
Towards a New Generation of Hormone Therapies: Design, Synthesis and Biological Evaluation of Novel 1,2,3-Triazoles as Estrogen-Positive Breast Cancer Therapeutics and Non-Steroidal Aromatase Inhibitors



Abstract
1. Introduction
2. Results and Discussion
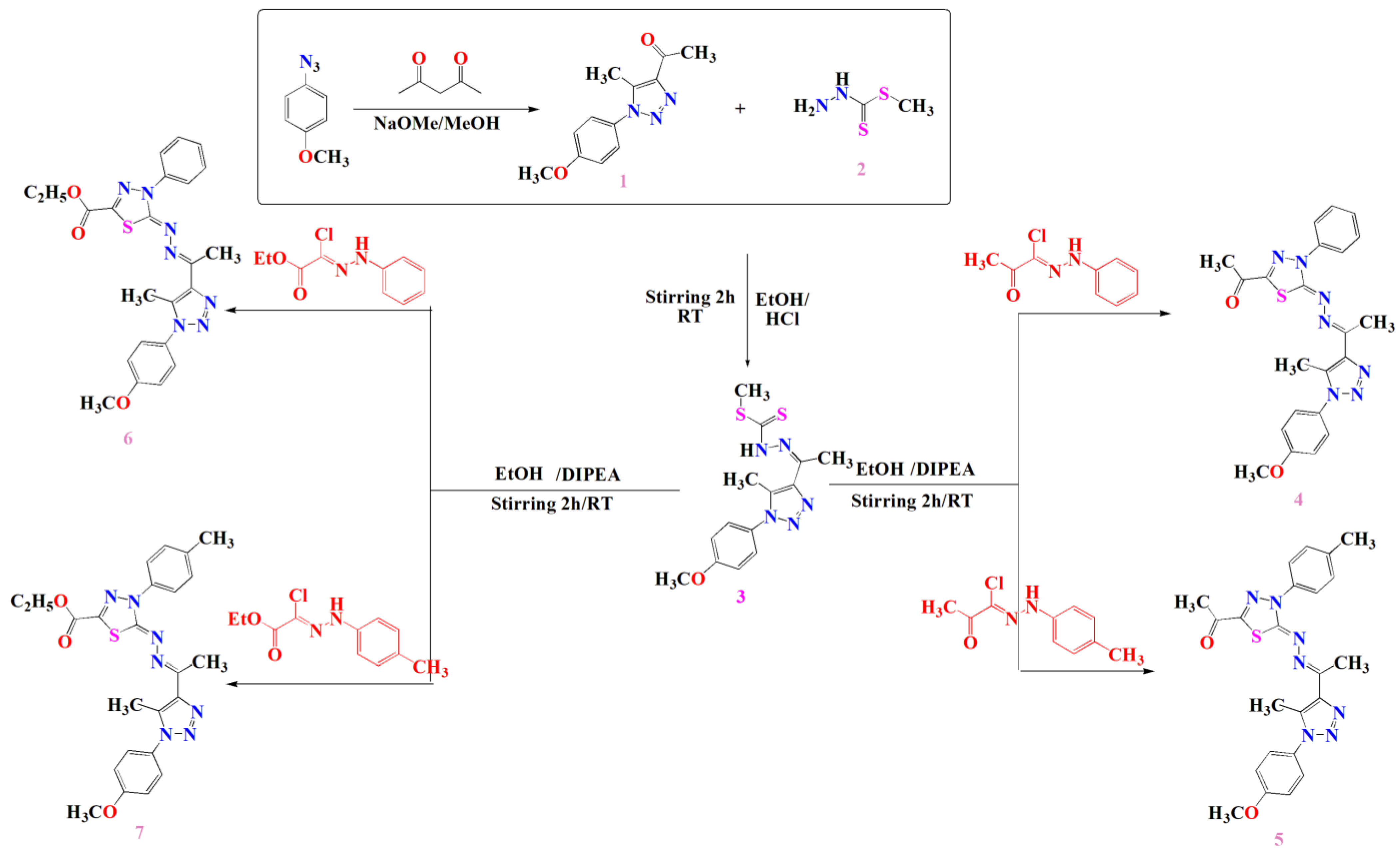
2.1. Chemical Synthesis and Characterization of the Compound 1, 3, 4, 5, 6 and 7
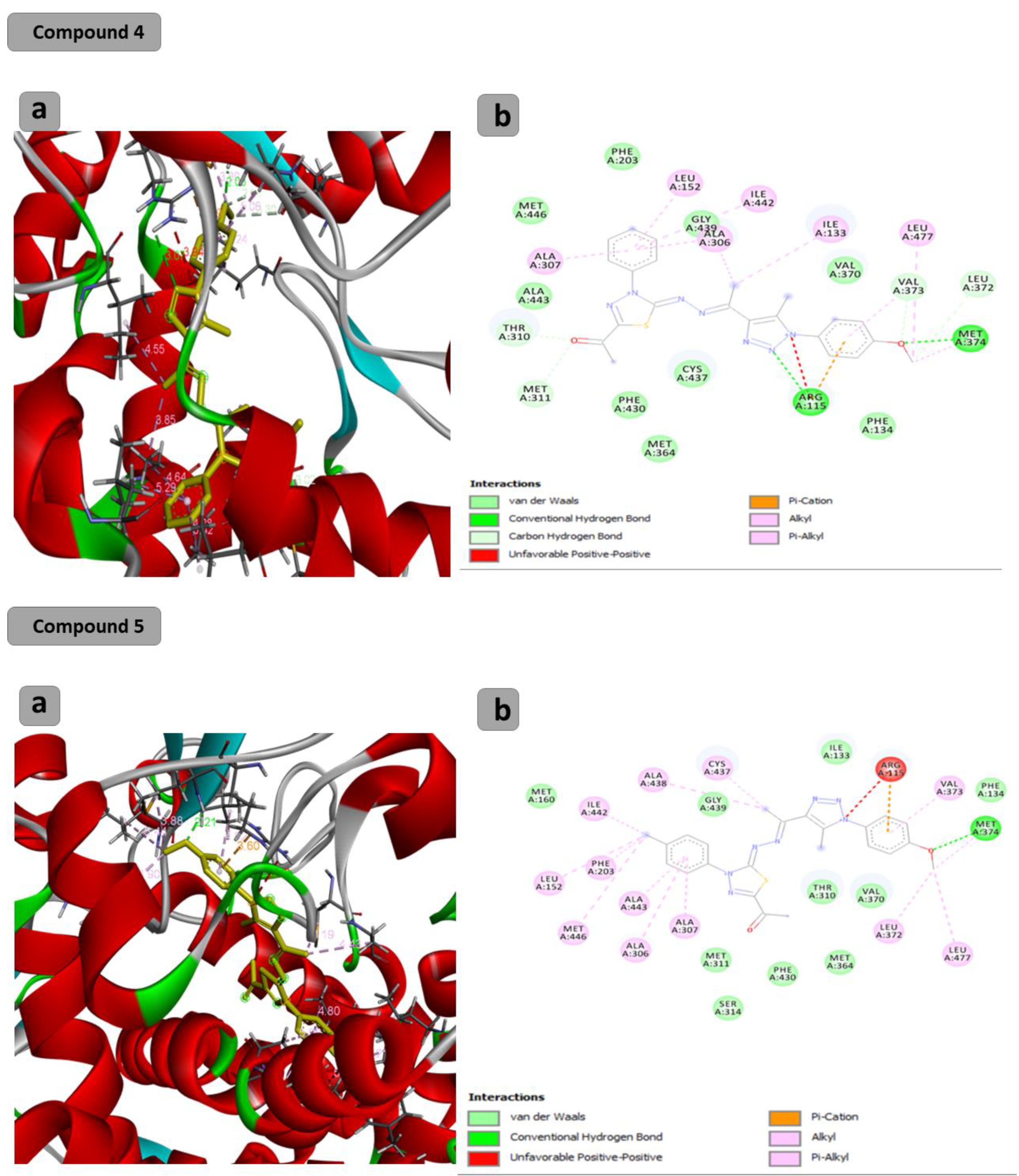
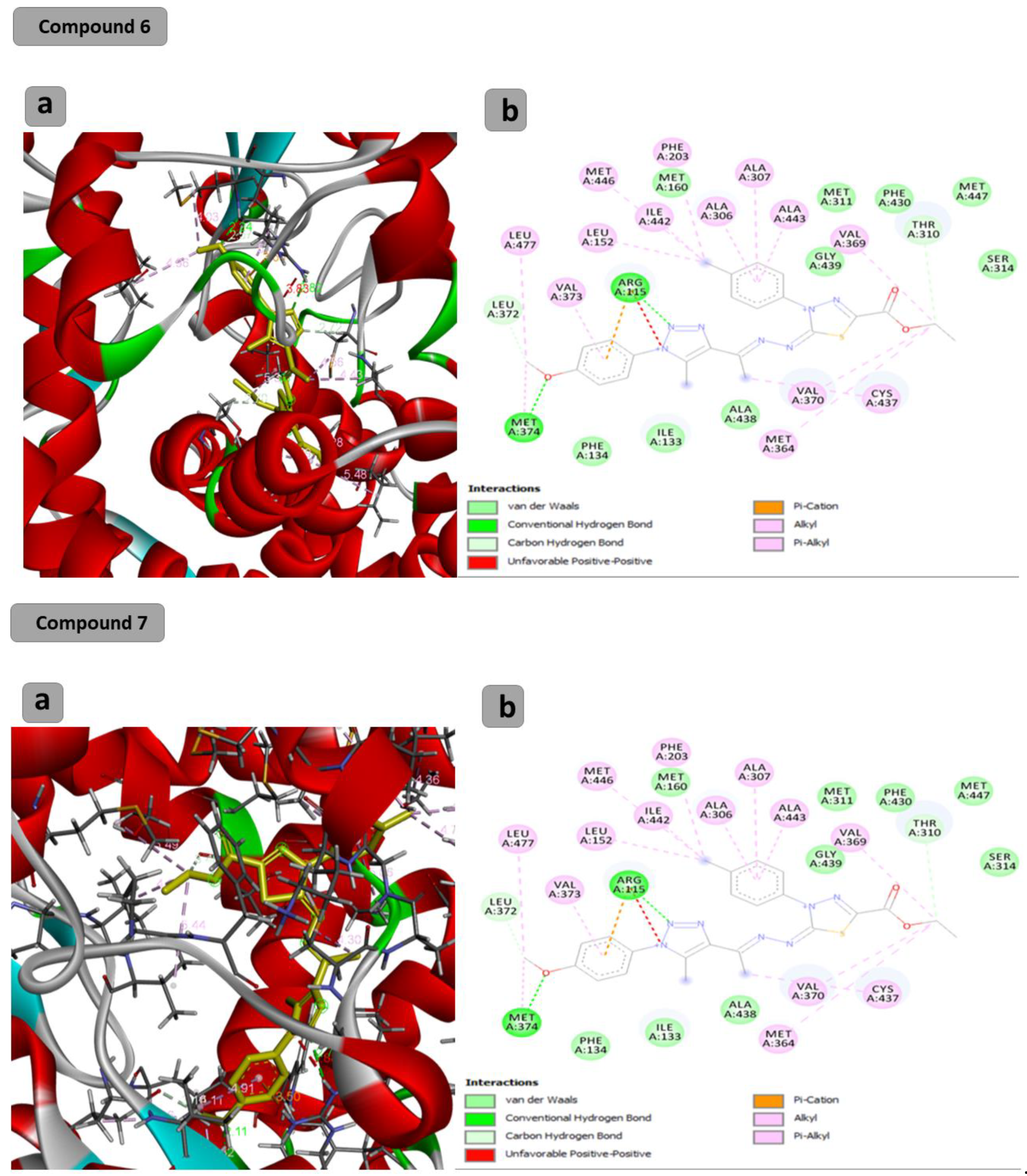
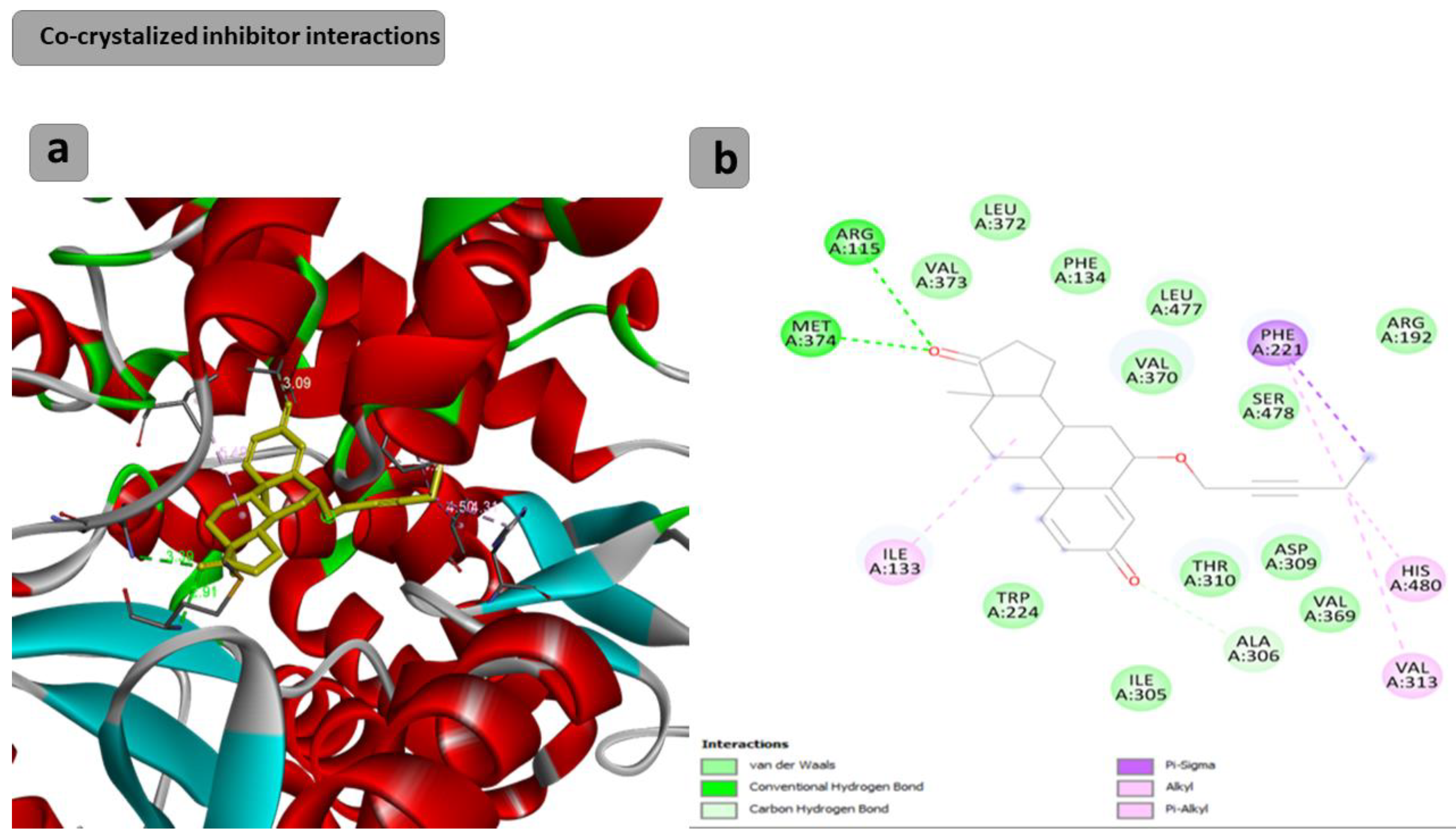
2.2. Computational Study
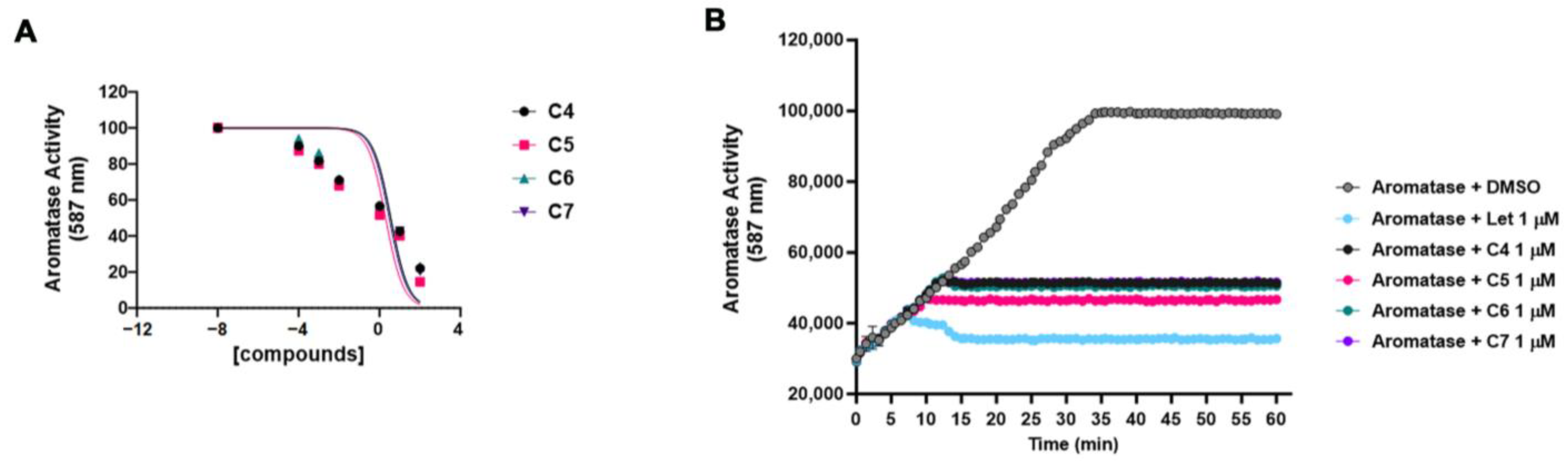
2.3. In Vitro Analysis of the Aromatase Activity Inhibition by C4, C5, C6 and C7
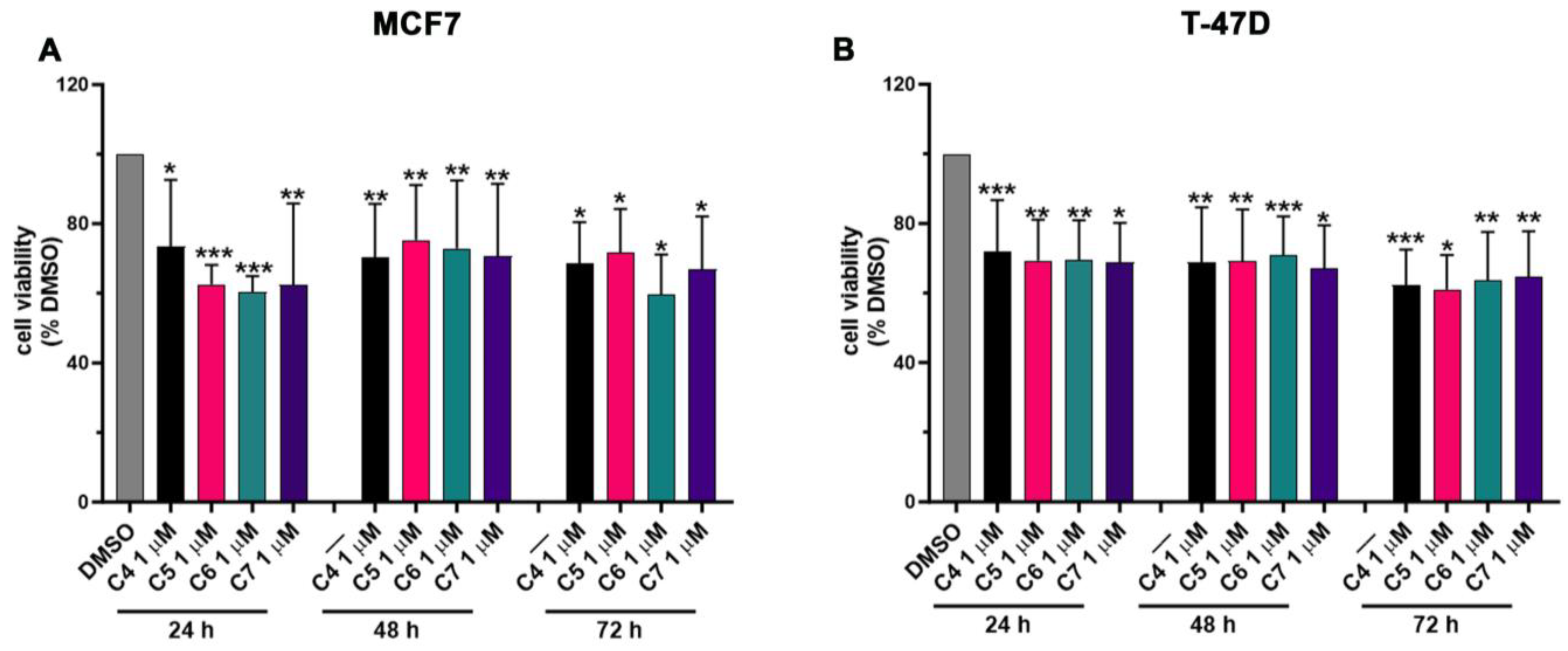
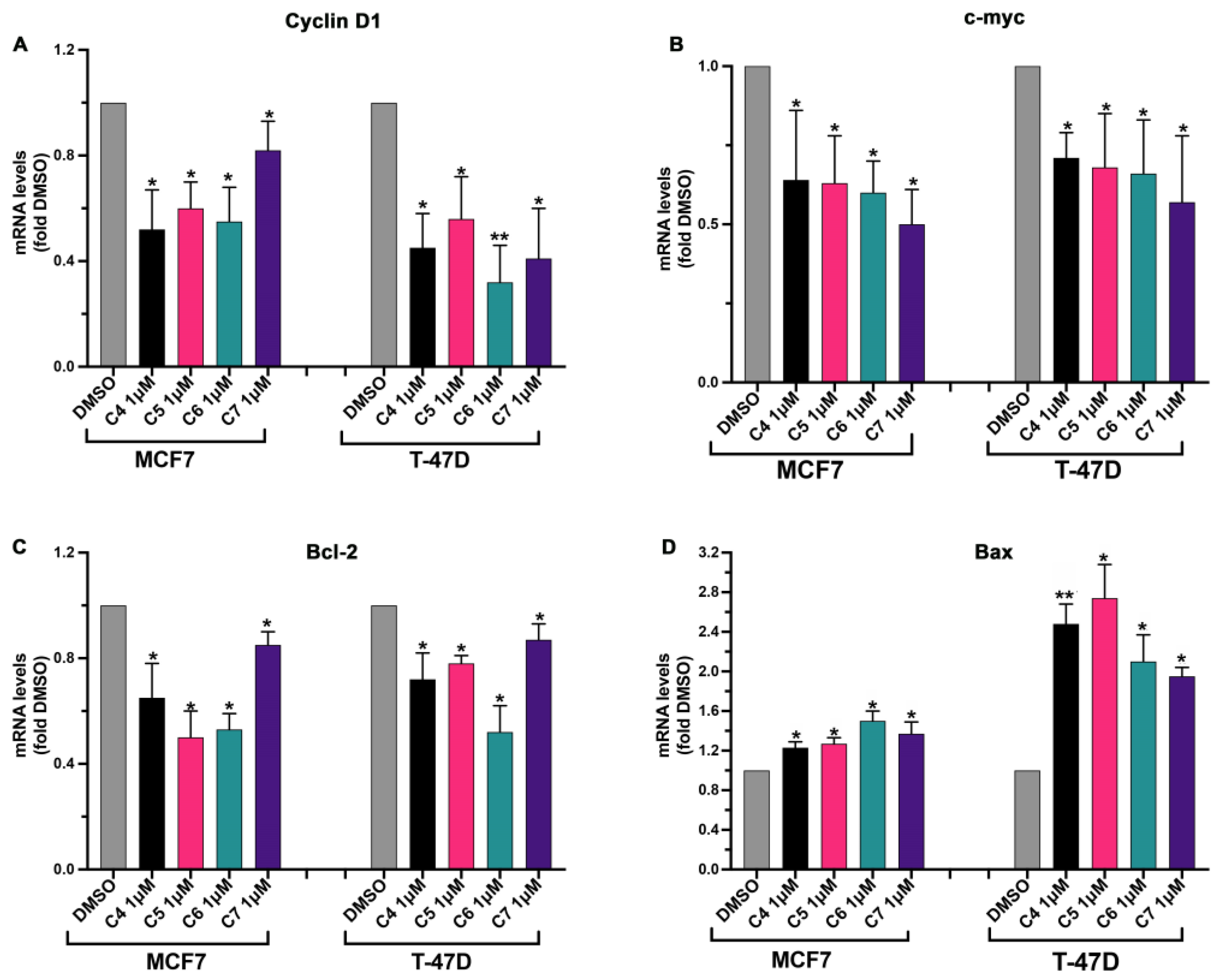
2.4. Anticancer Assays
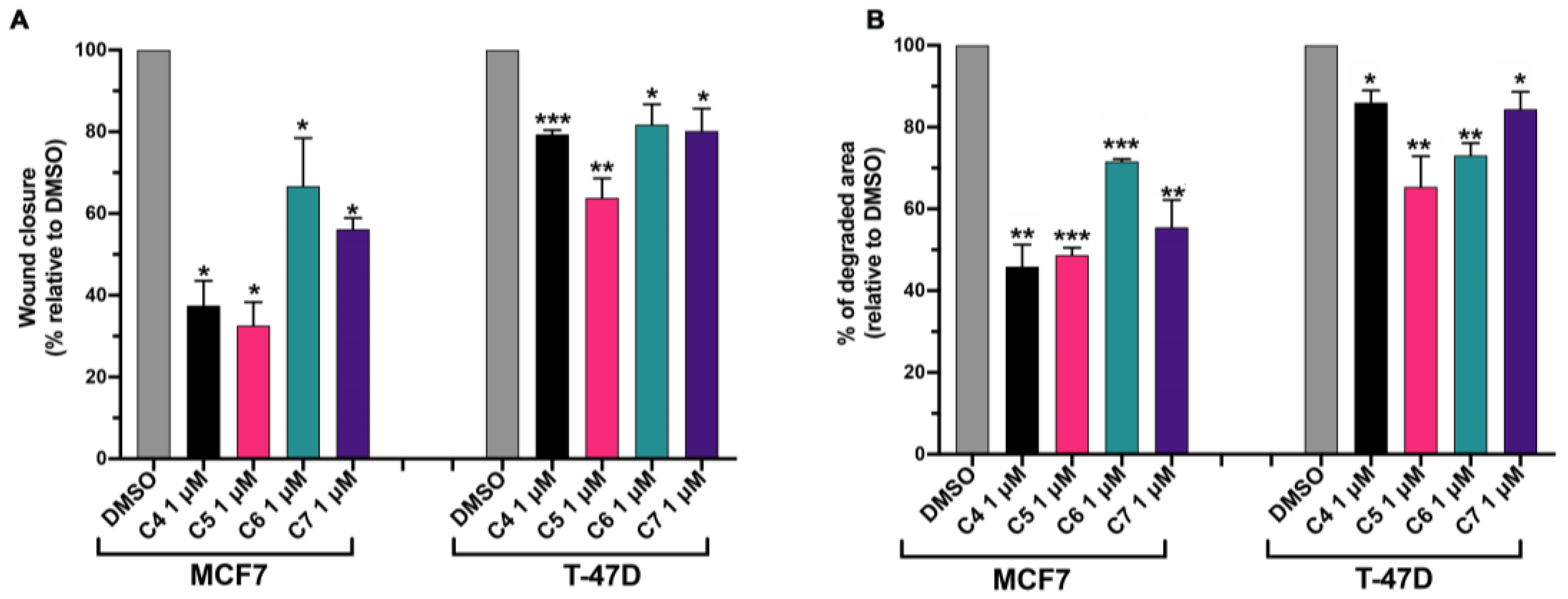
2.5. Compounds 4, 5, 6 and 7 Inhibit Cancer Cell Migration and Invasiveness
3. Materials and Methods
3.1. Chemistry Instrumentation
3.2. Synthesis
3.2.1. 1-(1-(4-Methoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethan-1-one (1)
3.2.2. Methyl 2-(1-(1-(4-Methoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene)hydrazine-1-carbodithioate (3)
3.2.3. General Procedures for Synthesis of Compounds 4–7
3.2.4. 1-(5-((1-(1-(4-Methoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene)hydrazono)-4-phenyl-4,5-dihydro-1,3,4-thiadiazol-2-yl)ethan-1-one (4)
3.2.5. 1-(5-((1-(1-(4-Methoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene)hydrazono)-4-(p-tolyl)-4,5-dihydro-1,3,4-thiadiazol-2-yl)ethan-1-one (5)
3.2.6. Ethyl 5-((1-(1-(4-Methoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene)hydrazono)-4-phenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate (6)
3.2.7. Ethyl 5-((1-(1-(4-Methoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl) ethylidene) hydrazono)-4-(p-tolyl)-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate (7)
3.3. Computational Study
3.4. Aromatase Enzymatic Activity Assay
3.5. Cell Culture
3.6. Anticancer Assay
3.6.1. MTT Assay
3.6.2. RNA Extraction and Real Time qPCR
3.6.3. Wound Healing Assay
3.6.4. Gelatin Degradation (Invasion) Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Safarinejad, M.R.; Shafiei, N.; Safarinejad, S. Quality of life and sexual functioning in young women with early-stage breast cancer 1 year after lumpectomy. Psycho-Oncology 2012, 22, 1242–1248. [Google Scholar] [CrossRef]
- Heemskerk-Gerritsen, B.A.M.; Brekelmans, C.T.M.; Menke-Pluymers, M.B.E.; van Geel, A.N.; Tilanus-Linthorst, M.M.A.; Bartels, C.C.M.; Tan, M.; Meijers-Heijboer, H.E.J.; Klijn, J.G.M.; Seynaeve, C. Prophylactic mastectomy in BRCA1/2 mutation carriers and women at risk of hereditary breast cancer: Long-term experiences at the Rotterdam Family Cancer Clinic. Ann. Surg. Oncol. 2007, 14, 3335–3344. [Google Scholar] [CrossRef]
- Afsharfard, A.; Mozaffar, M.; Orang, E.; Tahmasbpour, E. Trends in epidemiology, clinical and histopathological characteristics of breast cancer in Iran: Results of a 17 year study. Asian Pac. J. Cancer Prev. 2013, 14, 6905–6911. [Google Scholar] [CrossRef]
- Yedjou, C.G.; Sims, J.N.; Miele, L.; Noubissi, F.; Lowe, L.; Fonseca, D.D.; Alo, R.A.; Payton, M.; Tchounwou, P.B. Health and Racial Disparity in Breast Cancer. Adv. Exp. Med. Biol. 2019, 1152, 31–49. [Google Scholar] [CrossRef]
- Britt, K.L.; Cuzick, J.; Phillips, K.-A. Key steps for effective breast cancer prevention. Nat. Rev. Cancer 2020, 20, 417–436. [Google Scholar] [CrossRef]
- Centenera, M.M.; Hickey, T.E.; Jindal, S.; Ryan, N.K.; Ravindranathan, P.; Mohammed, H.; Robinson, J.L.; Schiewer, M.J.; Ma, S.; Kapur, P.; et al. A patient-derived explant (PDE) model of hormone-dependent cancer. Mol. Oncol. 2018, 12, 1608–1622. [Google Scholar] [CrossRef]
- Kur, P.; Kolasa-Wołosiuk, A.; Misiakiewicz-Has, K.; Wiszniewska, B. Sex hormone-dependent physiology and diseases of liver. Int. J. Environ. Res. Public Health 2020, 17, 2620. [Google Scholar] [CrossRef]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef]
- Arbabi, S.; Maier, R.V. Mitogen-activated protein kinases. Crit. Care Med. 2002, 30, S74–S79. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase path-ways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [PubMed]
- Rose, D.P.; Vona-Davis, L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr.-Relat. Cancer 2012, 19, R225–R241. [Google Scholar] [CrossRef]
- He, Z.; Bateman, A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J. Mol. Med. 2003, 81, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D.; Davidson, N.E. Estrogen Carcinogenesis in Breast Cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.C. Computational Investigations of Cytochrome P450 Aromatase Catalysis and Biological Evaluation of Isoflavone Aromatase Inhibitors. Ph. D. Thesis, The Ohio State University, Columbus, OH, USA, 2004. [Google Scholar]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of Estrogen Receptor-Positive Breast Cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- Almeida, C.F.; Oliveira, A.; Ramos, M.J.; Fernandes, P.A.; Teixeira, N.; Amaral, C. Estrogen receptor-positive (ER+) breast cancer treatment: Are multi-target compounds the next promising approach? Biochem. Pharmacol. 2020, 177, 113989. [Google Scholar] [CrossRef]
- Caciolla, J.; Bisi, A.; Belluti, F.; Rampa, A.; Gobbi, S. Reconsidering Aromatase for Breast Cancer Treatment: New Roles for an Old Target. Molecules 2020, 25, 5351. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Ratre, P.; Mishra, K.; Dubey, A.; Vyas, A.; Jain, A.; Thareja, S. Aromatase inhibitors for the treatment of breast cancer: A journey from the scratch. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Cancer Agents) 2020, 20, 1994–2004. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Ariazi, J.L.; Cordera, F.; Jordan, V.C. Estrogen Receptors as Therapeutic Targets in Breast Cancer. Curr. Top. Med. Chem. 2006, 6, 181–202. [Google Scholar] [CrossRef]
- Grilli, S. Tamoxifen (TAM): The dispute goes on. Ann.-Ist. Super. Di Sanita 2006, 42, 170. [Google Scholar]
- Mandlekar, S.; Kong, A.-N.T. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001, 6, 469–477. [Google Scholar] [CrossRef]
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk out-come in cancer. World J. Methodol. 2016, 6, 43. [Google Scholar] [CrossRef]
- Sukocheva, O.A.; Lukina, E.; Friedemann, M.; Menschikowski, M.; Hagelgans, A.; Aliev, G. The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: Current findings and future perspectives. Semin. Cancer Biol. 2020, 82, 35–59. [Google Scholar] [CrossRef] [PubMed]
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Ruiz Esparza-Garrido, R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer. Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef]
- Begam, A.J.; Jubie, S.; Nanjan, M. Estrogen receptor agonists/antagonists in breast cancer therapy: A critical review. Bioorg. Chem. 2017, 71, 257–274. [Google Scholar] [CrossRef]
- Chin, Y.; Beresford, M.; Ravichandran, D.; Makris, A. Exemestane after non-steroidal aromatase inhibitors for post-menopausal women with advanced breast cancer. Breast 2007, 16, 436–439. [Google Scholar] [CrossRef]
- Franik, S.; Eltrop, S.M.; Kremer, J.A.; Kiesel, L.; Farquhar, C. Aromatase inhibitors (letrozole) for subfertile women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2018, 2018, CD010287. [Google Scholar] [CrossRef]
- Ahlin, C.; Lundgren, C.; Embretsén-Varro, E.; Jirström, K.; Blomqvist, C.; Fjällskog, M.-L. High expression of cyclin D1 is associated to high proliferation rate and increased risk of mortality in women with ER-positive but not in ER-negative breast cancers. Breast Cancer Res. Treat. 2017, 164, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; De Francesco, E.M.; Lappano, R.; Muoio, M.G.; Manzella, L.; Maggiolini, M.; Belfiore, A. Microenvironmental Determinants of Breast Cancer Metastasis: Focus on the Crucial Interplay Between Estrogen and Insulin/Insulin-Like Growth Factor Signaling. Front. Cell Dev. Biol. 2020, 8, 608412. [Google Scholar] [CrossRef]
- T.M.S.M.O. Menu, info@rcsb.org. Available online: www.rcsb.org (accessed on 29 November 2023).
- Varone, A.; Amoruso, C.; Monti, M.; Patheja, M.; Greco, A.; Auletta, L.; Zannetti, A.; Corda, D. The phosphatase Shp1 interacts with and dephosphorylates cortactin to inhibit invadopodia function. Cell Commun. Signal. 2021, 19, 64. [Google Scholar] [CrossRef]
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase inhibitors: Role in postmenopausal breast cancer. Arch. Der Pharm. 2020, 353, e2000081. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Energy (Kcal/mol) | H Bonds | Residual Interactions |
|---|---|---|---|
| C1 | −6.9 | (2): Gly439-ala438 | Leu152-ala307-met446-ala306 phe203-ile442 |
| C3 | −7.0 | Gly439-ala438 | Leu152-ala307-met446-ala306-phe203-ile442-ile132-cys437-val373 |
| C4 | −9.6 | (2): Arg115-met374 | Leu152-ala307-ala306-Leu372-Leu377-arg115-ile133-ile442-met311 |
| C5 | −10.1 | (1): met374 | Leu152-ala307-met446-ala306-phe203-ile442-arg115-Leu372-Leu377-ala343-val373-cys437-ala438- |
| C6 | −9.7 | (2): Arg115-met374 | ala307-ala306-ala343-met364-cys437-Leu477-ala438-arg115-val373-ile442 |
| C7 | −9.8 | (2): Arg115-met374 | Leu152-ala307-met446-Leu372-Leu377-Leu372-Leu377-thr310-cys437-val370-met364-val370-ala443 |
| (6alpha,8alpha)-6-(pent-2-yn-1-yloxy)androsta-1,4-diene-3,17-dione (Inhibitor) | −11.1 | (2): Arg115-met374 | Phe220-His480-val313-ile133-ala306 |
| Compound | 24 h | 48 h | 72 h |
|---|---|---|---|
| C4 | 3.722 μM | 2.267 μM | 1.072 μM |
| C5 | 3.255 μM | 1.201 μM | 1.228 μM |
| C6 | 3.086 μM | 1.417 μM | 1.501 μM |
| C7 | 2.692 μM | 1.052 μM | 1.22 μM |
| Compound | 24 h | 48 h | 72 h |
|---|---|---|---|
| C4 | 3.177 μM | 1.685 μM | 1.67μM |
| C5 | 3.119 μM | 1.897 μM | 1.5 μM |
| C6 | 3.066 μM | 1.643 μM | 1.1 μM |
| C7 | 1.854 μM | 1.455 μM | 1.317 μM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashdan, H.R.M.; Abdelrahman, M.T.; De Luca, A.C.; Mangini, M. Towards a New Generation of Hormone Therapies: Design, Synthesis and Biological Evaluation of Novel 1,2,3-Triazoles as Estrogen-Positive Breast Cancer Therapeutics and Non-Steroidal Aromatase Inhibitors. Pharmaceuticals 2024, 17, 88. https://doi.org/10.3390/ph17010088
Rashdan HRM, Abdelrahman MT, De Luca AC, Mangini M. Towards a New Generation of Hormone Therapies: Design, Synthesis and Biological Evaluation of Novel 1,2,3-Triazoles as Estrogen-Positive Breast Cancer Therapeutics and Non-Steroidal Aromatase Inhibitors. Pharmaceuticals. 2024; 17(1):88. https://doi.org/10.3390/ph17010088
Chicago/Turabian StyleRashdan, Huda R. M., Mohamad T. Abdelrahman, Anna Chiara De Luca, and Maria Mangini. 2024. "Towards a New Generation of Hormone Therapies: Design, Synthesis and Biological Evaluation of Novel 1,2,3-Triazoles as Estrogen-Positive Breast Cancer Therapeutics and Non-Steroidal Aromatase Inhibitors" Pharmaceuticals 17, no. 1: 88. https://doi.org/10.3390/ph17010088
APA StyleRashdan, H. R. M., Abdelrahman, M. T., De Luca, A. C., & Mangini, M. (2024). Towards a New Generation of Hormone Therapies: Design, Synthesis and Biological Evaluation of Novel 1,2,3-Triazoles as Estrogen-Positive Breast Cancer Therapeutics and Non-Steroidal Aromatase Inhibitors. Pharmaceuticals, 17(1), 88. https://doi.org/10.3390/ph17010088