

In Vivo Effects of a GHR Synthesis Inhibitor During Prolonged Treatment in Dogs
 , , , and
, , , and
Abstract
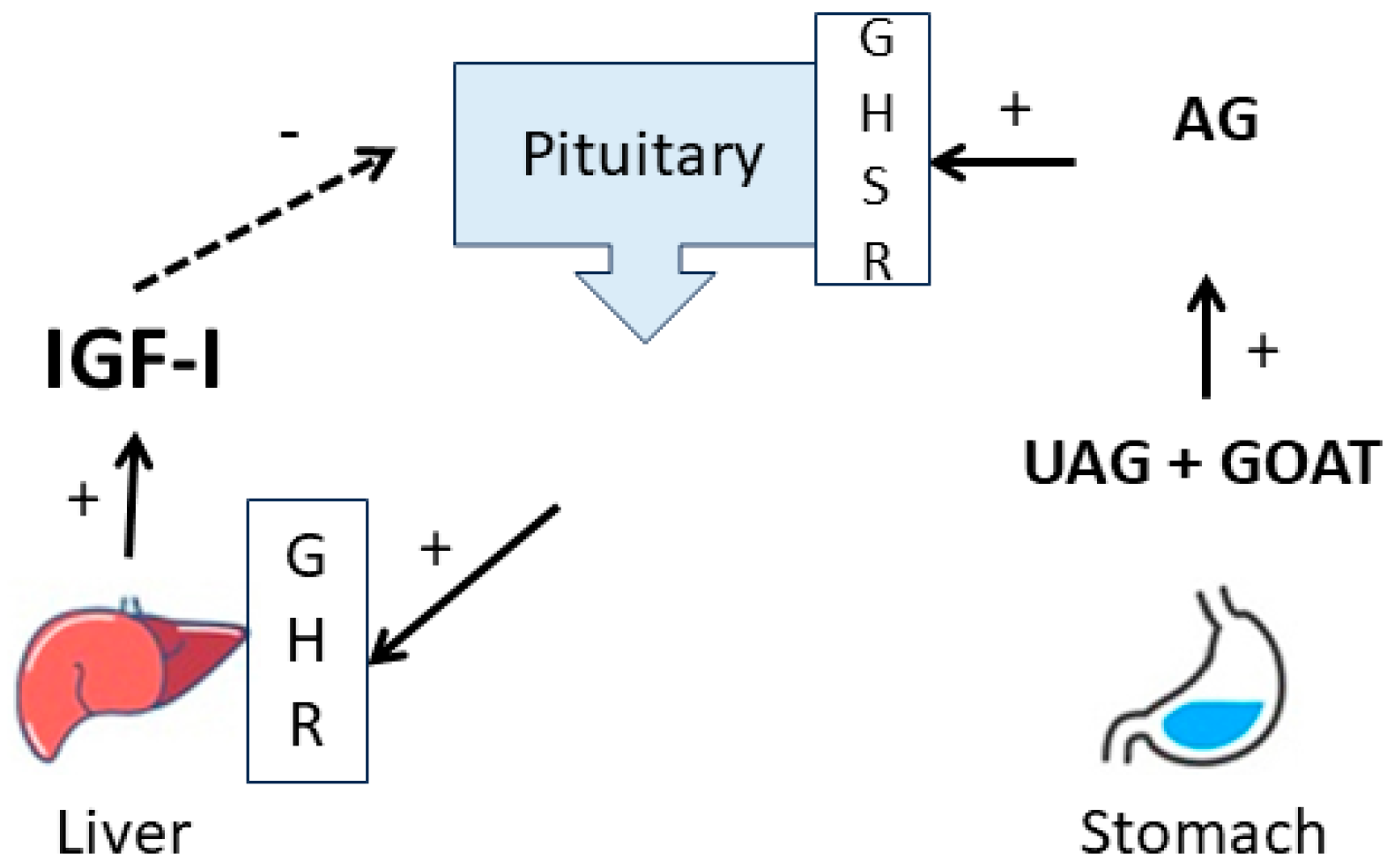
1. Introduction
2. Results
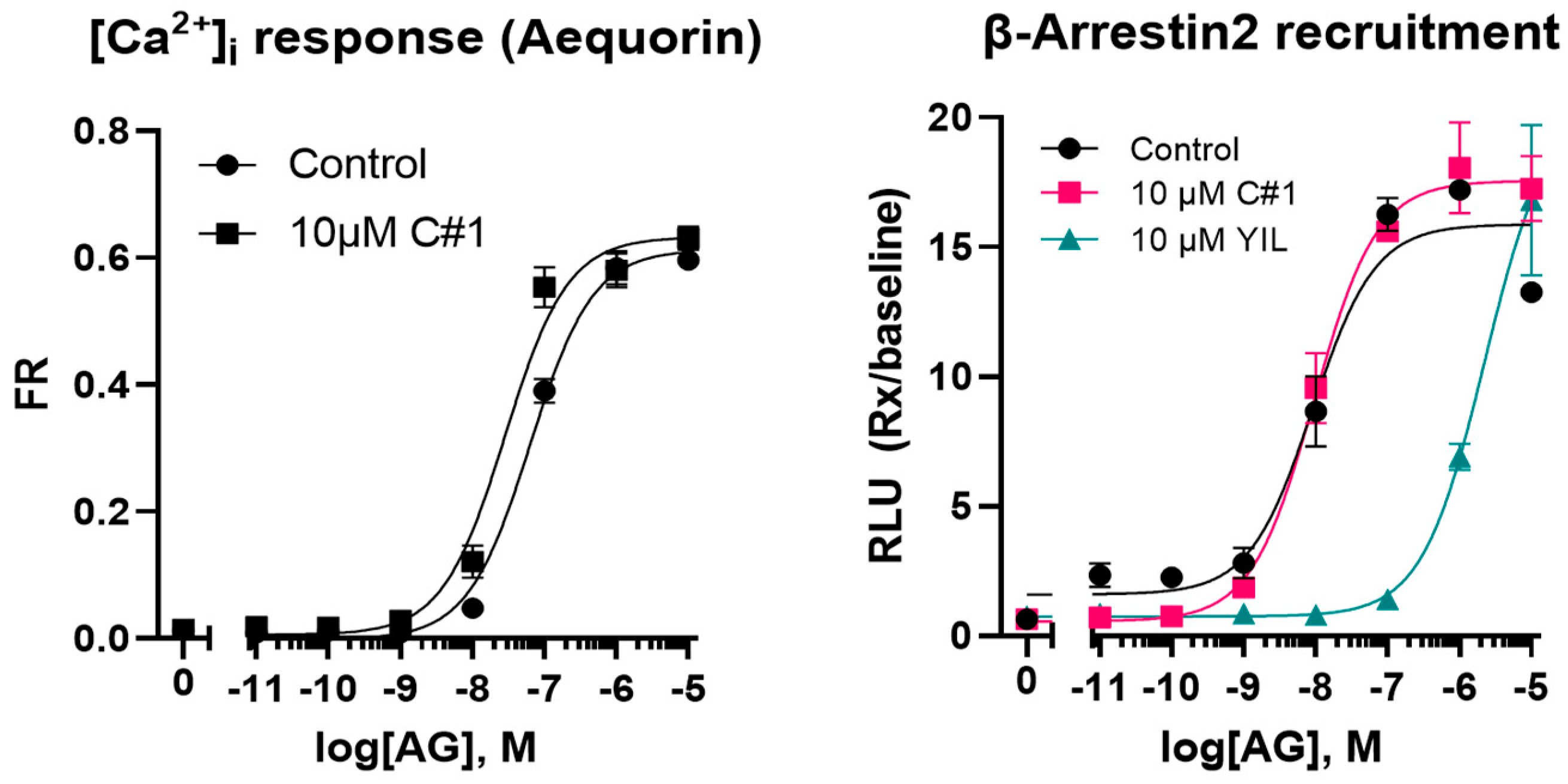
2.1. Physical and Metabolic Properties of C#1
2.2. Pilot Administration of C#1
2.3. Efficacy and Toxicity Trial During Prolonged Treatment of Female Beagle Dogs
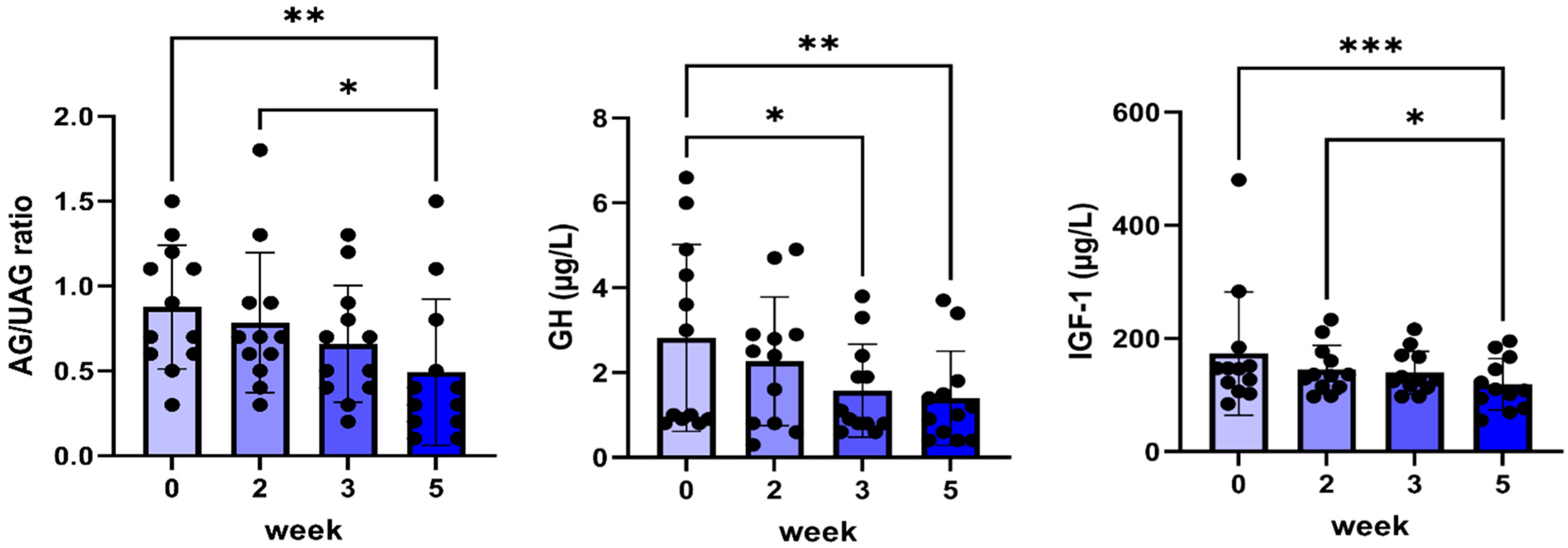
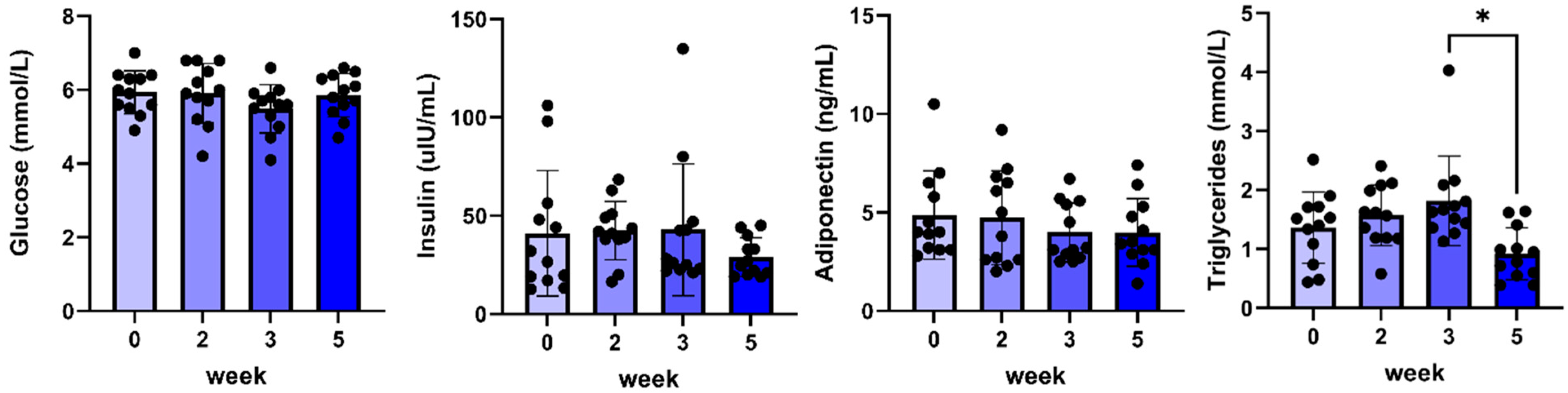
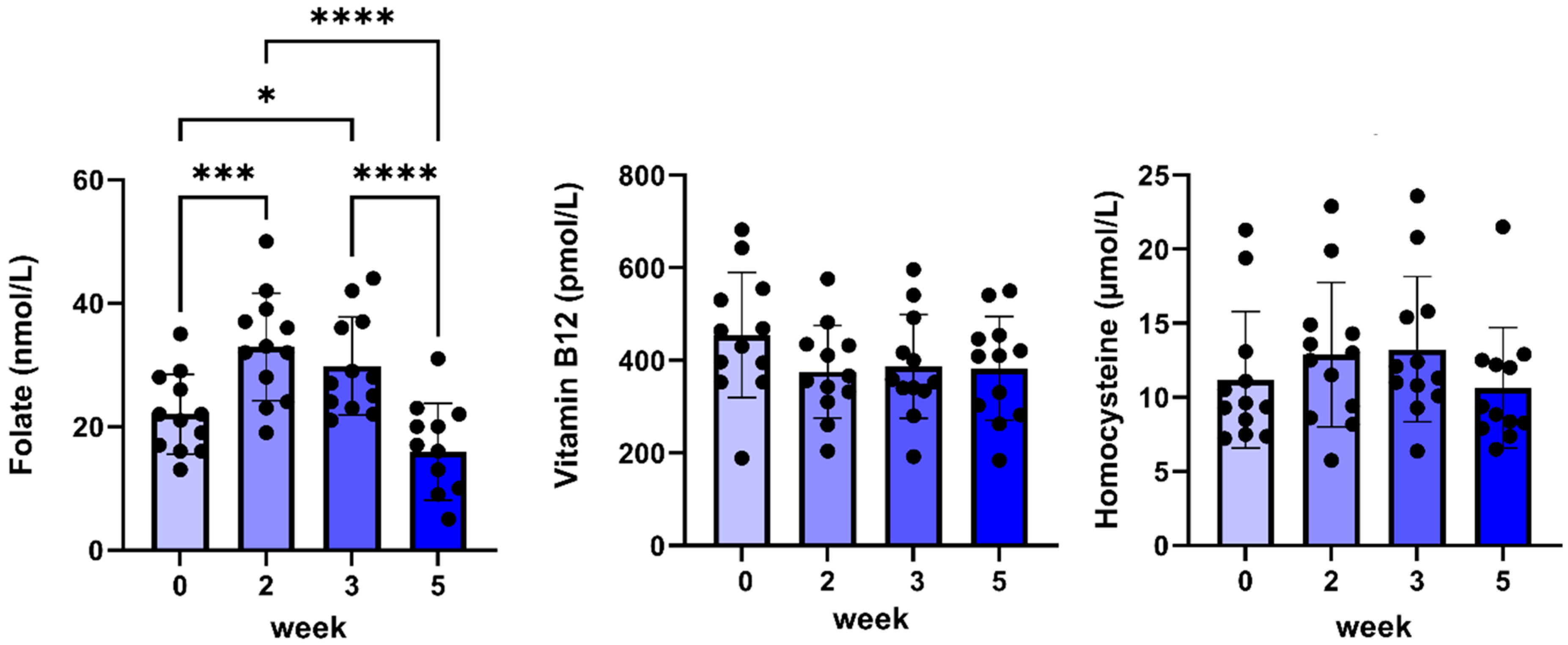
2.4. Results After 5 Weeks of C#1 Administration
2.5. Results from 90 Days of C#1 Administration
2.6. Pathology
3. Discussion
3.1. GHR Inhibition
3.2. DHFR Inhibition
3.3. Toxicity
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. Assays
4.4. LC/MS Analysis of C#1
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Apaydin, T.; Zonis, S.; Zhou, C.; Valencia, C.W.; Barrett, R.; Strous, G.J.; Mol, J.A.; Chesnokova, V.; Melmed, S. WIP1 is a novel specific target for growth hormone action. iScience 2023, 26, 108117. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. [Google Scholar] [CrossRef] [PubMed]
- Bergan-Roller, H.E.; Sheridan, M.A. The growth hormone signaling system: Insights into coordinating the anabolic and catabolic actions of growth hormone. Gen. Comp. Endocrinol. 2018, 258, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, Y.; Lee, C.M.; Müller, A.F.; Brooks, A.J. GHR signalling: Receptor activation and degradation mechanisms. Mol. Cell. Endocrinol. 2021, 520, 111075. [Google Scholar] [CrossRef] [PubMed]
- Eigenmann, J.E.; Eigenmann, R.Y. Radioimmunoassay of canine growth hormone. Eur. J. Endocrinol. 1981, 98, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Fleseriu, M.; Biller, B.M.K.; Freda, P.U.; Gadelha, M.R.; Giustina, A.; Katznelson, L.; Molitch, M.E.; Samson, S.L.; Strasburger, C.J.; van der Lely, A.J.; et al. A Pituitary Society update to acromegaly management guidelines. Pituitary 2021, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gauna, C.; van de Zande, B.; van Kerkwijk, A.; Themmen, A.P.; van der Lely, A.-J.; Delhanty, P.J. Unacylated ghrelin is not a functional antagonist but a full agonist of the type 1a growth hormone secretagogue receptor (GHS-R). Mol. Cell. Endocrinol. 2007, 274, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Kopchick, J.J.; Berryman, D.E.; Puri, V.; Lee, K.Y.; Jorgensen, J.O.L. The effects of growth hormone on adipose tissue: Old observations, new mechanisms. Nat. Rev. Endocrinol. 2020, 16, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kopchick, J.J.; Parkinson, C.; Stevens, E.C.; Trainer, P.J. Growth Hormone Receptor Antagonists: Discovery, Development, and Use in Patients with Acromegaly. Endocr. Rev. 2002, 23, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, S.; Honeth, G.; Ginestier, C.; Shinomiya, I.; Marlow, R.; Buchupalli, B.; Gazinska, P.; Brown, J.; Catchpole, S.; Liu, S.; et al. Growth Hormone Is Secreted by Normal Breast Epithelium upon Progesterone Stimulation and Increases Proliferation of Stem/Progenitor Cells. Stem Cell Rep. 2014, 2, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Flanagan, J.U.; Langley, R.J.; Hay, M.P.; Perry, J.K. Targeting growth hormone function: Strategies and therapeutic applications. Signal Transduct. Target. Ther. 2019, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, L.M.; Maas, P.; van Amersfoort, M.; Timmermans-Sprang, E.P.M.; Mensinga, A.; van der Vaart, E.; Malergue, F.; Viëtor, H.; Derksen, P.W.B.; Klumperman, J.; et al. Small molecules to regulate the GH/IGF1 axis by inhibiting the growth hormone receptor synthesis. Front. Endocrinol. 2022, 13, 926210. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Laron, Z. Insulin-like growth factors and aging: Lessons from Laron syndrome. Front. Endocrinol. 2023, 14, 1291812. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Sarfstein, R.; Nagaraj, K.; Laron, Z. Laron Syndrome Research Paves the Way for New Insights in Oncological Investigation. Cells 2020, 9, 2446. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Serby, M.D.; Zhao, H.; Kosogof, C.; Szczepankiewicz, B.G.; Liu, M.; Liu, B.; Hutchins, C.W.; Sarris, K.A.; Hoff, E.D.; et al. Discovery and Pharmacological Evaluation of Growth Hormone Secretagogue Receptor Antagonists. J. Med. Chem. 2006, 49, 4459–4469. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, Y.; Xu, G.; Fu, C. Growth hormone receptor promotes breast cancer progression via the BRAF/MEK/ERK signaling pathway. FEBS Open Bio 2020, 10, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Mol, J.A.; van Garderen, E.; Selman, P.J.; Wolfswinkel, J.; Rijinberk, A.; Rutteman, G.R. Growth hormone mRNA in mammary gland tumors of dogs and cats. J. Clin. Investig. 1995, 95, 2028–2034. [Google Scholar] [CrossRef] [PubMed]
- Strous, G.J.; Almeida, A.D.S.; Putters, J.; Schantl, J.; Sedek, M.; Slotman, J.A.; Nespital, T.; Hassink, G.C.; Mol, J.A. Growth Hormone Receptor Regulation in Cancer and Chronic Diseases. Front. Endocrinol. 2020, 11, 597573. [Google Scholar] [CrossRef] [PubMed]
- Timmermans-Sprang, E.P.M.; Gracanin, A.; Mol, J.A. Molecular Signaling of Progesterone, Growth Hormone, Wnt, and HER in Mammary Glands of Dogs, Rodents, and Humans: New Treatment Target Identification. Front. Veter. Sci. 2017, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- van der Krift, F.; Zijlmans, D.W.; Shukla, R.; Javed, A.; Koukos, P.I.; Schwarz, L.L.E.; Timmermans-Sprang, E.P.; Maas, P.E.; Gahtory, D.; Nieuwboer, M.v.D.; et al. A novel antifolate suppresses growth of FPGS-deficient cells and overcomes methotrexate resistance. Life Sci. Alliance 2023, 6, e202302058. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Week 0 | Week 2 | Week 3 | Week 5 | |
|---|---|---|---|---|
| Urea (mmol/L) | 5.4 ± 0.5 | 6.9 ± 0.4 * | 6.6 ± 0.3 | 5.4 ± 0.6 |
| Creatinine (µmol/L) | 56 ± 4 | 59 ± 3 | 56 ± 3 | 55 ± 3 |
| Glucose (mmol/L) | 5.9 ± 0.2 | 5.9 ± 0.2 | 5.5 ± 0.2 | 5.9 ± 0.2 |
| AF (U/L) | 151 ± 48 | 139 ± 36 | 128 ± 30 | 108 ± 23 |
| ALAT (U/L) | 57 ± 11 | 56 ± 11 | 59 ± 13 | 52 ± 11 |
| ASAT (U/L) | 28 ± 2 | 27 ± 1 | 27 ± 1 | 26 ± 2 |
| Bile acids (µmol/L) | 16 ± 7 | 30 ± 18 | 23 ± 12 | 35 ± 27 |
| Total bilirubin (µmol/L) | 3.4 ± 0.2 | 3.2 ± 0.3 | 3.3 ± 0.2 | 3.0 ± 0.3 * |
| Triglycerides (mmol/L) | 1.4 ± 0.2 | 1.6 ± 0.2 | 1.8 ± 0.2 | 0.9 ± 0.1 |
| Folic acid (nmol/L) | 22 ± 2 | 33 ± 3 * | 30 ± 2 * | 16 ± 2 |
| Vitamin B12 (pmol/L) | 455 ± 39 | 375 ± 29 | 387 ± 32 | 383 ± 32 |
| TLI (µg/L) | 30 ± 4 | 36 ± 3 * | 37 ± 3 * | 30 ± 3 |
| Week 0 | Week 2 | Week 3 | Week 5 | |
|---|---|---|---|---|
| Hemoglobin (mmol/L) | 9.9 ± 0.5 | 9.5 ± 0.4 | 10.1 ± 0.5 | 10.0 ± 0.4 |
| Hematocrit (L/L) | 0.46 ± 0.02 | 0.45 ± 0.02 | 0.48 ± 0.02 | 0.47 ± 0.02 |
| MCV (fl) | 68.0 ± 1.5 | 68.1 ± 1.3 | 69.1 ± 1.3 | 69.2 ± 1.4 |
| MCH (fmol) | 1.44 ± 0.03 | 1.45 ± 0.03 | 1.46 ± 0.03 | 1.48 ± 0.04 |
| RDW (%) | 14.3 ± 0.6 | 14.6 ± 0.6 | 15.1 ± 0.6 | 15.3 ± 0.6 |
| Reticulocytes (×109/L) | 0.66 ± 0.12 | 0.84 ± 0.16 | 1.05 ± 0.22 | 0.88 ± 0.18 |
| Leucocytes (×109/L) | 8.2 ± 0.6 | 8.0 ± 0.4 | 8.4 ± 0.5 | 8.3 ± 0.7 |
| Lymphocytes (×109/L) | 2.2 ± 0.3 | 2.0 ± 0.1 | 2.3 ± 0.2 | 2.1 ± 0.3 |
| Monocytes (×109/L) | 0.37 ± 0.03 | 0.40 ± 0.03 | 0.37 ± 0.02 | 0.33 ± 0.03 |
| Segments (×109/L) | 5.3 ± 0.5 | 5.3 ± 0.3 | 5.4 ± 0.3 | 5.5 ± 0.6 |
| Eosinophils (×109/L) | 0.41 ± 0.06 | 0.36 ± 0.07 | 0.37 ± 0.07 | 0.33 ± 0.07 |
| Thrombocytes (×109/L) | 412 ± 49 | 415 ± 56 | 456 ± 59 | 423 ± 54 |
| Dog | Main Conclusion |
|---|---|
| A | Mammary carcinoma (grade 2); lipoma Mild glomerulopathy and interstitial nephritis |
| B | Follicular cell carcinoma thyroid Benign mixed mammary tumor |
| C | Follicular cell carcinoma thyroid Nephritis |
| D | Suspected large cell lymphoma kidney |
| E | Moderate lymphoplasmacytic enteritis Mild membranous glomerulopathy |
| F | Slight gastritis Mild enteritis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmermans, E.P.M.; Blankevoort, J.; Grinwis, G.C.M.; Mesu, S.J.; Gehring, R.; Delhanty, P.J.D.; Maas, P.E.M.; Strous, G.J.; Mol, J.A. In Vivo Effects of a GHR Synthesis Inhibitor During Prolonged Treatment in Dogs. Pharmaceuticals 2024, 17, 1381. https://doi.org/10.3390/ph17101381
Timmermans EPM, Blankevoort J, Grinwis GCM, Mesu SJ, Gehring R, Delhanty PJD, Maas PEM, Strous GJ, Mol JA. In Vivo Effects of a GHR Synthesis Inhibitor During Prolonged Treatment in Dogs. Pharmaceuticals. 2024; 17(10):1381. https://doi.org/10.3390/ph17101381
Chicago/Turabian StyleTimmermans, Elpetra P. M., Joëlle Blankevoort, Guy C. M. Grinwis, Sietske J. Mesu, Ronette Gehring, Patric J. D. Delhanty, Peter E. M. Maas, Ger J. Strous, and Jan A. Mol. 2024. "In Vivo Effects of a GHR Synthesis Inhibitor During Prolonged Treatment in Dogs" Pharmaceuticals 17, no. 10: 1381. https://doi.org/10.3390/ph17101381
APA StyleTimmermans, E. P. M., Blankevoort, J., Grinwis, G. C. M., Mesu, S. J., Gehring, R., Delhanty, P. J. D., Maas, P. E. M., Strous, G. J., & Mol, J. A. (2024). In Vivo Effects of a GHR Synthesis Inhibitor During Prolonged Treatment in Dogs. Pharmaceuticals, 17(10), 1381. https://doi.org/10.3390/ph17101381