tablets after a single oral administration using a validated LC-MS/MS method. Drug Des. Dev. Ther. 2015, 9, 5315–5321. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Cho, S.M.; Choi, Y.W.; Lee, H.J.; Kwon, J.H.; Kim, S.W.; Kim, J.W.; Lee, S.; Hong, J.H. Comparison of pharmacokinetic characteristics of sildenafil citrate chewable tablets and film-coated tablets in healthy male subjects. Transl. Clin. Pharmacol. 2017, 25, 153–156. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Valenzuela, F.; Davila, G.; Ibañez, Y.; Garcia, L.; Crownover, P.H.; Gómez-Palacio, R.; Ovalle, J.F.; Velasco, C.; Malhotra, B.K. Relative bioavailability of chewable and conventional film-coated tablet formulations of sildenafil 100 mg in healthy volunteers under fasting conditions. J. Bioequivalence Bioavailab. 2011, 3, 207–210. [Google Scholar] [CrossRef]
- Marcelín-Jiménez, G.; Angeles-Moreno, A.P.; Contreras-Zavala, L.; García-González, A.; Ramírez-San Juan, E. Comparison of fasting bioavailability among 100-mg commercial, 100-mg generic, and 50-mg chewable generic sildenafil tablets in healthy male Mexican volunteers: A single-dose, 3-period, crossover study. Clin. Ther. 2012, 34, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Knorr, B.; Hartford, A.; Li, X.; Yang, A.Y.; Noonan, G.; Migoya, E. Bioequivalence of the 4-mg Oral Granules and Chewable Tablet Formulations of Montelukast. Arch. Drug Inf. 2010, 3, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, T.; Li, Y.; Du, B.; Weng, W. Formulation and evaluation of a montelukast sodium orally disintegrating tablet with a similar dissolution profile as the marketed product. Pharm. Dev. Technol. 2017, 22, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Kimaro, E.; Tibalinda, P.; Shedafa, R.; Temu, M.; Kaale, E. Formulation development of chewable albendazole tablets with improved dissolution rate. Heliyon 2019, 5, e02911. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Food and Drug Administration. FDA Guidance: Quality Attribute Considerations for Chewable Tablets Guidance for Industry; Food and Drug Administration, Center for Drug Evaluation and Research (CDER): Rockville, MD, USA, 2018.
- Kim, S.; Kim, M.; Kim, K. Intra-and inter-examiner reliability on the estimation of masticatory efficiency: By pulverizing peanuts with a sieve-system. J. Oral Med. Pain 2002, 27, 381–389. [Google Scholar]
- Lee, K.C. Development of New Evaluation Method of Masticatory Efficiency by Using Wax Cube. Ph.D. Thesis, Department of Dentistry, The Graduate School, Yonsei University, Seoul, Republic of Korea, 2008. [Google Scholar]
- Islam, M.S.; Narurkar, M.M. Solubility, stability and ionization behaviour of famotidine. J. Pharm. Pharmacol. 1993, 45, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Matsumoto, S.; Kaguragi, K.; Kurihara, K.; Tochigi, K.; Tomono, K. Determination and correlation of solubilities of famotidine in water +co-solvent mixed solvents. Fluid Phase Equilibria 2011, 302, 115–122. [Google Scholar] [CrossRef]
- Food and Drug Administration. FDA Guidance: Dissolution Testing of Immediate Release Solid Oral Dosage Forms; Food and Drug Administration, Center for Drug Evaluation and Research (CDER): Rockville, MD, USA, 1997.
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.P. In vitro dissolution profile comparison-statistics and analysis of the similarity factor, f2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. FDA Guidance: Draft Guidance on Loratadine; Food and Drug Administration: Rockville, MD, USA, 2022.
- Korean Pharmacopoeia XII, General Tests. Available online: https://www.mfds.go.kr/eng/brd/m_18/view.do?seq=71538 (accessed on 1 November 2024).



{kind=link}
{kind=link}
{kind=link}
| API * | Market ** | Pharmacological Efficacy | No. of Products |
|---|---|---|---|
| Alginic Acid 200/Carboxymethylcellulose Sodium 100 | Export only | Adjuvant therapy in weight loss | 1 |
| Cyproheptadine Orotate Hydrate 0.75/DL-Carnitine Hydrochloride 75/L-Lysine Hydrochloride 75/Cyanocobalamin 0.5 | OTC | Appetite stimulant | 1 |
| Dimenhydrinate 20/Scopolamine Hydrobromide Hydrate 0.125 | OTC | Antihistamine | 1 |
| Dried Aluminium Hydroxide Gel 80/Magnesium Silicate 20/Scopolia Extract 5 | Export only | Gastritis | 1 |
| Hydrotalcite 259/Glutamine 140/Soluble Azulene 2 | OTC | Gastritis | 1 |
| Lamotrigine 2, 5, 25, 50, 100 | ETC | Seizure | 5 |
| Meclizine Hydrochloride Hydrate 12.5/Scopolamine Hydrobromide Hydrate 0.125 | OTC | Antihistamine | 1 |
| Microencapsulated Acetaminophen 86 | OTC | Analgesics | 1 |
| Montelukast Sodium 4, 5 | ETC | Antihistamine | 115 |
| Montelukast Sodium 5.2/Levocetirizine Hydrochloride 5.0 | ETC | Antihistamine | 1 |
| Sildenafil 25, 50, 100 | ETC | Erectile dysfunction | 7 |
| Simethicone 80 | OTC | Gastrointestinal disturbance | 2 |
| Simethicone Powder 181.2/Loperamide Hydrochloride 2.0 | OTC | Gastrointestinal disturbance, Diarrhea | 1 |
| Sodium Alginate 200, 250 | ETC | Gastritis | 2 |
| Sucroferric Oxyhydroxide 2500 | ETC | Serum phosphate regulation | 1 |
| Tadalafil 5, 10, 20 | ETC | Erectile dysfunction | 1 |
| Parameter | UV Absorption in Dissolution Medium | ||||
|---|---|---|---|---|---|
| pH 1.2 | pH 4.0 | pH 6.8 | Water | ||
| System suitability | Mean | 0.0029 | 0.0079 | 0.0018 | 0.0017 |
| SD | 0.0003 | 0.0002 | 0.0002 | 0.0004 | |
| RSD (%) | 0.10 | 0.07 | 0.08 | 0.18 | |
| Specificity | Blank | −0.0001 | 0.0001 | 0.0000 | 0.0001 |
| Placebo | 0.0016 | 0.0005 | 0.0011 | 0.0013 | |
| Parameters | Dissolution Medium | |||
|---|---|---|---|---|
| pH 1.2 | pH 4.0 | pH 6.8 | Water | |
| Slope | 0.0310 | 0.0303 | 0.0252 | 0.0244 |
| y-intercept | 0.0010 | −0.0001 | 0.0012 | 0.0057 |
| SD of y-intercept | 0.0005 | 0.0004 | 0.0012 | 0.0003 |
| R2, coefficient of determination | 1.0000 | 1.0000 | 0.9999 | 0.9978 |
| Range (µg/mL) | 0.509~12.212 (4.6~109.9% §) | 0.511~12.252 (4.6~110.3%) | 0.503~12.064 (4.5~108.6%) | 1.223~12.229 (11.0~110.1%) |
| Detection limit * (µg/mL) | 0.045 | 0.047 | 0.157 | 0.042 |
| Quantitation limit * (µg/mL) | 0.145 | 0.144 | 0.478 | 0.130 |
| Accuracy (Recovery) (%) | 99.041 ± 2.070 | 98.386±1.413 | 99.432 ± 1.439 | 99.278 ± 2.310 |
| Precision (%) | ||||
| repeatability | 99.959 ± 0.471 | 99.515 ± 0.826 | 100.784 ± 0.853 | 99.929 ± 2.005 |
| reproducibility | 100.266 ± 0.513 | 99.825 ± 0.665 | 100.162 ± 0.879 | 100.766 ± 1.658 |
| Dissolution Medium | Pulverized Reference Tablet (PCT) vs. Test Tablet (FT) | Chewable Reference Tablet (CT) vs. Test Tablet (FT) | Chewable Reference Tablet (CT) vs. Pulverized Tablet (PCT) | |||
|---|---|---|---|---|---|---|
| f1 | f2 | f1 | f2 | f1 | f2 | |
| pH 1.2 | 8.48 | 56.81 | 38.02 | 25.27 | 38.02 | 28.53 |
| pH 4.0 | 10.03 | 53.10 | 27.76 | 36.05 | 27.76 | 31.02 |
| pH 6.8 | 5.64 | 67.58 | 61.36 | 27.04 | 56.77 | 28.24 |
| Water | 4.28 | 75.95 | 48.25 | 34.19 | 45.18 | 36.24 |
| Pharmaceutical Tests | API | Comparison | Reference |
|---|---|---|---|
| in vivo | pyrantel | either to swallow whole, to chew and swallow, or to swallow previously pulverized tablets | Wesche 1994 [6] |
| raltegravir | administered after crushing | Krogstad 2021 [7] | |
| albendazole | crushed to a fine powder by mortar and pestle | Sawatdee 2019 [8] | |
| montelukast | intact chewable tablet with 240 mL water | Cánovas 2011 [9] | |
| montelukast | chewable tablet with warm water 240 mL | Zhu 2023 [10] | |
| montelukast | chewable tablet with 240 mL water | Zaid 2015 [11] | |
| sildenafil | chewable tablet with 50 mL water | Yoo 2017 [12] | |
| sildenafil | chewed until full disintegration and then swallowed with or without 250 mL water | Valenzuela 2011 [13] | |
| sildenafil | chewed and immediately swallowed | Marcelín-Jiménez 2012 [14] | |
| montelukast | intact chewable tablet | Knorr 2010 [15] | |
| in vitro dissolution | verapamil | whole tablet, crushed tablet using a commercial tablet crusher, or tablet chewed in the mouth and then expelled into dissolution fluid | El-Gazayerly 2004 [4] |
| lanthanum carbonate | whole tablet, crushed tablet, or the tablet fraction of less than 200 μm | Yang 2013 [3] | |
| montelukast | intact chewable tablet compared to ODT | Chen 2015 [16] | |
| albendazole | intact chewable tablet | Sawatdee 2019 [8] | |
| albendazole | intact chewable tablet | Kimaro 2019 [17] | |
| ciprofloxacin | intact chewable tablet | Usmani 2023 [2] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-Y.; Choi, S.-U. Pharmaceutical Equivalence of Film-Coated and Chewable Tablets: A Comparative Dissolution Study Using Pulverized Chewable Tablets. Pharmaceuticals 2024, 17, 1525. https://doi.org/10.3390/ph17111525
Park S-Y, Choi S-U. Pharmaceutical Equivalence of Film-Coated and Chewable Tablets: A Comparative Dissolution Study Using Pulverized Chewable Tablets. Pharmaceuticals. 2024; 17(11):1525. https://doi.org/10.3390/ph17111525
Chicago/Turabian StylePark, Suck-Yong, and Sung-Up Choi. 2024. "Pharmaceutical Equivalence of Film-Coated and Chewable Tablets: A Comparative Dissolution Study Using Pulverized Chewable Tablets" Pharmaceuticals 17, no. 11: 1525. https://doi.org/10.3390/ph17111525
APA StylePark, S.-Y., & Choi, S.-U. (2024). Pharmaceutical Equivalence of Film-Coated and Chewable Tablets: A Comparative Dissolution Study Using Pulverized Chewable Tablets. Pharmaceuticals, 17(11), 1525. https://doi.org/10.3390/ph17111525