
Epigenetic Regulation in Myocardial Fibroblasts and Its Impact on Cardiovascular Diseases



Abstract
1. Introduction
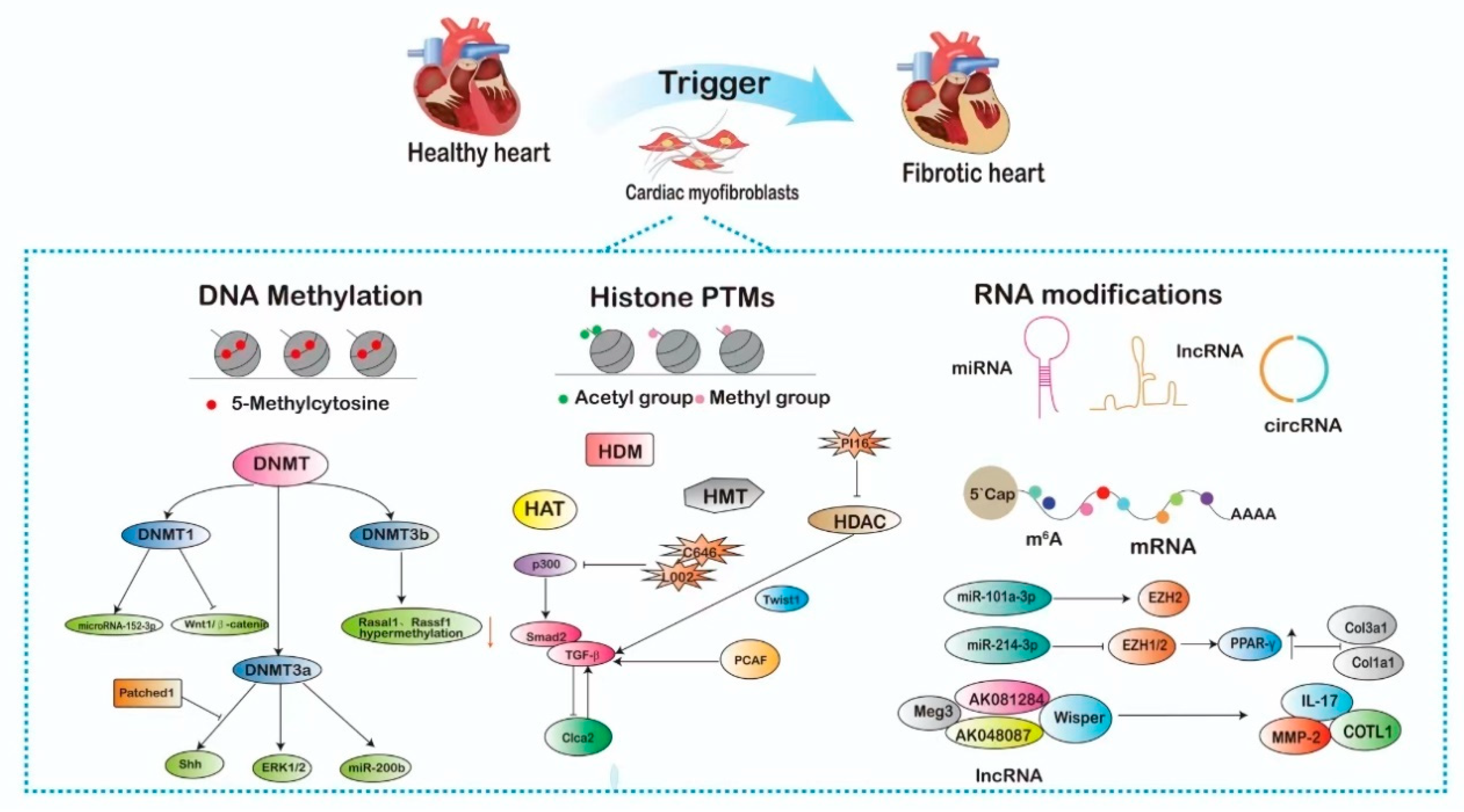
2. Epigenetic Mechanisms Influencing the Regulation of Cardiac Fibroblasts
2.1. DNA Methylation-Mediated Activation of Cardiac Fibroblast
2.2. Histone Post-Translational Modifications-Mediated Activation of Cardiac Fibroblast
2.2.1. Histone Acetylation and Deacetylation in CFs
2.2.2. Histone Methylation and Demethylation in CFs
2.3. Non-Coding RNA-Mediated Activation of Cardiac Fibroblasts
2.3.1. miRNAs in CFs
2.3.2. LncRNAs in CFs
2.3.3. CircRNAs in CFs
2.4. RNA Modifications-Mediated Activation of Cardiac Fibroblast
3. Epigenetic Therapies and Cardiovascular Diseases
3.1. DNA Methylation Inhibitors
3.2. Histone Deacetylase Inhibitors
3.3. Histone Methyltransferase Inhibitors
3.4. Non-Coding RNA Modulators
4. Conclusions and Future Prospective
Author Contributions
Funding
Conflicts of Interest
References
- Kavey, R.-E.W.; Daniels, S.R.; Lauer, R.M.; Atkins, D.L.; Hayman, L.L.; Taubert, K. American Heart Association Guidelines for Primary Prevention of Atherosclerotic Cardiovascular Disease Beginning in Childhood. Circulation 2003, 107, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, L.; Lei, W.; Chua, H.Z.; Li, Z.; Huang, X.; Wang, Q.; Li, N.; Zhang, H. Traditional Chinese medicine as a therapeutic option for cardiac fibrosis: Pharmacology and mechanisms. Biomed. Pharmacother. 2021, 142, 111979. [Google Scholar] [CrossRef] [PubMed]
- Preda, A.; Liberale, L.; Montecucco, F. Imaging techniques for the assessment of adverse cardiac remodeling in metabolic syndrome. Heart Fail. Rev. 2022, 27, 1883–1897. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-P.; Chang-Lee, S.N.; Day, C.H.; Ho, T.-J.; Viswanadha, V.P.; Chung, L.-C.; Hwang, J.-M.; Jong, G.-P.; Kuo, W.-W.; Huang, C.-Y. Secondhand smoke exposure enhances cardiac fibrosis effects on the aging rat hearts. Acta Cardiol. Sin. 2016, 32, 594. [Google Scholar]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The origin of fibroblasts and mechanism of cardiac fibrosis. J. Cell. Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef]
- Liu, M.; de Juan Abad, B.L.; Cheng, K. Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies. Adv. Drug Deliv. Rev. 2021, 173, 504–519. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- Gao, Y.; Chu, M.; Hong, J.; Shang, J.; Xu, D. Hypoxia induces cardiac fibroblast proliferation and phenotypic switch: A role for caveolae and caveolin-1/PTEN mediated pathway. J. Thorac. Dis. 2014, 6, 1458. [Google Scholar]
- Shao, J.; Liu, J.; Zuo, S. Roles of epigenetics in cardiac fibroblast activation and fibrosis. Cells 2022, 11, 2347. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Xie, D.; Xu, D. Epigenetic Regulation of Fibroblasts and Crosstalk between Cardiomyocytes and Non-Myocyte Cells in Cardiac Fibrosis. Biomolecules 2023, 13, 1382. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Song, Z.-Y.; Ding, X.-S.; Yang, J.-J.; Shi, K.-H.; Li, J. Epigenetic signatures in cardiac fibrosis, special emphasis on DNA methylation and histone modification. Heart Fail. Rev. 2018, 23, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P. Epigenetics: Principles and practice. Dig. Dis. 2011, 29, 130–135. [Google Scholar] [CrossRef]
- Komal, S.; Zhang, L.-R.; Han, S.-N. Potential regulatory role of epigenetic RNA methylation in cardiovascular diseases. Biomed. Pharmacother. 2021, 137, 111376. [Google Scholar] [CrossRef]
- Tammen, S.A.; Friso, S.; Choi, S.-W. Epigenetics: The link between nature and nurture. Mol. Aspects Med. 2013, 34, 753–764. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications: Basic Mechanisms and Role in Cardiovascular Disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Sum, H.; Brewer, A.C. Epigenetic modifications as therapeutic targets in atherosclerosis: A focus on DNA methylation and non-coding RNAs. Front. Cardiovasc. Med. 2023, 10, 1183181. [Google Scholar] [CrossRef]
- Ferrari, S.; Pesce, M. Cell-based mechanosensation, epigenetics, and non-coding RNAs in progression of cardiac fibrosis. Int. J. Mol. Sci. 2019, 21, 28. [Google Scholar] [CrossRef]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell. Cardiol. 2016, 93, 162–174. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Foo, R.; Anene-Nzelu, C.G.; Travers, J.G.; Vagnozzi, R.J.; Weber, N.; Thum, T. Emerging epigenetic therapies of cardiac fibrosis and remodelling in heart failure: From basic mechanisms to early clinical development. Cardiovasc. Res. 2022, 118, 3482–3498. [Google Scholar] [CrossRef]
- Parry, A.; Rulands, S.; Reik, W. Active turnover of DNA methylation during cell fate decisions. Nat. Rev. Genet. 2021, 22, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- Tao, H.; Shi, P.; Zhao, X.; Xuan, H.; Gong, W.; Ding, X. DNMT1 deregulation of SOCS3 axis drives cardiac fibroblast activation in diabetic cardiac fibrosis. J. Cell. Physiol. 2021, 236, 3481–3494. [Google Scholar] [CrossRef]
- Shang, L.; Pin, L.; Zhu, S.; Zhong, X.; Zhang, Y.; Shun, M.; Liu, Y.; Hou, M. Plantamajoside attenuates isoproterenol-induced cardiac hypertrophy associated with the HDAC2 and AKT/GSK-3β signaling pathway. Chem. Biol. Interact. 2019, 307, 21–28. [Google Scholar] [CrossRef]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.; Chen, S.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef]
- Kopinke, D.; Norris, A.M.; Mukhopadhyay, S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. In Proceedings of the Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 110, pp. 89–103. [Google Scholar]
- Zhao, K.; Weng, L.; Xu, T.; Yang, C.; Zhang, J.; Ni, G.; Guo, X.; Tu, J.; Zhang, D.; Sun, W.; et al. Low-intensity pulsed ultrasound prevents prolonged hypoxia-induced cardiac fibrosis through HIF-1α/DNMT3a pathway via a TRAAK-dependent manner. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1500–1514. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yang, J.-J.; Chen, Z.-W.; Xu, S.-S.; Zhou, X.; Zhan, H.-Y.; Shi, K.-H. DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology 2014, 323, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-S.; Ding, J.-F.; Shi, P.; Shi, K.-H.; Tao, H. DNMT1-Induced miR-152-3p Suppression Facilitates Cardiac Fibroblast Activation in Cardiac Fibrosis. Cardiovasc. Toxicol. 2021, 21, 984–999. [Google Scholar] [CrossRef]
- Qin, R.-H.; Tao, H.; Ni, S.-H.; Shi, P.; Dai, C.; Shi, K.-H. microRNA-29a inhibits cardiac fibrosis in Sprague-Dawley rats by downregulating the expression of DNMT3A. Anatol. J. Cardiol. Kardiyol. Derg. 2018, 20, 198–205. [Google Scholar]
- Tao, H.; Dai, C.; Ding, J.-F.; Yang, J.-J.; Ding, X.-S.; Xu, S.-S.; Shi, K.-H. Epigenetic aberrations of miR-369-5p and DNMT3A control Patched1 signal pathway in cardiac fibrosis. Toxicology 2018, 410, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-D.; Qin, R.-H.; Yang, J.-J.; Xu, S.-S.; Tao, H.; Ding, X.-S.; Shi, K.-H. DNMT3A controls miR-200b in cardiac fibroblast autophagy and cardiac fibrosis. Inflamm. Res. 2018, 67, 681–690. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Y. Role of Mammalian DNA Methyltransferases in Development. Annu. Rev. Biochem. 2020, 89, 135–158. [Google Scholar] [CrossRef]
- Kusano, K.F.; Pola, R.; Murayama, T.; Curry, C.; Kawamoto, A.; Iwakura, A.; Shintani, S.; Ii, M.; Asai, J.; Tkebuchava, T. Sonic hedgehog myocardial gene therapy: Tissue repair through transient reconstitution of embryonic signaling. Nat. Med. 2005, 11, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Chen, S.; Zhou, J.; Li, J.; Cheng, Y. Epigenetics-based therapeutics for myocardial fibrosis. Life Sci. 2021, 271, 119186. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Tao, H.; Xu, W.; Qu, W.; Gao, H.; Zhang, J.; Cheng, X.; Liu, N.; Chen, J.; Xu, G.-L.; Li, X. Loss of ten-eleven translocation 2 induces cardiac hypertrophy and fibrosis through modulating ERK signaling pathway. Hum. Mol. Genet. 2021, 30, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Spearman, A.D.; Ke, X.; Fu, Q.; Lane, R.H.; Majnik, A. Adverse maternal environment leads to cardiac fibrosis in adult male mice. Birth Defects Res. 2018, 110, 1551–1555. [Google Scholar] [CrossRef]
- Wang, Y.; Sano, S.; Yura, Y.; Ke, Z.; Sano, M.; Oshima, K.; Ogawa, H.; Horitani, K.; Min, K.-D.; Miura-Yura, E.; et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 2020, 5, e135204. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc. Res. 2015, 105, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Sarkar, S.; Tan, D. Histone variants and chromatin structure, update of advances. Comput. Struct. Biotechnol. J. 2022, 21, 299–311. [Google Scholar] [CrossRef]
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 2019, 26, 3–13. [Google Scholar] [CrossRef]
- Barcena-Varela, M.; Paish, H.; Alvarez, L.; Uriarte, I.; Latasa, M.U.; Santamaria, E.; Recalde, M.; Garate, M.; Claveria, A.; Colyn, L. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: Dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut 2021, 70, 388–400. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Song, K.; Tu, B.; Lin, L.-C.; Sun, H.; Zhou, Y.; Li, R.; Shi, Y.; Yang, J.-J.; Zhang, Y.; et al. Crosstalk between oxidative stress and epigenetic marks: New roles and therapeutic implications in cardiac fibrosis. Redox Biol. 2023, 65, 102820. [Google Scholar] [CrossRef]
- Feng, G.; Bajpai, G.; Ma, P.; Koenig, A.; Bredemeyer, A.; Lokshina, I.; Lai, L.; Förster, I.; Leuschner, F.; Kreisel, D.; et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation 2022, 145, 765–782. [Google Scholar] [CrossRef]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.-H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.-Y.; Hindmarch, C.C.T.; et al. Epigenetic Metabolic Reprogramming of Right Ventricular Fibroblasts in Pulmonary Arterial Hypertension: A Pyruvate Dehydrogenase Kinase-Dependent Shift in Mitochondrial Metabolism Promotes Right Ventricular Fibrosis. Circ. Res. 2020, 126, 1723–1745. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Gallinari, P.; Marco, S.D.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The roles of histone deacetylases and their inhibitors in cancer therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Lim, Y.; Jeong, A.; Kwon, D.-H.; Lee, Y.-U.; Kim, Y.-K.; Ahn, Y.; Kook, T.; Park, W.-J.; Kook, H. P300/CBP-associated factor activates cardiac fibroblasts by SMAD2 acetylation. Int. J. Mol. Sci. 2021, 22, 9944. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Varga, J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell. Physiol. 2007, 213, 663–671. [Google Scholar] [CrossRef]
- Travers, J.G.; Tharp, C.A.; Rubino, M.; McKinsey, T.A. Therapeutic targets for cardiac fibrosis: From old school to next-gen. J. Clin. Investig. 2022, 132, e148554. [Google Scholar] [CrossRef]
- Shao, T.; Xue, Y.; Fang, M. Epigenetic repression of chloride channel accessory 2 transcription in cardiac fibroblast: Implication in cardiac fibrosis. Front. Cell Dev. Biol. 2021, 9, 771466. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Murphy, S.B.; Kishore, R.; Vaughan, D.E. Global gene expression profiling in PAI-1 knockout murine heart and kidney: Molecular basis of cardiac-selective fibrosis. PLoS ONE 2013, 8, e63825. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Kee, H.J.; Bai, L.; Kim, M.-K.; Kee, S.-J.; Jeong, M.H. Selective HDAC8 inhibition attenuates isoproterenol-induced cardiac hypertrophy and fibrosis via p38 MAPK pathway. Front. Pharmacol. 2021, 12, 677757. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.S.; Bagchi, R.A.; Felisbino, M.B.; Hirsch, R.A.; Smith, H.E.; Riching, A.S.; Enyart, B.Y.; Koch, K.A.; Cavasin, M.A.; Alexanian, M.; et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res. 2019, 125, 662–677. [Google Scholar] [CrossRef]
- He, Z.; Jiao, H.; An, Q.; Zhang, X.; Zengyangzong, D.; Xu, J.; Liu, H.; Ma, L.; Zhao, W. Discovery of novel 4-phenylquinazoline-based BRD4 inhibitors for cardiac fibrosis. Acta Pharm. Sin. B 2022, 12, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Chen, Y.; Wang, B.; Zhang, X.; Yan, Y.; Zhou, L.; Zhang, Y.; Xie, Y. MFGE8 attenuates Ang-II-induced atrial fibrosis and vulnerability to atrial fibrillation through inhibition of TGF-β1/Smad2/3 pathway. J. Mol. Cell. Cardiol. 2020, 139, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; van de Leemput, J.; Han, Z. The roles of histone lysine methyltransferases in heart development and disease. J. Cardiovasc. Dev. Dis. 2023, 10, 305. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, N.; Xu, J. miR-101a-3p overexpression prevents acetylcholine-CaCl 2 -induced atrial fibrillation in rats via reduction of atrial tissue fibrosis, involving inhibition of EZH2. Mol. Med. Rep. 2021, 24, 740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, D.; Ji, T.-F.; Shi, L.; Yu, J.-L. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-κB signaling pathway in a rat model. Oncotarget 2017, 8, 17347. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, C.; Li, J.; Che, J.; Yang, X.; Xian, Y.; Li, X.; Cao, C. Long non-coding RNA MALAT1 promotes cardiac remodeling in hypertensive rats by inhibiting the transcription of MyoD. Aging 2019, 11, 8792. [Google Scholar] [CrossRef]
- Qian, W.; Zheng, Z.; Nie, J.; Liu, L.; Meng, X.; Sun, H.; Xiao, F.; Kang, T. LncRNA SNHG12 alleviates hypertensive vascular endothelial injury through miR-25-3p/SIRT6 pathway. J. Leukoc. Biol. 2021, 110, 651–661. [Google Scholar] [CrossRef]
- Nicholson, T.B.; Chen, T. LSD1 demethylates histone and non-histone proteins. Epigenetics 2009, 4, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.-L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.-M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021, 129, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, Y.; Zhang, B.; Liang, X.; Li, X. The JMJD family histone demethylases in crosstalk between inflammation and cancer. Front. Immunol. 2022, 13, 881396. [Google Scholar] [CrossRef]
- Long, F.; Wang, Q.; Yang, D.; Zhu, M.; Wang, J.; Zhu, Y.; Liu, X. Targeting JMJD3 histone demethylase mediates cardiac fibrosis and cardiac function following myocardial infarction. Biochem. Biophys. Res. Commun. 2020, 528, 671–677. [Google Scholar] [CrossRef]
- Rubio, K.; Molina-Herrera, A.; Pérez-González, A.; Hernández-Galdámez, H.V.; Piña-Vázquez, C.; Araujo-Ramos, T.; Singh, I. EP300 as a molecular integrator of fibrotic transcriptional programs. Int. J. Mol. Sci. 2023, 24, 12302. [Google Scholar] [CrossRef]
- Rai, R.; Verma, S.K.; Kim, D.; Ramirez, V.; Lux, E.; Li, C.; Sahoo, S.; Wilsbacher, L.D.; Vaughan, D.E.; Quaggin, S.E.; et al. A novel acetyltransferase p300 inhibitor ameliorates hypertension-associated cardio-renal fibrosis. Epigenetics 2017, 12, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Marcu, M.G.; Jung, Y.-J.; Lee, S.; Chung, E.-J.; Lee, M.-J.; Trepel, J.; Neckers, L. Curcumin is an inhibitor of p300 histone acetylatransferase. Med. Chem. 2006, 2, 169–174. [Google Scholar]
- Hutzen, B.; Friedman, L.; Sobo, M.; Lin, L.; Cen, L.; De Angelis, S.; Yamakoshi, H.; Shibata, H.; Iwabuchi, Y.; Lin, J. Curcumin analogue GO-Y030 inhibits STAT3 activity and cell growth in breast and pancreatic carcinomas. Int. J. Oncol. 2009, 35, 867–872. [Google Scholar]
- Love, I.M.; Sekaric, P.; Shi, D.; Grossman, S.R.; Androphy, E.J. The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 2012, 11, 2458–2466. [Google Scholar] [CrossRef]
- Hao, S.; Sui, X.; Wang, J.; Zhang, J.; Pei, Y.; Guo, L.; Liang, Z. Secretory products from epicardial adipose tissue induce adverse myocardial remodeling after myocardial infarction by promoting reactive oxygen species accumulation. Cell Death Dis. 2021, 12, 848. [Google Scholar] [CrossRef]
- Yuan, Z.; Rezai-Zadeh, N.; Zhang, X.; Seto, E. Histone Deacetylase Activity Assay. In Chromatin Protocols; Chellappan, S.P., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; Volume 523, pp. 279–293. ISBN 978-1-58829-873-7. [Google Scholar]
- Wang, Y.; Abrol, R.; Mak, J.Y.W.; Das Gupta, K.; Ramnath, D.; Karunakaran, D.; Fairlie, D.P.; Sweet, M.J. Histone deacetylase 7: A signalling hub controlling development, inflammation, metabolism and disease. FEBS J. 2023, 290, 2805–2832. [Google Scholar] [CrossRef]
- Williams, S.M.; Golden-Mason, L.; Ferguson, B.S.; Schuetze, K.B.; Cavasin, M.A.; Demos-Davies, K.; Yeager, M.E.; Stenmark, K.R.; McKinsey, T.A. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J. Mol. Cell. Cardiol. 2014, 67, 112–125. [Google Scholar] [CrossRef]
- Kee, H.J.; Bae, E.H.; Park, S.; Lee, K.E.; Suh, S.H.; Kim, S.W.; Jeong, M.H. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive rats via regulation of HDAC6/HDAC8 enzyme activity. Kidney Blood Press. Res. 2013, 37, 229–239. [Google Scholar] [CrossRef]
- Deng, M.; Yang, S.; Ji, Y.; Lu, Y.; Qiu, M.; Sheng, Y.; Sun, W.; Kong, X. Overexpression of peptidase inhibitor 16 attenuates angiotensin II–induced cardiac fibrosis via regulating HDAC1 of cardiac fibroblasts. J. Cell. Mol. Med. 2020, 24, 5249–5259. [Google Scholar] [CrossRef]
- Huynh, T.V.; Rethi, L.; Chung, C.; Yeh, Y.; Kao, Y.; Chen, Y. Class I HDAC modulates angiotensin II–induced fibroblast migration and mitochondrial overactivity. Eur. J. Clin. Investig. 2022, 52, e13712. [Google Scholar] [CrossRef]
- Yoon, S.; Kook, T.; Min, H.-K.; Kwon, D.-H.; Cho, Y.K.; Kim, M.; Shin, S.; Joung, H.; Jeong, S.H.; Lee, S. PP2A negatively regulates the hypertrophic response by dephosphorylating HDAC2 S394 in the heart. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Gillette, T.G. HDAC Inhibition in the Heart: Erasing Hidden Fibrosis. Circulation 2021, 143, 1891–1893. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Zhang, X.; Zhang, M.; Hu, X.; Yin, H. Molecular mechanisms of histone deacetylases and inhibitors in renal fibrosis progression. Front. Mol. Biosci. 2022, 9, 986405. [Google Scholar] [CrossRef]
- Liu, W.; Yuan, Q.; Cao, S.; Wang, G.; Liu, X.; Xia, Y.; Bian, Y.; Xu, F.; Chen, Y. Acetylation Mechanisms andTargeted Therapies in Cardiac Fibrosis. Pharmacol. Res. 2023, 193, 106815. [Google Scholar] [CrossRef]
- Alam, H.; Gu, B.; Lee, M.G. Histone methylation modifiers in cellular signaling pathways. Cell. Mol. Life Sci. 2015, 72, 4577–4592. [Google Scholar] [CrossRef]
- Phillips, T. The role of methylation in gene expression. Nat. Educ. 2008, 1, 116. [Google Scholar]
- Lee, H.-T.; Oh, S.; Yoo, H.; Kwon, Y.-W. The key role of DNA methylation and histone acetylation in epigenetics of atherosclerosis. J. Lipid Atheroscler. 2020, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fan, Z.; Shliaha, P.V.; Miele, M.; Hendrickson, R.C.; Jiang, X.; Helin, K. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 2023, 615, 339–348. [Google Scholar] [CrossRef]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhang, R.; Mo, B.; Chen, L.; Liu, L.; Yu, Y.; Cao, W.; Fang, G.; Wan, Y.; Gu, Y. EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J. Mol. Cell. Cardiol. 2019, 135, 119–133. [Google Scholar] [CrossRef]
- Tsou, P.-S.; Campbell, P.; Amin, M.A.; Coit, P.; Miller, S.; Fox, D.A.; Khanna, D.; Sawalha, A.H. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc. Natl. Acad. Sci. USA 2019, 116, 3695–3702. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.-S.; Tang, C.-M.; Xiao, Z.; Zhu, J.-N.; Lin, Q.-X.; Fu, Y.-H.; Hu, Z.-Q.; Zhang, Z.; Yang, M.; Zheng, X.-L. Targeting EZH1 and EZH2 contributes to the suppression of fibrosis-associated genes by miR-214-3p in cardiac myofibroblasts. Oncotarget 2016, 7, 78331. [Google Scholar] [CrossRef]
- Ge, Z.; Yin, C.; Li, Y.; Tian, D.; Xiang, Y.; Li, Q.; Tang, Y.; Zhang, Y. Long noncoding RNA NEAT1 promotes cardiac fibrosis in heart failure through increased recruitment of EZH2 to the Smad7 promoter region. J. Transl. Med. 2022, 20, 7. [Google Scholar] [CrossRef]
- Wang, J.; Lu, F.; Ren, Q.; Sun, H.; Xu, Z.; Lan, R.; Liu, Y.; Ward, D.; Quan, J.; Ye, T. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011, 71, 7238–7249. [Google Scholar] [CrossRef]
- Zhang, Q.-J.; Tran, T.A.T.; Wang, M.; Ranek, M.J.; Kokkonen-Simon, K.M.; Gao, J.; Luo, X.; Tan, W.; Kyrychenko, V.; Liao, L. Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat. Commun. 2018, 9, 5230. [Google Scholar] [CrossRef]
- Qin, J.; Guo, N.; Tong, J.; Wang, Z. Function of histone methylation and acetylation modifiers in cardiac hypertrophy. J. Mol. Cell. Cardiol. 2021, 159, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue Inhibitor of Matrix Metalloproteinase-1 Promotes Myocardial Fibrosis by Mediating CD63–Integrin β1 Interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef]
- Kim, J.; Jang, S.; Kim, C.; An, J.; Kang, E.; Choi, K. Tip60 regulates myoblast differentiation by enhancing the transcriptional activity of MyoD via their physical interactions. FEBS J. 2011, 278, 4394–4404. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, M.; Rotili, D.; Patsilinakos, A.; Forgione, M.; Tomaselli, D.; Alby, F.; Arimondo, P.B.; Mai, A.; Ragno, R. Disruptor of telomeric silencing 1-like (DOT1L): Disclosing a new class of non-nucleoside inhibitors by means of ligand-based and structure-based approaches. J. Comput. Aided Mol. Des. 2018, 32, 435–458. [Google Scholar] [CrossRef]
- Li, F.; Li, L.; Zhang, J.; Yang, X.; Liu, Y. Histone methyltransferase DOT1L mediates the TGF-β1/Smad3 signaling pathway through epigenetic modification of SYK in myocardial infarction. Hum. Cell 2022, 35, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Bao, S.; Li, R.; Sun, H.; Peng, Y. Noncoding RNAs and Cardiac Fibrosis. Rev. Cardiovasc. Med. 2023, 24, 63. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, J. Non-coding RNAs in melanoma: Biological functions and potential clinical applications. Mol. Ther.-Oncolytics 2021, 22, 219–231. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
- Yang, X.; Yu, T.; Zhang, S. MicroRNA-489 suppresses isoproterenol-induced cardiac fibrosis by downregulating histone deacetylase 2. Exp. Ther. Med. 2020, 19, 2229–2235. [Google Scholar] [CrossRef]
- Li, C.; Li, J.; Xue, K.; Zhang, J.; Wang, C.; Zhang, Q.; Chen, X.; Gao, C.; Yu, X.; Sun, L. MicroRNA-143-3p promotes human cardiac fibrosis via targeting sprouty3 after myocardial infarction. J. Mol. Cell. Cardiol. 2019, 129, 281–292. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Y.; Ding, Y.; Bi, X.; Yuan, J.; Zhou, H.; Wang, P.; Zhang, L.; Ye, J. MicroRNA-99b-3p promotes angiotensin II-induced cardiac fibrosis in mice by targeting GSK-3β. Acta Pharmacol. Sin. 2021, 42, 715–725. [Google Scholar] [CrossRef]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Lu, Z.; Fu, X.; Ma, S.; Lu, X. MicroRNA 27b promotes cardiac fibrosis by targeting the FBW7/Snail pathway. Aging 2019, 11, 11865. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Yang, Y.; Kong, F.; Kong, Q.; Shan, C. MiR-143-3p inhibits the proliferation, cell migration and invasion of human breast cancer cells by modulating the expression of MAPK7. Biochimie 2018, 147, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Caudle, Y.; Shaikh, A.; Yao, B.; Yin, D. Inhibition of microRNA-23b prevents polymicrobial sepsis-induced cardiac dysfunction by modulating TGIF1 and PTEN. Biomed. Pharmacother. 2018, 103, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Ramanujam, D.; Schön, A.P.; Beck, C.; Vaccarello, P.; Felician, G.; Dueck, A.; Esfandyari, D.; Meister, G.; Meitinger, T.; Schulz, C.; et al. MicroRNA-21–Dependent Macrophage-to-Fibroblast Signaling Determines the Cardiac Response to Pressure Overload. Circulation 2021, 143, 1513–1525. [Google Scholar] [CrossRef]
- Li, G.; Shao, Y.; Guo, H.C.; Zhi, Y.; Qiao, B.; Ma, K.; Du, J.; Lai, Y.Q.; Li, Y. MicroRNA-27b-3p down-regulates FGF1 and aggravates pathological cardiac remodelling. Cardiovasc. Res. 2022, 118, 2139–2151. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, Y.; Chen, Y.; Li, J.; Zhang, Z.; Sun, Y.; Shen, H.; Zhao, Z.; Huang, Z.; Zhang, W.; et al. Inhibition of MicroRNA-9-5p Protects Against Cardiac Remodeling Following Myocardial Infarction in Mice. Hum. Gene Ther. 2019, 30, 286–301. [Google Scholar] [CrossRef]
- Nishiga, M.; Horie, T.; Kuwabara, Y.; Nagao, K.; Baba, O.; Nakao, T.; Nishino, T.; Hakuno, D.; Nakashima, Y.; Nishi, H.; et al. MicroRNA-33 Controls Adaptive Fibrotic Response in the Remodeling Heart by Preserving Lipid Raft Cholesterol. Circ. Res. 2017, 120, 835–847. [Google Scholar] [CrossRef]
- Zhang, X.; Fernández-Hernando, C. miR-33 Regulation of Adaptive Fibrotic Response in Cardiac Remodeling. Circ. Res. 2017, 120, 753–755. [Google Scholar] [CrossRef]
- Huang, Y.; Qi, Y.; Du, J.-Q.; Zhang, D. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin. Ther. Targets 2014, 18, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Y.; Zhu, H.; Hu, J.; Xie, Z. MiR-34a/miR-93 target c-Ski to modulate the proliferaton of rat cardiac fibroblasts and extracellular matrix deposition in vivo and in vitro. Cell. Signal. 2018, 46, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bounds, K.R.; Chatterjee, P.; Gupta, S. MicroRNA-130a, a Potential Antifibrotic Target in Cardiac Fibrosis. J. Am. Heart Assoc. 2017, 6, e006763. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Pan, J.; Wen, L.; Gong, B.; Li, J.; Gao, H.; Tan, W.; Liang, S.; Zhang, H.; Wang, X. MiR-144-3p enhances cardiac fibrosis after myocardial infarction by targeting PTEN. Front. Cell Dev. Biol. 2019, 7, 249. [Google Scholar] [CrossRef]
- Shen, J.; Xing, W.; Gong, F.; Wang, W.; Yan, Y.; Zhang, Y.; Xie, C.; Fu, S. MiR-150-5p retards the progression of myocardial fibrosis by targeting EGR1. Cell Cycle 2019, 18, 1335–1348. [Google Scholar] [CrossRef]
- Verjans, R.; Peters, T.; Beaumont, F.J.; Van Leeuwen, R.; Van Herwaarden, T.; Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload–Induced Heart Failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef]
- Chiasson, V.; Takano, A.P.C.; Guleria, R.S.; Gupta, S. Deficiency of MicroRNA miR-1954 Promotes Cardiac Remodeling and Fibrosis. J. Am. Heart Assoc. 2019, 8, e012880. [Google Scholar] [CrossRef]
- Yuan, X.; Pan, J.; Wen, L.; Gong, B.; Li, J.; Gao, H.; Tan, W.; Liang, S.; Zhang, H.; Wang, X. MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J. Cell. Mol. Med. 2020, 24, 227–237. [Google Scholar] [CrossRef]
- Rawal, S.; Munasinghe, P.E.; Nagesh, P.T.; Lew, J.K.S.; Jones, G.T.; Williams, M.J.; Davis, P.; Bunton, D.; Galvin, I.F.; Manning, P. Down-regulation of miR-15a/b accelerates fibrotic remodelling in the Type 2 diabetic human and mouse heart. Clin. Sci. 2017, 131, 847–863. [Google Scholar] [CrossRef]
- Ge, Z.-W.; Zhu, X.-L.; Wang, B.-C.; Hu, J.-L.; Sun, J.-J.; Wang, S.; Chen, X.-J.; Meng, S.-P.; Liu, L.; Cheng, Z.-Y. MicroRNA-26b relieves inflammatory response and myocardial remodeling of mice with myocardial infarction by suppression of MAPK pathway through binding to PTGS2. Int. J. Cardiol. 2019, 280, 152–159. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, L.; Lu, L.; Jiang, P.; Sun, H.; Wang, H. Inhibition of miR-29 by TGF-beta-Smad3 signaling through dual mechanisms promotes transdifferentiation of mouse myoblasts into myofibroblasts. PLoS ONE 2012, 7, e33766. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, S.; Wa, M.; Liu, Z.; Hu, S. MicroRNA-101 Protects Against Cardiac Remodeling Following Myocardial Infarction via Downregulation of Runt-Related Transcription Factor 1. J. Am. Heart Assoc. 2019, 8, e013112. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, S.; Shang, J.; Jiang, Y.; Dai, Y.; Xu, B.; Yu, Y.; Liang, Z.; Yang, Y. LncRNA: Shedding light on mechanisms and opportunities in fibrosis and aging. Ageing Res. Rev. 2019, 52, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; Lei, W.; Wu, H.; Wu, J.; Yang, Z.; Yan, S.; Lu, X.-A.; Li, J.; Xia, X.; Han, X. LncRNA-Safe contributes to cardiac fibrosis through Safe-Sfrp2-HuR complex in mouse myocardial infarction. Theranostics 2019, 9, 7282. [Google Scholar] [CrossRef]
- Zhang, F.; Fu, X.; Kataoka, M.; Liu, N.; Wang, Y.; Gao, F.; Liang, T.; Dong, X.; Pei, J.; Hu, X. Long noncoding RNA Cfast regulates cardiac fibrosis. Mol. Ther. Nucleic Acids 2021, 23, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Zhang, J.-G.; Qin, R.-H.; Dai, C.; Shi, P.; Yang, J.-J.; Deng, Z.-Y.; Shi, K.-H. LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology 2017, 386, 11–18. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, Y.; Hu, Y.; Guan, J.; Xu, L.; Xiao, W.; Zhong, Q.; Ren, C.; Lu, J.; Liang, J.; et al. Long noncoding RNA Crnde attenuates cardiac fibrosis via Smad3-Crnde negative feedback in diabetic cardiomyopathy. FEBS J. 2019, 286, 1645–1655. [Google Scholar] [CrossRef]
- Kong, C.; Lyu, D.; He, C.; Li, R.; Lu, Q. Dioscin elevates lncRNA MANTIS in therapeutic angiogenesis for heart diseases. Aging Cell 2021, 20, e13392. [Google Scholar] [CrossRef]
- Kenneweg, F.; Bang, C.; Xiao, K.; Boulanger, C.M.; Loyer, X.; Mazlan, S.; Schroen, B.; Hermans-Beijnsberger, S.; Foinquinos, A.; Hirt, M.N. Long noncoding RNA-enriched vesicles secreted by hypoxic cardiomyocytes drive cardiac fibrosis. Mol. Ther.-Nucleic Acids 2019, 18, 363–374. [Google Scholar] [CrossRef]
- Sun, F.; Zhuang, Y.; Zhu, H.; Wu, H.; Li, D.; Zhan, L.; Yang, W.; Yuan, Y.; Xie, Y.; Yang, S. LncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. J. Mol. Cell. Cardiol. 2019, 133, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Qin, Y.; Lv, J.; Wang, Y.; Che, H.; Chen, X.; Jiang, Y.; Li, A.; Sun, X.; Yue, E. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018, 9, 1000. [Google Scholar] [CrossRef] [PubMed]
- Micheletti, R.; Plaisance, I.; Abraham, B.J.; Sarre, A.; Ting, C.-C.; Alexanian, M.; Maric, D.; Maison, D.; Nemir, M.; Young, R.A.; et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci. Transl. Med. 2017, 9, eaai9118. [Google Scholar] [CrossRef]
- Piccoli, M.-T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast–Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.-F.; Sun, H.; Song, K.; Zhou, Y.; Tu, B.; Shi, K.-H.; Lu, D.; Xu, S.-S.; Tao, H. IGFBP3 epigenetic promotion induced by METTL3 boosts cardiac fibroblast activation and fibrosis. Eur. J. Pharmacol. 2023, 942, 175494. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Sun, H.; Tu, B.; Zhou, Y.; Lin, L.-C.; Liu, Z.-Y.; Li, R.; Yang, J.-J.; Zhang, Y.; Zhao, J.-Y. WTAP boosts lipid oxidation and induces diabetic cardiac fibrosis by enhancing AR methylation. Iscience 2023, 26, 107931. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Liu, K.; Ouyang, S.; Liu, Z.; He, F.; Wu, J. Changes in N6-methyladenosine modification modulate diabetic cardiomyopathy by reducing myocardial fibrosis and myocyte hypertrophy. Front. Cell Dev. Biol. 2021, 9, 702579. [Google Scholar] [CrossRef]
- Meng, Y.; Xi, T.; Fan, J.; Yang, Q.; Ouyang, J.; Yang, J. The inhibition of FTO attenuates the antifibrotic effect of leonurine in rat cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2024, 693, 149375. [Google Scholar] [CrossRef]
- Yang, K.; Zhao, Y.; Hu, J.; Gao, R.; Shi, J.; Wei, X.; Chen, J.; Hu, K.; Sun, A.; Ge, J. ALKBH5 induces fibroblast-to-myofibroblast transformation during hypoxia to protect against cardiac rupture after myocardial infarction. J. Adv. Res. 2024, 61, 193–209. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, X.; Jia, L.; Mondal, A.K.; Diallo, A.; Hawkins, G.A.; Das, S.K.; Parks, J.S.; Yu, L.; Shi, H. Inhibiting DNA methylation by 5-Aza-2′-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology 2014, 155, 4925–4938. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.-Y.; Li, T.-T.; Wang, J.; Jiang, Y.; Zhao, Y.; Jin, X.-X.; Xue, G.-L.; Yang, Y.; Zhang, X.-F. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J. Mol. Cell. Cardiol. 2018, 115, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Du, Y.; Shu, Y.; Gao, M.; Sun, F.; Luo, S.; Yang, T.; Zhan, L.; Yuan, Y.; Chu, W. MIAT is a pro-fibrotic long non-coding RNA governing cardiac fibrosis in post-infarct myocardium. Sci. Rep. 2017, 7, 42657. [Google Scholar] [CrossRef]
- Leisegang, M.S. LET’s sponge: How the lncRNA PFL promotes cardiac fibrosis. Theranostics 2018, 8, 874. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Pan, Z.; Zhao, X.; Liu, L.; Sun, J.; Su, X.; Xu, C.; Zhou, Y.; Zhao, D.; Xu, B. LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics 2018, 8, 1180. [Google Scholar] [CrossRef]
- Guo, J.; Chen, L.-W.; Huang, Z.-Q.; Guo, J.-S.; Li, H.; Shan, Y.; Chen, Z.-R.; Yan, Y.-M.; Zhu, J.-N.; Guo, H.-M.; et al. Suppression of the Inhibitory Effect of circ_0036176-Translated Myo9a-208 on Cardiac Fibroblast Proliferation by miR-218-5p. J. Cardiovasc. Transl. Res. 2022, 15, 548–559. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Zhou, S.; Dain, L.; Mei, L.; Zhu, G. Circular RNA: An Emerging Frontier in RNA Therapeutic Targets, RNA Therapeutics, and Mrna Vaccines. J. Control. Release 2022, 348, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Komal, S.; Gohar, A.; Althobaiti, S.; Ahmad Khan, I.; Cui, L.-G.; Zhang, L.-R.; Han, S.-N.; Shakeel, M. ALKBH5 inhibitors as a potential treatment strategy in heart failure—Inferences from gene expression profiling. Front. Cardiovasc. Med. 2023, 10, 1194311. [Google Scholar] [CrossRef]
- Li, X.; Mu, B.; Li, X.; Bie, Z. circCELF1 Inhibits Myocardial Fibrosis by Regulating the Expression of DKK2 Through FTO/m6A and miR-636. J Cardiovasc. Transl. Res. 2022, 15, 998–1009. [Google Scholar] [CrossRef]
- Zhuang, Y.; Li, T.; Hu, X.; Xie, Y.; Pei, X.; Wang, C.; Li, Y.; Liu, J.; Tian, Z.; Zhang, X.; et al. MetBil as a novel molecular regulator in ischemia-induced cardiac fibrosis via METTL3-mediated m6A modification. FASEB J. 2023, 37, e22797. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, H.; Huang, S.; Yin, L.; Wang, F.; Luo, P.; Huang, H. Epigenetic regulation in cardiovascular disease: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2022, 7, 200. [Google Scholar] [CrossRef]
- Stenzig, J.; Schneeberger, Y.; Löser, A.; Peters, B.S.; Schaefer, A.; Zhao, R.-R.; Ng, S.L.; Höppner, G.; Geertz, B.; Hirt, M.N.; et al. Pharmacological inhibition of DNA methylation attenuates pressure overload-induced cardiac hypertrophy in rats. J. Mol. Cell. Cardiol. 2018, 120, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.-H.; Chen, Y.-C.; Cheng, C.-C.; Lee, T.-I.; Chen, Y.-J.; Chen, S.-A. Tumor necrosis factor-α decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit. Care Med. 2010, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Connelly, J.J.; Cherepanova, O.A.; Doss, J.F.; Karaoli, T.; Lillard, T.S.; Markunas, C.A.; Nelson, S.; Wang, T.; Ellis, P.D.; Langford, C.F. Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum. Mol. Genet. 2013, 22, 5107–5120. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Dasgupta, C.; Chen, M.; Zhang, K.; Buchholz, J.; Xu, Z.; Zhang, L. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc. Res. 2014, 101, 373–382. [Google Scholar] [CrossRef]
- Guay, S.-P.; Légaré, C.; Houde, A.-A.; Mathieu, P.; Bossé, Y.; Bouchard, L. Acetylsalicylic acid, aging and coronary artery disease are associated with ABCA1 DNA methylation in men. Clin. Epigenetics 2014, 6, 14. [Google Scholar] [CrossRef]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef]
- Liu, F.; Levin, M.D.; Petrenko, N.B.; Lu, M.M.; Wang, T.; Yuan, L.J.; Stout, A.L.; Epstein, J.A.; Patel, V.V. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J. Mol. Cell. Cardiol. 2008, 45, 715–723. [Google Scholar] [CrossRef]
- Seki, M.; LaCanna, R.; Powers, J.C.; Vrakas, C.; Liu, F.; Berretta, R.; Chacko, G.; Holten, J.; Jadiya, P.; Wang, T.; et al. Class I Histone Deacetylase Inhibition for the Treatment of Sustained Atrial Fibrillation. J. Pharmacol. Exp. Ther. 2016, 358, 441–449. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Hill, J.A. Inhibition of Hypertrophy Is a Good Therapeutic Strategy in Ventricular Pressure Overload. Circulation 2015, 131, 1435–1447. [Google Scholar] [CrossRef]
- Han, H.; Feng, X.; He, T.; Wu, Y.; He, T.; Yue, Z.; Zhou, W. Discussion on structure classification and regulation function of histone deacetylase and their inhibitor. Chem. Biol. Drug Des. 2024, 103, e14366. [Google Scholar] [CrossRef] [PubMed]
- Papait, R.; Cattaneo, P.; Kunderfranco, P.; Greco, C.; Carullo, P.; Guffanti, A.; Viganò, V.; Stirparo, G.G.; Latronico, M.V.G.; Hasenfuss, G.; et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 20164–20169. [Google Scholar] [CrossRef] [PubMed]
- Gorica, E.; Mohammed, S.A.; Ambrosini, S.; Calderone, V.; Costantino, S.; Paneni, F. Epi-drugs in heart failure. Front. Cardiovasc. Med. 2022, 9, 923014. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Uludag, M.O.; Usanmaz, S.E.; Ayaloglu-Butun, F.; Akcali, K.C.; Demirel-Yilmaz, E. Resveratrol affects histone 3 lysine 27 methylation of vessels and blood biomarkers in DOCA salt-induced hypertension. Mol. Biol. Rep. 2015, 42, 35–42. [Google Scholar] [CrossRef]
- Komal, S.; Yin, J.-J.; Wang, S.-H.; Huang, C.-Z.; Tao, H.-L.; Dong, J.-Z.; Han, S.-N.; Zhang, L.-R. MicroRNAs: Emerging biomarkers for atrial fibrillation. J. Cardiol. 2019, 74, 475–482. [Google Scholar] [CrossRef]
- Batkai, S.; Genschel, C.; Viereck, J.; Rump, S.; Bär, C.; Borchert, T.; Traxler, D.; Riesenhuber, M.; Spannbauer, A.; Lukovic, D.; et al. CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur. Heart J. 2021, 42, 192–201. [Google Scholar] [CrossRef]
- Ruan, W.; Zhao, F.; Zhao, S.; Zhang, L.; Shi, L.; Pang, T. Knockdown of long noncoding RNA MEG3 impairs VEGF-stimulated endothelial sprouting angiogenesis via modulating VEGFR2 expression in human umbilical vein endothelial cells. Gene 2018, 649, 32–39. [Google Scholar] [CrossRef]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef]
- Liao, J.; He, Q.; Li, M.; Chen, Y.; Liu, Y.; Wang, J. LncRNA MIAT: Myocardial infarction associated and more. Gene 2016, 578, 158–161. [Google Scholar] [CrossRef]
- Haemmig, S.; Yang, D.; Sun, X.; Das, D.; Ghaffari, S.; Molinaro, R.; Chen, L.; Deng, Y.; Freeman, D.; Moullan, N.; et al. Long noncoding RNA SNHG12 integrates a DNA-PK-mediated DNA damage response and vascular senescence. Sci. Transl. Med. 2020, 12, eaaw1868. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, X.; Hamblin, M.H.; Yin, K.-J. Long non-coding RNA Malat1 regulates angiogenesis in hindlimb ischemia. Int. J. Mol. Sci. 2018, 19, 1723. [Google Scholar] [CrossRef] [PubMed]
- Man, H.S.J.; Sukumar, A.N.; Lam, G.C.; Turgeon, P.J.; Yan, M.S.; Ku, K.H.; Dubinsky, M.K.; Ho, J.J.D.; Wang, J.J.; Das, S.; et al. Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc. Natl. Acad. Sci. USA 2018, 115, 2401–2406. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.; Zhang, J.; Li, C.; Li, J.; Wang, C.; Zhang, Q.; Chen, X.; Yu, X.; Sun, L.; Yu, X. The role and mechanism of transforming growth factor beta 3 in human myocardial infarction-induced myocardial fibrosis. J. Cell. Mol. Med. 2019, 23, 4229–4243. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef]
- Zhang, Z.; Kurashima, Y. Two sides of the coin: Mast cells as a key regulator of allergy and acute/chronic inflammation. Cells 2021, 10, 1615. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.P.; Lindsey, M.L. Cardiac fibroblast activation post-myocardial infarction: Current knowledge gaps. Trends Pharmacol. Sci. 2017, 38, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Garvin, A.M.; Hale, T.M. State of change: Epigenetic and mitochondrial regulation of cardiac fibroblast activation. Curr. Opin. Physiol. 2022, 28, 100557. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef]
- Li, P.; Wang, Q.-S.; Zhai, Y.; Xiong, R.-P.; Chen, X.; Liu, P.; Peng, Y.; Zhao, Y.; Ning, Y.-L.; Yang, N. Ski mediates TGF-β1-induced fibrosarcoma cell proliferation and promotes tumor growth. J. Cancer 2020, 11, 5929. [Google Scholar] [CrossRef]

{kind=link}
| Epigenetic Modification | Epigenetic Modifiers | Fibrosis Model | Targets | Cardiac Function | References |
|---|---|---|---|---|---|
| DNA methylation | DNMT1 | Diabetic cardiomyopathy; thoracic aortic constriction; isoproterenol | SOCS3, microRNA- 152-3p | Pro-fibrotic activation, CFs autophagy, CF proliferation | [27,31] |
| DNMT3a | Thoracic aortic constriction | TRAAK/RASSF1A, Ras/ERK1/2/miR-200b | Pro-fibrotic, CF activation and proliferation, CF activation/pro-fibrotic | [30,31,34] | |
| DNMT3b | Hypoxia; thoracic aortic constriction | HIF-1α/Rasal1, Rassf1 | Pro-fibrotic, CF activation/pro-fibrotic, CF activation | [33,34] | |
| DNA demethylation | TET2 | Angiotensin II; TET2 KO | IL-6, Rasal1, | anti-fibrotic, inflammatory response, anti-fibrotic, protection of cardiomyocyte | [39,41] |
| TET3 | Thoracic aortic constriction, | Hspa1b | Anti-fibrotic, EndMT | [43] | |
| Histone acetylation | p300 | High glucose; angiotensin II; Thoracic aortic constriction | Smad2, H3K9, GATA4 | Pro-fibrotic, collagen production/ Pro-fibrotic, CF activation and type I collagen synthesis/ Pro-fibrotic, collagen production | [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59] |
| PCAF (p300/CBP-associated factor) | Isoproterenol-induce rat fibrotic model | Smad2 | Pro-fibrotic, CF activation | [55] | |
| Histone deacetylation | HDAC1 | Myocardial infarction; thoracic aortic constriction; angiotensin II | Clca2, p53 | Pro-fibrotic, CF activation and proliferation | [60,61] |
| HDAC2 | Isoproterenol | PPP2CA, α-SMA | Pro-fibrotic, α-SMA synthesis/pro-fibrotic, CF activation | [28,62] | |
| HDAC3 | Diabetic cardiomyopathy; | DUSP5 | Pro-fibrotic, fibrosis markers and collagen accumulation | [29] | |
| HDAC8 | isoproterenol-induce fibrotic model; | P38 MAPK | Pro-fibrotic, markers of fibrosis | [63] | |
| BRD4 | thoracic aortic constriction | Sertad4/Meox1 | Pro-fibrotic, CF activation and proliferation | [64,65] | |
| Histone methylation | EZH1/2, | angiotensin II | PPAR-γ | Pro-fibrotic, Col1a1 and Col3a1 synthesis | [66] |
| EZH2 | High-Fat; angiotensin II; diabetic cardiomyopathy; thoracic aortic constriction | H3K27me2/3, ACTA2, lncRNA-ANRIL, Smad7 | Anti-fibrotic, suppression of pro-fibrotic genes/pro-fibrotic, CF activation and migration/pro-fibrotic, increased expression of FN, Col1α4/pro-fibrotic, CF activation | [67,68,69] | |
| SUV39H1 | SHRs | MyoD | Pro-fibrotic, CF proliferation and collagen accumulation | [70] | |
| DOT1L, | Myocardial infarction | SYK | Pro-fibrotic, CF activation | [71] | |
| Histone demethylation | LSD1, | Thoracic aortic constriction | TGF-β | Pro-fibrotic, CF activation and collagen secretion | [72] |
| KDM3A | Thoracic aortic constriction | Timp1 | Pro-fibrotic, CF activation | [73] | |
| KDM3C | Angiotensin II | Timp1 | Pro-fibrotic, CF activation | [74] | |
| KDM6B | Angiotensin II | β-catenin | Pro-fibrotic, ECM deposition | [75] |
| Epigenetic Modification | Epigenetic Modifiers | Fibrosis Model | Targets | Cardiac Function | References |
|---|---|---|---|---|---|
| miRNA | miR-23b | Myocardial infarction; angiotensin II; thoracic aortic constriction | FBW7, p53, SPRY3, FGF1, ZEB1, SMAD2, TGIF1, PTEN | Pro-fibrotic, CF proliferation and collagen production | [117] |
| miR-27b | Myocardial infarction | FBW | Pro-fibrotic, CF proliferation and collagen production | [115] | |
| miR-125b | Angiotensin II | Apelin, p53 | Pro-fibrotic, CF proliferation | [114] | |
| miR-99b-3p | Angiotensin II | GSK-3β | Pro-fibrotic, CF proliferation and migration | [113] | |
| miR-143-3p | Myocardial infarction | SPRY3 | Pro-fibrotic, CF activation, proliferation, and migration | [112] | |
| miR-21 | Thoracic aortic constriction | Sprouty1/2 (Spry1/2) | Pro-fibrotic, CF activation | [118] | |
| miR-27b-3p | Thoracic aortic constriction; angiotensin II | Fgf1 | Pro-fibrotic, mitochondrial oxidative phosphorylation | [119] | |
| miR-9- 5p | Myocardial infarction | Follistatin-like 1 (FSL1) | Attenuated fibrosis and inflammatory response | [120] | |
| miR-33 | Transverse aortic constriction | ABCA1 | Cardiac fibrosis, transverse aortic constriction | [121] | |
| miR- 34a | Myocardial infarction | Smad4 | Cardiac fibrosis progression | [123] | |
| miR-93 | Myocardial fibrosis | c-Ski | Myocardial fibrosis | [124] | |
| miR-130a | AngII-infused model | PPARγ | Cardiac fibrosis, myofibroblasts differentiation | [125] | |
| miR-144-3p | Myocardial infarction | PTEN | Promotes cell proliferation, migration, and collagen production | [126] | |
| miR-150-5p | Myocardial fibrosis | EGR1 | Myocardial fibrosis | [127] | |
| miR-221/222 | Angiotensin II | SMAD2 | Anti-fibrotic, CF activation, and proliferation | [128] | |
| miR-1954 | Angiotensin II | THBS1 | Anti-fibrotic, attenuation inflammation | [129] | |
| miR-590-3p | Myocardial infarction | ZEB1 | Anti-fibrotic, CF activation, proliferation, and migration | [130] | |
| miR-15a/b | Diabetic and non-diabetic patients undergoing coronary artery bypass graft surgery | CTFG | Diastolic dysfunction, fibrosis | [131] | |
| miR-26b | Myocardial infarction | Smad2/3 | Inflammatory reaction, myocardial injury, fibrosis and myocardial cell apoptosis | [132] | |
| miR-29 | Smad 3+/+, Smad 3−/−, Smad 7+/+, Smad 7−/− | FBN-1, MMP | Myogenic differentiation, transdifferentiation of myoblasts into myofibroblasts | [133,134] | |
| miR-101 | Myocardial infarction | RUNX1 | Myocardial fibrosis, cardiomyocyte apoptosis | [135] | |
| lncRNA | lncRNA AK081284 | Diabetic cardiomyopathy | IL-17 | Pro-fibrotic, CF proliferation, and collagen production | [136] |
| lncRNA AK137033 | Myocardial infarction | Sfrp2 | Pro-fibrotic, CF activation, and proliferation | [137] | |
| lncRNA AK048087 | Myocardial infarction/ Angiotensin II | COTL1 | Pro-fibrotic, CF activation, and proliferation | [138] | |
| lnc GASS | Isoproterenol | MiR-21 | Anti-fibrotic, CF proliferation | [139] | |
| lncRNA Crnde | Diabetic cardiomyopathy | Smad3 | Anti-fibrotic, CF activation | [140] | |
| lncRNA MANTIS | Myocardial infarction | Sox18, Smad3/6 | Anti-fibrotic, vascular neogenesis | [141] | |
| lnc Neat1 | Myocardial infarction | Neat1, P53 HIF2A | Fibroblast and cardiomyocyte survival and functions | [142] | |
| lnc PCFL | Myocardial infarction | miR-378 | Pro-fibrotic, CF proliferation, and collagen production | [143] | |
| lnc Kcnq1ot1 | Streptozotocin (STZ)-induced diabetic (C57BL/6 mice) | Kcnq1ot1, miR-214-3p, caspase-1, TGF-β1 | Ameliorated pyroptosis and fibrosis | [144] | |
| lnc Wisper | Thoracic aortic constriction | TIA1-related protein | Pro-fibrotic, CF proliferation | [145] | |
| lncMeg3 | Thoracic aortic constriction | MMP-2 | Pro-fibrotic, ECM deposition | [146] | |
| RNA modification (m6A) | METTL3 | Myocardial infarction | MetBil, Fibrosis-related genes | Pro-fibrotic, CF activation, and proliferation | [147] |
| METTL3 | ISO-induced cardiac fibrosis | IGFBP3 | Promote cell activation, migration | [148] | |
| WTAP/YTHDF2 | Cardiac fibrosis | Decr1 | Diabetic cardiac fibrosis | [149] | |
| FTO | Myocardial infarction; diabetic cardiomyopathy | Serca2a/CD36, Slc5a33 | Anti-fibrotic, CF activation, proliferation, and migration/Anti-fibrotic, collagen deposition suppression | [148] | |
| ALKBH5 | Myocardial infarction | ErbB4 | Fibroblast activation | [150,151,152] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komal, S.; Gao, Y.; Wang, Z.-M.; Yu, Q.-W.; Wang, P.; Zhang, L.-R.; Han, S.-N. Epigenetic Regulation in Myocardial Fibroblasts and Its Impact on Cardiovascular Diseases. Pharmaceuticals 2024, 17, 1353. https://doi.org/10.3390/ph17101353
Komal S, Gao Y, Wang Z-M, Yu Q-W, Wang P, Zhang L-R, Han S-N. Epigenetic Regulation in Myocardial Fibroblasts and Its Impact on Cardiovascular Diseases. Pharmaceuticals. 2024; 17(10):1353. https://doi.org/10.3390/ph17101353
Chicago/Turabian StyleKomal, Sumra, Yuan Gao, Zhi-Mo Wang, Qing-Wen Yu, Pei Wang, Li-Rong Zhang, and Sheng-Na Han. 2024. "Epigenetic Regulation in Myocardial Fibroblasts and Its Impact on Cardiovascular Diseases" Pharmaceuticals 17, no. 10: 1353. https://doi.org/10.3390/ph17101353
APA StyleKomal, S., Gao, Y., Wang, Z.-M., Yu, Q.-W., Wang, P., Zhang, L.-R., & Han, S.-N. (2024). Epigenetic Regulation in Myocardial Fibroblasts and Its Impact on Cardiovascular Diseases. Pharmaceuticals, 17(10), 1353. https://doi.org/10.3390/ph17101353