
Amyotrophic Lateral Sclerosis: Insights and New Prospects in Disease Pathophysiology, Biomarkers and Therapies


Abstract
1. Introduction
2. Pathophysiology of ALS
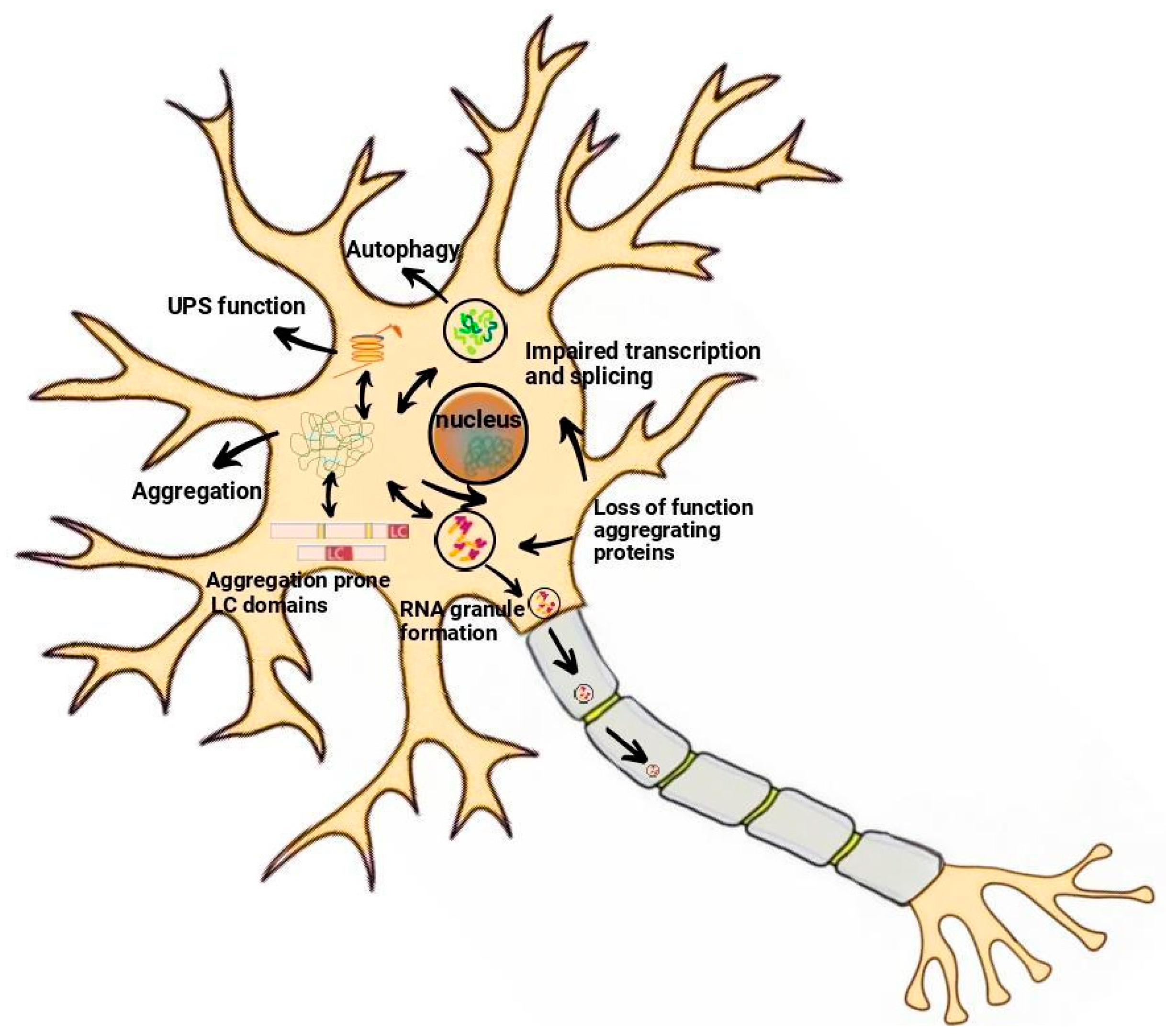
2.1. Protein Aggregation
2.2. Neuroinflammation
2.3. Oxidative Stress
2.4. Excitotoxicity
3. Biomarkers for Amyotrophic Lateral Sclerosis
4. Emerging Therapeutic Targets for ALS
4.1. Inhibiting Protein Aggregation by Small Molecules
4.2. Modulation of Neuroinflammatory Pathways
4.3. Antioxidant Therapies and Mitochondrial Protection
4.4. Gene Therapy Approaches
4.5. Stem Cell-Based Therapies for Neuroprotection and Regeneration
5. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Nigel Leigh, P. Amyotrophic Lateral Sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Blumenau, H. Basic Epidemiology of ALS. Eukaryon 2024, 20. [Google Scholar]
- Forsgren, L.; Almay, B.G.; Holmgren, G.; Wall, S. Epidemiology of Motor Neuron Disease in Northern Sweden. Acta Neurol. Scand. 1983, 68, 20–29. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The Epidemiology of Amyotrophic Lateral Sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 138, pp. 225–238. [Google Scholar]
- Manjaly, Z.R.; Scott, K.M.; Abhinav, K.; Wijesekera, L.; Ganesalingam, J.; Goldstein, L.H.; Janssen, A.; Dougherty, A.; Willey, E.; Stanton, B.R.; et al. The Sex Ratio in Amyotrophic Lateral Sclerosis: A Population Based Study. Amyotroph. Lateral Scler. 2010, 11, 439–442. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Piccininni, M. Amyotrophic Lateral Sclerosis Descriptive Epidemiology: The Origin of Geographic Difference. Neuroepidemiology 2019, 52, 93–103. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Sawada, H. Clinical Efficacy of Edaravone for the Treatment of Amyotrophic Lateral Sclerosis. Expert Opin. Pharmacother. 2017, 18, 735–738. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Özdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and Diagnostic Implications of Cortical Dysfunction in ALS. Nat. Rev. Neurol. 2016, 12, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of Endoplasmic Reticulum Proteostasis in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and Mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2413. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Braak, H.; Del Tredici, K.; Lemon, R.; Ludolph, A.C.; Kiernan, M.C. Cortical Influences Drive Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 917–924. [Google Scholar] [CrossRef]
- Van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef]
- Zakharova, M.N.; Abramova, A.A. Lower and Upper Motor Neuron Involvement and Their Impact on Disease Prognosis in Amyotrophic Lateral Sclerosis. Neural Regen. Res. 2022, 17, 65–73. [Google Scholar] [CrossRef]
- Lichtenstein, T.; Sprenger, A.; Weiss, K.; Große Hokamp, N.; Maintz, D.; Schlamann, M.; Fink, G.R.; Lehmann, H.C.; Henning, T.D. MRI DTI and PDFF as Biomarkers for Lower Motor Neuron Degeneration in ALS. Front. Neurosci. 2021, 15, 682126. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the Mechanisms Involved in Motor Neuron Degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef]
- Wu, D.-C.; Ré, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The Inflammatory NADPH Oxidase Enzyme Modulates Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Zeineddine, R.; Farrawell, N.E.; Lambert-Smith, I.A.; Yerbury, J.J. Addition of Exogenous SOD1 Aggregates Causes TDP-43 Mislocalisation and Aggregation. Cell Stress Chaperones 2017, 22, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Roos, P.M.; Vesterberg, O.; Nordberg, M. Metals in Motor Neuron Diseases. Exp. Biol. Med. 2006, 231, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.O.; Baccarelli, A. Metals and Neurotoxicology. J. Nutr. 2007, 137, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.N.T.; Lim, N.K.H.; Grubman, A.; Li, Q.-X.; Volitakis, I.; White, A.R.; Crouch, P.J. Increased Metal Content in the TDP-43(A315T) Transgenic Mouse Model of Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2014, 6, 15. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical Perspective on Oxidative Stress in Sporadic Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Filipi, T.; Hermanova, Z.; Tureckova, J.; Vanatko, O.; Anderova, A.M. Glial Cells-The Strategic Targets in Amyotrophic Lateral Sclerosis Treatment. J. Clin. Med. Res. 2020, 9, 261. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Maris, C.; Dominguez, C.; Allain, F.H.-T. The RNA Recognition Motif, a Plastic RNA-Binding Platform to Regulate Post-Transcriptional Gene Expression. FEBS J. 2005, 272, 2118–2131. [Google Scholar] [CrossRef] [PubMed]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-Binding Proteins: Modular Design for Efficient Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Cléry, A.; Blatter, M.; Allain, F.H.-T. RNA Recognition Motifs: Boring? Not Quite. Curr. Opin. Struct. Biol. 2008, 18, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.-H.; Chiang, C.-H.; Wang, Y.-T.; Doudeva, L.G.; Yuan, H.S. The Crystal Structure of TDP-43 RRM1-DNA Complex Reveals the Specific Recognition for UG- and TG-Rich Nucleic Acids. Nucleic Acids Res. 2014, 42, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H.-T. Molecular Basis of UG-Rich RNA Recognition by the Human Splicing Factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv. Genet. 2015, 91, 1–53. [Google Scholar]
- Moreno, F.; Rabinovici, G.D.; Karydas, A.; Miller, Z.; Hsu, S.C.; Legati, A.; Fong, J.; Schonhaut, D.; Esselmann, H.; Watson, C.; et al. A Novel Mutation P112H in the TARDBP Gene Associated with Frontotemporal Lobar Degeneration without Motor Neuron Disease and Abundant Neuritic Amyloid Plaques. Acta Neuropathol. Commun. 2015, 3, 19. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Grauffel, C.; Wu, L.-S.; Kuo, P.-H.; Doudeva, L.G.; Lim, C.; Shen, C.-K.J.; Yuan, H.S. Structural Analysis of Disease-Related TDP-43 D169G Mutation: Linking Enhanced Stability and Caspase Cleavage Efficiency to Protein Accumulation. Sci. Rep. 2016, 6, 21581. [Google Scholar] [CrossRef]
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical Diagnosis and Management of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef]
- Byrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of Familial Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 623–627. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The Genetics and Neuropathology of Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in Prion-like Domains in hnRNPA2B1 and hnRNPA1 Cause Multisystem Proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Nahm, M.; Lim, S.M.; Kim, Y.-E.; Park, J.; Noh, M.-Y.; Lee, S.; Roh, J.E.; Hwang, S.-M.; Park, C.-K.; Kim, Y.H.; et al. ANXA11 Mutations in ALS Cause Dysregulation of Calcium Homeostasis and Stress Granule Dynamics. Sci. Transl. Med. 2020, 12, eaax3993. [Google Scholar] [CrossRef]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-Wide Association Analyses Identify New Risk Variants and the Genetic Architecture of Amyotrophic Lateral Sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Yuva-Aydemir, Y.; Almeida, S.; Gao, F.-B. Insights into C9ORF72-Related ALS/FTD from Drosophila and iPSC Models. Trends Neurosci. 2018, 41, 457–469. [Google Scholar] [CrossRef]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense Oligonucleotide Therapy for Neurodegenerative Disease. J. Clin. Investig. 2006, 116, 2290–2296. [Google Scholar] [CrossRef]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef]
- Cao, B.; Gu, X.; Wei, Q.; Li, C.; Chen, Y.; Ou, R.; Hou, Y.; Zhang, L.; Li, T.; Song, W.; et al. Mutation Screening and Burden Analysis of GLT8D1 in Chinese Patients with Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2021, 101, 298.e17–298.e21. [Google Scholar] [CrossRef]
- Forsberg, K.; Graffmo, K.; Pakkenberg, B.; Weber, M.; Nielsen, M.; Marklund, S.; Brännström, T.; Andersen, P.M. Misfolded SOD1 Inclusions in Patients with Mutations in C9orf72 and Other ALS/FTD-Associated Genes. J. Neurol. Neurosurg. Psychiatry 2019, 90, 861–869. [Google Scholar] [CrossRef]
- Shu, S.; Lei, X.; Liu, F.; Cui, B.; Liu, Q.; Ding, Q.; Liu, M.S.; Li, X.G.; Cui, L.; Zhang, X. Mutation Screening of NEK1 in Chinese ALS Patients. Neurobiol. Aging 2018, 71, 267.e1–267.e4. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN Mutations Cause Autosomal Recessive Juvenile Amyotrophic Lateral Sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Soo, K.Y.; Halloran, M.; Sundaramoorthy, V.; Parakh, S.; Toth, R.P.; Southam, K.A.; McLean, C.A.; Lock, P.; King, A.; Farg, M.A.; et al. Rab1-Dependent ER-Golgi Transport Dysfunction Is a Common Pathogenic Mechanism in SOD1, TDP-43 and FUS-Associated ALS. Acta Neuropathol. 2015, 130, 679–697. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Glial Activation in Motor Neuron Disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as Determinants of Disease Progression in Inherited Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The Role of Neuroinflammation in Neurodegenerative Diseases: Current Understanding and Future Therapeutic Targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in Immunoregulation and Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Yang, H.; Wu, L.; Deng, H.; Chen, Y.; Zhou, H.; Liu, M.; Wang, S.; Zheng, L.; Zhu, L.; Lv, X. Anti-Inflammatory Protein TSG-6 Secreted by Bone Marrow Mesenchymal Stem Cells Attenuates Neuropathic Pain by Inhibiting the TLR2/MyD88/NF-κB Signaling Pathway in Spinal Microglia. J. Neuroinflamm. 2020, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Radandish, M.; Khalilian, P.; Esmaeil, N. The Role of Distinct Subsets of Macrophages in the Pathogenesis of MS and the Impact of Different Therapeutic Agents on These Populations. Front. Immunol. 2021, 12, 667705. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.C.; Couch, Y.; Sibson, N.; Turner, M.R. Inflammation and Neurovascular Changes in Amyotrophic Lateral Sclerosis. Mol. Cell. Neurosci. 2013, 53, 34–41. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a Neuroprotective to a Neurotoxic Microglial Phenotype in a Mouse Model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Ferri, A.; Valle, C.; Carrì, M.T. Mitochondria and ALS: Implications from Novel Genes and Pathways. Mol. Cell. Neurosci. 2013, 55, 44–49. [Google Scholar] [CrossRef]
- Barber, S.C.; Shaw, P.J. Oxidative Stress in ALS: Key Role in Motor Neuron Injury and Therapeutic Target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A Mitochondrial Origin for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis through CHCHD10 Involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- McGeer, E.G.; Olney, J.W.; McGeer, P.L. Kainic Acid as a Tool in Neurobiology; Raven Press: Randburg, South Africa, 1978; ISBN 9780890042793. [Google Scholar]
- Doble, A. The Role of Excitotoxicity in Neurodegenerative Disease: Implications for Therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Jin, L.; Dykes-Hoberg, M.; Kuncl, R.W. Chronic Inhibition of Glutamate Uptake Produces a Model of Slow Neurotoxicity. Proc. Natl. Acad. Sci. USA 1993, 90, 6591–6595. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The Role of Excitotoxicity in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ji, Y.; Wang, W.; Zhang, L.; Chen, Z.; Yu, M.; Shen, Y.; Ding, F.; Gu, X.; Sun, H. Amyotrophic Lateral Sclerosis: Molecular Mechanisms, Biomarkers, and Therapeutic Strategies. Antioxidants 2021, 10, 1012. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tejerina, D.; Llaurado, A.; Sotoca, J.; Lopez-Diego, V.; Vidal Taboada, J.M.; Salvado, M.; Juntas-Morales, R. Biofluid Biomarkers in the Prognosis of Amyotrophic Lateral Sclerosis: Recent Developments and Therapeutic Applications. Cells 2023, 12, 1180. [Google Scholar] [CrossRef]
- Sturmey, E.; Malaspina, A. Blood Biomarkers in ALS: Challenges, Applications and Novel Frontiers. Acta Neurol. Scand. 2022, 146, 375–388. [Google Scholar] [CrossRef]
- Lee, M.K.; Cleveland, D.W. Neuronal Intermediate Filaments. Annu. Rev. Neurosci. 1996, 19, 187–217. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as Biomarkers in Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Benatar, M.; Zhang, L.; Wang, L.; Granit, V.; Statland, J.; Barohn, R.; Swenson, A.; Ravits, J.; Jackson, C.; Burns, T.M.; et al. Validation of Serum Neurofilaments as Prognostic and Potential Pharmacodynamic Biomarkers for ALS. Neurology 2020, 95, e59–e69. [Google Scholar] [CrossRef]
- Steinacker, P.; Huss, A.; Mayer, B.; Grehl, T.; Grosskreutz, J.; Borck, G.; Kuhle, J.; Lulé, D.; Meyer, T.; Oeckl, P.; et al. Diagnostic and Prognostic Significance of Neurofilament Light Chain NF-L, but Not Progranulin and S100B, in the Course of Amyotrophic Lateral Sclerosis: Data from the German MND-Net. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 112–119. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022, 19, 1248–1258. [Google Scholar] [CrossRef]
- Staats, K.A.; Borchelt, D.R.; Tansey, M.G.; Wymer, J. Blood-Based Biomarkers of Inflammation in Amyotrophic Lateral Sclerosis. Mol. Neurodegener. 2022, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Kyheng, M.; Garçon, G.; Rolland, A.S.; Blasco, H.; Gelé, P.; Timothée Lenglet, T.; Veyrat-Durebex, C.; Corcia, P.; et al. A Ferroptosis–based Panel of Prognostic Biomarkers for Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 2918. [Google Scholar] [CrossRef] [PubMed]
- Lunetta, C.; Lizio, A.; Maestri, E.; Sansone, V.A.; Mora, G.; Miller, R.G.; Appel, S.H.; Chiò, A. Serum C-Reactive Protein as a Prognostic Biomarker in Amyotrophic Lateral Sclerosis. JAMA Neurol. 2017, 74, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; De Schaepdryver, M.; Floeter, M.K.; De Vocht, J.; Lamaire, N.; D’Hondt, A.; Traynor, B.; Poesen, K.; Van Damme, P. Prognostic Relationship of Neurofilaments, CHIT1, YKL-40 and MCP-1 in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 681–682. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Staff, N.P.; Bornschlegl, S.; Butler, G.W.; Maas, M.L.; Kazamel, M.; Zubair, A.; Gastineau, D.A.; Windebank, A.J.; Dietz, A.B. Comprehensive Immune Profiling Reveals Substantial Immune System Alterations in a Subset of Patients with Amyotrophic Lateral Sclerosis. PLoS ONE 2017, 12, e0182002. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Dupuis, L.; Kiernan, M.C. Paradox of Amyotrophic Lateral Sclerosis and Energy Metabolism. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1013–1014. [Google Scholar] [CrossRef]
- Ingre, C.; Chen, L.; Zhan, Y.; Termorshuizen, J.; Yin, L.; Fang, F. Lipids, Apolipoproteins, and Prognosis of Amyotrophic Lateral Sclerosis. Neurology 2020, 94, e1835–e1844. [Google Scholar] [CrossRef]
- Sol, J.; Jové, M.; Povedano, M.; Sproviero, W.; Domínguez, R.; Piñol-Ripoll, G.; Romero-Guevara, R.; Hye, A.; Al-Chalabi, A.; Torres, P.; et al. Lipidomic Traits of Plasma and Cerebrospinal Fluid in Amyotrophic Lateral Sclerosis Correlate with Disease Progression. Brain Commun. 2021, 3, fcab143. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS Is Associated with Greater Functional Decline and Shorter Survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Chen, X.; Li, S.; Shang, H. Abnormal Serum Iron-Status Indicator Changes in Amyotrophic Lateral Sclerosis (ALS) Patients: A Meta-Analysis. Front. Neurol. 2020, 11, 380. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Bovio, G.; Canosa, A.; Bertuzzo, D.; Galmozzi, F.; Cugnasco, P.; Clerico, M.; De Mercanti, S.; Bersano, E.; et al. Amyotrophic Lateral Sclerosis Outcome Measures and the Role of Albumin and Creatinine: A Population-Based Study. JAMA Neurol. 2014, 71, 1134–1142. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Garofalo, D.C.; Santella, R.M.; Sorenson, E.J.; Oskarsson, B.; Fernandes, J.A.M., Jr.; Andrews, H.; Hupf, J.; Gilmore, M.; Heitzman, D.; et al. Plasma Creatinine and Oxidative Stress Biomarkers in Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-P.; Wei, Q.-Q.; Ou, R.-W.; Hou, Y.-B.; Zhang, L.-Y.; Yuan, X.-Q.; Yao, Y.-Q.; Jia, D.-S.; Zhang, Q.; Li, W.-X.; et al. Creatine Kinase in the Diagnosis and Prognostic Prediction of Amyotrophic Lateral Sclerosis: A Retrospective Case-Control Study. Neural Regen. Res. 2021, 16, 591–595. [Google Scholar] [PubMed]
- Kläppe, U.; Chamoun, S.; Shen, Q.; Finn, A.; Evertsson, B.; Zetterberg, H.; Blennow, K.; Press, R.; Samuelsson, K.; Månberg, A.; et al. Cardiac Troponin T Is Elevated and Increases Longitudinally in ALS Patients. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Weskamp, K.; Barmada, S.J. TDP43 and RNA Instability in Amyotrophic Lateral Sclerosis. Brain Res. 2018, 1693, 67–74. [Google Scholar] [CrossRef]
- Ichiyanagi, N.; Fujimori, K.; Yano, M.; Ishihara-Fujisaki, C.; Sone, T.; Akiyama, T.; Okada, Y.; Akamatsu, W.; Matsumoto, T.; Ishikawa, M.; et al. Establishment of In Vitro FUS-Associated Familial Amyotrophic Lateral Sclerosis Model Using Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 496–510. [Google Scholar] [CrossRef]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational Analysis Reveals the FUS Homolog TAF15 as a Candidate Gene for Familial Amyotrophic Lateral Sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156B, 285–290. [Google Scholar] [CrossRef]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.-J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the Role of the FUS/TLS-Related Gene EWSR1 in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 Intermediate-Length Polyglutamine Expansions Are Associated with Increased Risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Honda, H.; Hamasaki, H.; Wakamiya, T.; Koyama, S.; Suzuki, S.O.; Fujii, N.; Iwaki, T. Loss of hnRNPA1 in ALS Spinal Cord Motor Neurons with TDP-43-Positive Inclusions. Neuropathology 2015, 35, 37–43. [Google Scholar] [CrossRef]
- Kao, C.S.; van Bruggen, R.; Kim, J.R.; Chen, X.X.L.; Chan, C.; Lee, J.; Cho, W.I.; Zhao, M.; Arndt, C.; Maksimovic, K.; et al. Selective Neuronal Degeneration in MATR3 S85C Knock-in Mouse Model of Early-Stage ALS. Nat. Commun. 2020, 11, 5304. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhao, Y.; Zhou, X.; Luan, J.; Cui, Y.; Han, J. Comparison of the Extraction and Determination of Serum Exosome and miRNA in Serum and the Detection of miR-27a-3p in Serum Exosome of ALS Patients. Intractable Rare Dis. Res. 2018, 7, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Duan, Y.; Duan, G.; Wang, Q.; Zhang, K.; Deng, X.; Qian, B.; Gu, J.; Ma, Z.; Zhang, S.; et al. Stress Induces Dynamic, Cytotoxicity-Antagonizing TDP-43 Nuclear Bodies via Paraspeckle LncRNA NEAT1-Mediated Liquid-Liquid Phase Separation. Mol. Cell 2020, 79, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Errichelli, L.; Dini Modigliani, S.; Laneve, P.; Colantoni, A.; Legnini, I.; Capauto, D.; Rosa, A.; De Santis, R.; Scarfò, R.; Peruzzi, G.; et al. FUS Affects Circular RNA Expression in Murine Embryonic Stem Cell-Derived Motor Neurons. Nat. Commun. 2017, 8, 14741. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Malik, R.; Meng, H.; Wongkongkathep, P.; Corrales, C.I.; Sepanj, N.; Atlasi, R.S.; Klärner, F.-G.; Schrader, T.; Spencer, M.J.; Loo, J.A.; et al. The Molecular Tweezer CLR01 Inhibits Aberrant Superoxide Dismutase 1 (SOD1) Self-Assembly in Vitro and in the G93A-SOD1 Mouse Model of ALS. J. Biol. Chem. 2019, 294, 3501–3513. [Google Scholar] [CrossRef]
- Samanta, N.; Ruiz-Blanco, Y.B.; Fetahaj, Z.; Gnutt, D.; Lantz, C.; Loo, J.A.; Sanchez-Garcia, E.; Ebbinghaus, S. Superoxide Dismutase 1 Folding Stability as a Target for Molecular Tweezers in SOD1-Related Amyotrophic Lateral Sclerosis. Chembiochem 2022, 23, e202200396. [Google Scholar] [CrossRef]
- Koh, S.-H.; Lee, S.M.; Kim, H.Y.; Lee, K.-Y.; Lee, Y.J.; Kim, H.-T.; Kim, J.; Kim, M.-H.; Hwang, M.S.; Song, C.; et al. The Effect of Epigallocatechin Gallate on Suppressing Disease Progression of ALS Model Mice. Neurosci. Lett. 2006, 395, 103–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Benmohamed, R.; Huang, H.; Chen, T.; Voisine, C.; Morimoto, R.I.; Kirsch, D.R.; Silverman, R.B. Arylazanylpyrazolone Derivatives as Inhibitors of Mutant Superoxide Dismutase 1 Dependent Protein Aggregation for the Treatment of Amyotrophic Lateral Sclerosis. J. Med. Chem. 2013, 56, 2665–2675. [Google Scholar] [CrossRef]
- Uechi, H.; Sridharan, S.; Nijssen, J.; Bilstein, J.; Iglesias-Artola, J.M.; Kishigami, S.; Casablancas-Antras, V.; Poser, I.; Martinez, E.J.; Boczek, E.; et al. Small Molecule Modulation of a Redox-Sensitive Stress Granule Protein Dissolves Stress Granules with Beneficial Outcomes for Familial Amyotrophic Lateral Sclerosis Models. bioRxiv 2024, bioRxiv:721001. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, H.; Liang, W.; Yang, Y.; Cong, C.; Wang, Y.; Wang, S.; Wang, X.; Wang, D.; Huo, D.; et al. Diphenyl Diselenide Protects Motor Neurons through Inhibition of Microglia-Mediated Inflammatory Injury in Amyotrophic Lateral Sclerosis. Pharmacol. Res. 2021, 165, 105457. [Google Scholar] [CrossRef] [PubMed]
- Ibarburu, S.; Kovacs, M.; Varela, V.; Rodríguez-Duarte, J.; Ingold, M.; Invernizzi, P.; Porcal, W.; Arévalo, A.P.; Perelmuter, K.; Bollati-Fogolín, M.; et al. A Nitroalkene Benzoic Acid Derivative Targets Reactive Microglia and Prolongs Survival in an Inherited Model of ALS via NF-κB Inhibition. Neurotherapeutics 2021, 18, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-Mediated Neuroinflammation in CNS Diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Krauser, J.A.; Jin, Y.; Walles, M.; Pfaar, U.; Sutton, J.; Wiesmann, M.; Graf, D.; Pflimlin-Fritschy, V.; Wolf, T.; Camenisch, G.; et al. Phenotypic and Metabolic Investigation of a CSF-1R Kinase Receptor Inhibitor (BLZ945) and Its Pharmacologically Active Metabolite. Xenobiotica 2015, 45, 107–123. [Google Scholar] [CrossRef]
- Martínez-Muriana, A.; Mancuso, R.; Francos-Quijorna, I.; Olmos-Alonso, A.; Osta, R.; Perry, V.H.; Navarro, X.; Gomez-Nicola, D.; López-Vales, R. CSF1R Blockade Slows the Progression of Amyotrophic Lateral Sclerosis by Reducing Microgliosis and Invasion of Macrophages into Peripheral Nerves. Sci. Rep. 2016, 6, 25663. [Google Scholar] [CrossRef]
- Crisafulli, S.G.; Brajkovic, S.; Cipolat Mis, M.S.; Parente, V.; Corti, S. Therapeutic Strategies Under Development Targeting Inflammatory Mechanisms in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 2789–2813. [Google Scholar] [CrossRef]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; NYGC ALS Consortium; Fagegaltier, D.; Harris, B.T.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Alsuntangled Group; Richard, B. ALSUntangled 55: Vitamin E (α-Tocopherol). Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 154–160. [Google Scholar]
- Desnuelle, C.; Dib, M.; Garrel, C.; Favier, A. A Double-Blind, Placebo-Controlled Randomized Clinical Trial of α-Tocopherol (vitamin E) in the Treatment of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2001, 2, 9–18. [Google Scholar] [CrossRef]
- Ascherio, A.; Weisskopf, M.G.; O’reilly, E.J.; Jacobs, E.J.; McCullough, M.L.; Calle, E.E.; Cudkowicz, M.; Thun, M.J. Vitamin E Intake and Risk of Amyotrophic Lateral Sclerosis. Ann. Neurol. 2005, 57, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Isonaka, R.; Hiruma, H.; Katakura, T.; Kawakami, T. Inhibition of Superoxide Dismutase Selectively Suppresses Growth of Rat Spinal Motor Neurons: Comparison with Phosphorylated Neurofilament-Containing Spinal Neurons. Brain Res. 2011, 1425, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; O’Reilly, É.J.; Fondell, E.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Intakes of Vitamin C and Carotenoids and Risk of Amyotrophic Lateral Sclerosis: Pooled Results from 5 Cohort Studies. Ann. Neurol. 2013, 73, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Krishnaraj, R.N.; Kumari, S.S.S.; Mukhopadhyay, S.S. Antagonistic Molecular Interactions of Photosynthetic Pigments with Molecular Disease Targets: A New Approach to Treat AD and ALS. J. Recept. Signal Transduct. Res. 2016, 36, 67–71. [Google Scholar] [CrossRef]
- Mantilla, C.B.; Ermilov, L.G. The Novel TrkB Receptor Agonist 7,8-Dihydroxyflavone Enhances Neuromuscular Transmission. Muscle Nerve 2012, 45, 274–276. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, S.Y.; Wang, X.D.; Jiang, H.Q.; Yang, Y.Q.; Wang, Y.; Cheng, J.L.; Zhang, C.T.; Liang, W.W.; Feng, H.L. Fisetin Exerts Antioxidant and Neuroprotective Effects in Multiple Mutant hSOD1 Models of Amyotrophic Lateral Sclerosis by Activating ERK. Neuroscience 2018, 379, 152–166. [Google Scholar] [CrossRef]
- Ip, P.; Sharda, P.R.; Cunningham, A.; Chakrabartty, S.; Pande, V.; Chakrabartty, A. Quercitrin and Quercetin 3-β-D-Glucoside as Chemical Chaperones for the A4V SOD1 ALS-Causing Mutant. Protein Eng. Des. Sel. 2017, 30, 431–440. [Google Scholar] [CrossRef]
- Jiang, H.; Tian, X.; Guo, Y.; Duan, W.; Bu, H.; Li, C. Activation of Nuclear Factor Erythroid 2-Related Factor 2 Cytoprotective Signaling by Curcumin Protect Primary Spinal Cord Astrocytes against Oxidative Toxicity. Biol. Pharm. Bull. 2011, 34, 1194–1197. [Google Scholar] [CrossRef]
- Lu, J.; Duan, W.; Guo, Y.; Jiang, H.; Li, Z.; Huang, J.; Hong, K.; Li, C. Mitochondrial Dysfunction in Human TDP-43 Transfected NSC34 Cell Lines and the Protective Effect of Dimethoxy Curcumin. Brain Res. Bull. 2012, 89, 185–190. [Google Scholar] [CrossRef]
- Dong, H.; Xu, L.; Wu, L.; Wang, X.; Duan, W.; Li, H.; Li, C. Curcumin Abolishes Mutant TDP-43 Induced Excitability in a Motoneuron-like Cellular Model of ALS. Neuroscience 2014, 272, 141–153. [Google Scholar] [CrossRef]
- Ghasemi, F.; Bagheri, H.; Barreto, G.E.; Read, M.I.; Sahebkar, A. Effects of Curcumin on Microglial Cells. Neurotox. Res. 2019, 36, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol Improves Motoneuron Function and Extends Survival in SOD1G93A ALS Mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef]
- Wang, H.; O’Reilly, É.J.; Weisskopf, M.G.; Logroscino, G.; McCullough, M.L.; Schatzkin, A.; Kolonel, L.N.; Ascherio, A. Vitamin E Intake and Risk of Amyotrophic Lateral Sclerosis: A Pooled Analysis of Data from 5 Prospective Cohort Studies. Am. J. Epidemiol. 2011, 173, 595–602. [Google Scholar] [CrossRef]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 Administration Increases Brain Mitochondrial Concentrations and Exerts Neuroprotective Effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Pattee, G.L. Creatine and Coenzyme Q10 in the Treatment of ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1 (Suppl. 4), 17–20. [Google Scholar] [CrossRef]
- Beal, M.F. Oxidatively Modified Proteins in Aging and Disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Bartels, C.; Pölking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Hüther, G.; et al. Reduced Oxidative Damage in ALS by High-Dose Enteral Melatonin Treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef]
- Yoshino, H. Edaravone for the Treatment of Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2019, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, G.M.; Gowing, G.; Latter, J.; Chen, M.; Vit, J.-P.; Staggenborg, K.; Avalos, P.; Alkaslasi, M.; Ferraiuolo, L.; Likhite, S.; et al. Delayed Disease Onset and Extended Survival in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis after Suppression of Mutant SOD1 in the Motor Cortex. J. Neurosci. 2014, 34, 15587–15600. [Google Scholar] [CrossRef]
- Miller, T.M.; Kaspar, B.K.; Kops, G.J.; Yamanaka, K.; Christian, L.J.; Gage, F.H.; Cleveland, D.W. Virus-Delivered Small RNA Silencing Sustains Strength in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2005, 57, 773–776. [Google Scholar] [CrossRef]
- Rizvanov, A.A.; Mukhamedyarov, M.A.; Palotás, A.; Islamov, R.R. Retrogradely Transported siRNA Silences Human Mutant SOD1 in Spinal Cord Motor Neurons. Exp. Brain Res. 2009, 195, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Biferi, M.G.; Cohen-Tannoudji, M.; Cappelletto, A.; Giroux, B.; Roda, M.; Astord, S.; Marais, T.; Bos, C.; Voit, T.; Ferry, A.; et al. A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Mol. Ther. 2017, 25, 2038–2052. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, L.H.; Rothstein, J.D.; Shaw, P.J.; Babu, S.; Benatar, M.; Bucelli, R.C.; Genge, A.; Glass, J.D.; Hardiman, O.; Libri, V.; et al. Safety, tolerability, and pharmacokinetics of antisense oligonucleotide BIIB078 in adults with C9orf72-associated amyotrophic lateral sclerosis: A phase 1, randomised, double blinded, placebo-controlled, multiple ascending dose study. Lancet Neurol. 2024, 23, 901–912. [Google Scholar] [CrossRef]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An Antisense Oligonucleotide against SOD1 Delivered Intrathecally for Patients with SOD1 Familial Amyotrophic Lateral Sclerosis: A Phase 1, Randomised, First-in-Man Study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo Genome Editing Improves Motor Function and Extends Survival in a Mouse Model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The Deletion of Mutant SOD1 via CRISPR/Cas9/sgRNA Prolongs Survival in an Amyotrophic Lateral Sclerosis Mouse Model. Gene Ther. 2020, 27, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Pribadi, M.; Yang, Z.; Kim, T.S.; Swartz, E.W.; Huang, A.Y.; Chen, J.A.; Dokuru, D.; Baek, J.; Gao, F.; Fua, A.T.; et al. CRISPR-Cas9 Targeted Deletion of the C9orf72 Repeat Expansion Mutation Corrects Cellular Phenotypes in Patient-Derived iPS Cells. bioRxiv 2016, bioRxiv:051193. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Yang, D.; Pribadi, M.; Kim, T.S.; Krishnan, G.; Choi, S.Y.; Lee, S.; Coppola, G.; Gao, F.-B. Partial Inhibition of the Overactivated Ku80-Dependent DNA Repair Pathway Rescues Neurodegeneration in C9ORF72-ALS/FTD. Proc. Natl. Acad. Sci. USA 2019, 116, 9628–9633. [Google Scholar] [CrossRef]
- Zhao, C.-P.; Zhang, C.; Zhou, S.-N.; Xie, Y.-M.; Wang, Y.-H.; Huang, H.; Shang, Y.-C.; Li, W.-Y.; Zhou, C.; Yu, M.-J.; et al. Human Mesenchymal Stromal Cells Ameliorate the Phenotype of SOD1-G93A ALS Mice. Cytotherapy 2007, 9, 414–426. [Google Scholar] [CrossRef]
- Suzuki, M.; McHugh, J.; Tork, C.; Shelley, B.; Hayes, A.; Bellantuono, I.; Aebischer, P.; Svendsen, C.N. Direct Muscle Delivery of GDNF with Human Mesenchymal Stem Cells Improves Motor Neuron Survival and Function in a Rat Model of Familial ALS. Mol. Ther. 2008, 16, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Fagioli, F.; Boccaletti, R.; Mareschi, K.; Oliveri, G.; Olivieri, C.; Pastore, I.; Marasso, R.; Madon, E. Stem Cell Therapy in Amyotrophic Lateral Sclerosis: A Methodological Approach in Humans. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2003, 4, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Mareschi, K.; Ferrero, I.; Vassallo, E.; Oliveri, G.; Boccaletti, R.; Testa, L.; Livigni, S.; Fagioli, F. Autologous Mesenchymal Stem Cells: Clinical Applications in Amyotrophic Lateral Sclerosis. Neurol. Res. 2006, 28, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Mareschi, K.; Ferrero, I.; Miglioretti, M.; Stecco, A.; Servo, S.; Carriero, A.; Monaco, F.; Fagioli, F. Mesenchymal Stromal Cell Transplantation in Amyotrophic Lateral Sclerosis: A Long-Term Safety Study. Cytotherapy 2012, 14, 56–60. [Google Scholar] [CrossRef]
- Petrou, P.; Kassis, I.; Yaghmour, N.E.; Ginzberg, A.; Karussis, D. A Phase II Clinical Trial with Repeated Intrathecal Injections of Autologous Mesenchymal Stem Cells in Patients with Amyotrophic Lateral Sclerosis. Front. Biosci. 2021, 26, 693–706. [Google Scholar]
- Cudkowicz, M.E.; Lindborg, S.R.; Goyal, N.A.; Miller, R.G.; Burford, M.J.; Berry, J.D.; Nicholson, K.A.; Mozaffar, T.; Katz, J.S.; Jenkins, L.J.; et al. A Randomized Placebo-Controlled Phase 3 Study of Mesenchymal Stem Cells Induced to Secrete High Levels of Neurotrophic Factors in Amyotrophic Lateral Sclerosis. Muscle Nerve 2022, 65, 291–302. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Gene/Protein | Role of Gene/Protein | Association with ALS | Reference |
|---|---|---|---|
| Annexin A11 (ANXA11) | Phosphoinositide-binding protein involved in RNA transport and lysosome interaction | Mutations impair RNA transport, triggering neuronal apoptosis. The abnormal protein aggregation linked to ALS | [45] |
| C21orf2 | Interacts with NEK1, involved in microtubule assembly, DNA repair, and mitochondrial function | Interrelated with C9orf72 and NEK1 mechanisms, thereby the mutation is associated with ALS | [46] |
| C9orf72 | Involved in vesicle transport, lysosomal homeostasis, mTORC1 signaling, and autophagy | The repeated expansion of GGGGCC causes neuroinflammation leading to familial ALS (fALS) | [47] |
| CHCHD10 | Mitochondrial protein linked to clinical lineage of ALS-FTD (frontotemporal dementia) | Mitochondrial dysfunction may play a role in ALS and FTD pathogenesis | [48] |
| Cyclin F (CCNF) | Part of E3 ubiquitin-protein ligase complex | Mutations increase TDP-43 aggregates, leading to ALS | [49] |
| Glycosyltransferase 8 domain 1 (GLT8D1) | Encodes glycosyltransferase, potentially toxic when mutated | Mutation of glycosyltransferase is associated with FALS development | [50] |
| Kinesin family member 5A (KIF5A) | Member of kinesin family, involved in cargo transport | Mutation in the C-terminal cargo-binding tail domain of KIF5A | [51] |
| NIMA-related kinase 1 (NEK1) | Kinase involved in cell cycle progression and mitosis | Variants of this kinase areassociated with fALS risk | [52] |
| SPG11 (spatacsin) | Key role in axon maintenance, synaptic vesicle transport, and autophagy | Mutations cause juvenile ALS with slower progression than adult ALS | [53] |
| Superoxide Dismutase 1 (SOD1) | Powerful antioxidant enzyme that protects against superoxide free radicals | 170 mutations linked to ALS; misfolded SOD1 spreads in a prion-like manner; causes motor neuron death and enhanced apoptosis. Early ER–Golgi transport dysfunction in mice | [54] |
| TANK-binding kinase 1 (TBK1) | Member of IκB kinase family, involved in innate immune signaling | Linked to TDP-43 proteinopathies. Insufficient TBK1 function causes ALS and FTD | [55] |
| Therapy | Advantages | Disadvantages/Limitations | References | |
|---|---|---|---|---|
| Small Molecules | Molecular tweezers—CLR01 | Reduces the aggregation of SOD1 in the spinal cord of mouse models. | Did not improve the motor function of the mice. | [108] |
| Epigallocatechingallate (EGCG) | Significant delay in the onset of symptoms and prolonged survival in ALS mice. | Extensive testing in ALS models is still required for therapeutic efficacy. | [110] | |
| Pyrazolone derivatives | Improved motor function and extended survival in ALS mouse models. | [111] | ||
| Lipoamide | Modulates stress granule proteins such as FUS and TDP-43. | [112] | ||
| Microglial Activation Inhibitors | Diphenyl diselenide (DPDS) | Suppresses microglial activation by inhibiting the NLRP3 inflammasome and the IκB/NF-κB pathways. | Understanding the regulation and release of microglial-associated inhibitors is crucial for assessing therapeutic potential. | [113] |
| Nitroalkene Benzoic Acid Derivative (BANA) | Reduced reactive microglia and prolonged survival in SOD1G93A mice. | [114] | ||
| RIPK1—SAR443820 | Reduces microglial inflammation. | [115] | ||
| CSF1R kinase inhibitor, BLZ945 | Depletes microglia and enhances remyelination in the cortex and striatum in mice. | [116,117] | ||
| GW2580 | Reduces microglial proliferation and slows disease progression. | [118] | ||
| Drugs—masitinib, ibudilast, and NP001 | Target microglia by inhibiting the production of molecules that play a crucial role in the inflammatory response. | [119] | ||
| Antioxidant Therapies and Mitochondrial Protection | Vitamin E | Delays the onset of disease in the SOD1 mutant mouse model. Enhances the systemic antioxidant defense mechanisms in patients with ALS. Reduced risk of mortality in ALS patients. | Need for more studies due to the limited availability of results, which are often contradictory, inconclusive, or statistically insignificant. | [121,122,123] |
| Plant pigments—astaxanthin and lycopene | Antioxidant properties. | [124] | ||
| Carotenoids | Prevent ALS and/or delay its onset. Therapeutic molecule for treating neuroinflammation and apoptosis in ALS patients | [125,126] | ||
| Flavonoids—Fisetin, and quercetin | Improves motor deficits and reduces ROS levels. Inhibits SOD1 aggregation and enhances stability | [127,128,129] | ||
| Salvianolic acid A, 7,8-dihydroxyflavone | Inhibits SOD1 aggregation and enhances stability | [129] | ||
| Curcumin | Reduces oxidative stress, inflammation, and protein aggregation | [130,131,132,133] | ||
| Resveratrol | Upregulates sirtuin 1 (SIRT1), delays ALS onset, and enhances motor neuron survival. | [134,135] | ||
| Coenzyme Q10 | Extends survival in ALS mice and increases brain mitochondrial levels. | [136,137,138] | ||
| Melatonin | Delayed disease progression and improved survival rates in the SOD1G93A transgenic mouse model. | [111,139] | ||
| Gene Therapy | AAV-mediated siRNA delivery | Reduced SOD1 levels in ALS. | Challenges such as in vivo instability, siRNA specificity, and potential toxicity remain for RNAi-based gene therapy. | [141,142,143,144] |
| Antisense oligonucleotides (ASOs) | Slowed disease progression in animal models. | [48] | ||
| CRISPR-Cas9 | Prevented disease progression in SOD1 mice using AAV-SaCas9-sgRNA. Improved life expectancy by 54.6%. | Ethical and safety concerns. | [148,149] | |
| Stem Cell-based Therapies | MSC-SOD1G93A mice model | Delay in motor neuron degeneration, improved motor function, and extended lifespan. | Limited migration into CNS. | [152] |
| Autologous bone marrow MSCs | Linear decline in FVC and ALSFRS was noted. | Absence of control group and small sample size. | [155] | |
| Autologous BM-MSCs | Showed temporary improvements in ALSFRS-R score with good safety profiles. | Limited sample size and the heterogeneity of individual disease progression. | [157] | |
| NurOwn® autologous MSCs | Safe, increase of neurotrophic factors. | The trial did not meet its primary outcome. | [158] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Khayri, J.M.; Ravindran, M.; Banadka, A.; Vandana, C.D.; Priya, K.; Nagella, P.; Kukkemane, K. Amyotrophic Lateral Sclerosis: Insights and New Prospects in Disease Pathophysiology, Biomarkers and Therapies. Pharmaceuticals 2024, 17, 1391. https://doi.org/10.3390/ph17101391
Al-Khayri JM, Ravindran M, Banadka A, Vandana CD, Priya K, Nagella P, Kukkemane K. Amyotrophic Lateral Sclerosis: Insights and New Prospects in Disease Pathophysiology, Biomarkers and Therapies. Pharmaceuticals. 2024; 17(10):1391. https://doi.org/10.3390/ph17101391
Chicago/Turabian StyleAl-Khayri, Jameel M., Mamtha Ravindran, Akshatha Banadka, Chendanda Devaiah Vandana, Kushalva Priya, Praveen Nagella, and Kowshik Kukkemane. 2024. "Amyotrophic Lateral Sclerosis: Insights and New Prospects in Disease Pathophysiology, Biomarkers and Therapies" Pharmaceuticals 17, no. 10: 1391. https://doi.org/10.3390/ph17101391
APA StyleAl-Khayri, J. M., Ravindran, M., Banadka, A., Vandana, C. D., Priya, K., Nagella, P., & Kukkemane, K. (2024). Amyotrophic Lateral Sclerosis: Insights and New Prospects in Disease Pathophysiology, Biomarkers and Therapies. Pharmaceuticals, 17(10), 1391. https://doi.org/10.3390/ph17101391