


Paclitaxel and Therapeutic Drug Monitoring with Microsampling in Clinical Practice
 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Method Validation
2.2. Patient Samples
3. Discussion
4. Limitations
5. Materials and Methods
5.1. Sample Collection
5.2. Chemicals and Reagents
5.3. Equipment and Conditions
5.4. Preparation of Calibrators and Quality Control (QC) Samples
5.5. Preparation and Extraction Procedure for Plasma, Whole Blood, and VAMSTM Samples for LC-MS/MS Analysis
5.6. Method Comparison and Statistical Analysis
5.7. Validation Protocol
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Sparano, J.A.; Wang, M.; Martino, S.; Jones, V.; Perez, E.A.; Saphner, T.; Wolff, A.C.; Sledge, G.W., Jr.; Wood, W.C.; Davidson, N.E. Weekly Paclitaxel in the Adjuvant Treatment of Breast Cancer. N. Engl. J. Med. 2008, 358, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Seidman, A.D.; Berry, D.; Cirrincione, C.; Harris, L.; Muss, H.; Marcom, P.K.; Gipson, G.; Burstein, H.; Lake, D.; Shapiro, C.L.; et al. Randomized Phase III Trial of Weekly Compared with Every-3-Weeks Paclitaxel for Metastatic Breast Cancer, with Trastuzumab for all HER-2 Overexpressors and Random Assignment to Trastuzumab or Not in HER-2 Nonoverexpressors: Final Results of Cancer and Leukemia Group B Protocol 9840. J. Clin. Oncol. 2008, 26, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W.; Neuberg, D.; Bernardo, P.; Ingle, J.N.; Martino, S.; Rowinsky, E.K.; Wood, W.C. Phase III Trial of Doxorubicin, Paclitaxel, and the Combination of Doxorubicin and Paclitaxel as Front-Line Chemotherapy for Metastatic Breast Cancer: An Intergroup Trial (E1193). J. Clin. Oncol. 2003, 21, 588–592. [Google Scholar] [CrossRef] [PubMed]
- du Bois, A.; Lück, H.J.; Meier, W.; Adams, H.P.; Möbus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schröder, W.; et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Natl. Cancer Inst. 2003, 95, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.K.; Ledermann, J.A.; Colombo, N.; du Bois, A.; Delaloye, J.F.; Kristensen, G.B.; Wheeler, S.; Swart, A.M.; Qian, W.; Torri, V.; et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: The ICON4/AGO-OVAR-2.2 trial. Lancet 2003, 361, 2099–2106. [Google Scholar] [PubMed]
- Belani, C.P.; Lee, J.S.; Socinski, M.A.; Robert, F.; Waterhouse, D.; Rowland, K.; Ansari, R.; Lilenbaum, R.; Natale, R.B. Randomized phase III trial comparing cisplatin–etoposide to carboplatin–paclitaxel in advanced or metastatic non-small cell lung cancer. Ann. Oncol. 2005, 16, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Forastiere, A.A.; Shank, D.; Neuberg, D.; Taylor, S.G.; DeConti, R.C.; Adams, G. Final report of a phase II evaluation of paclitaxel in patients with advanced squamous cell carcinoma of the head and neck: An Eastern Cooperative Oncology Group trial (PA390). Cancer 1998, 82, 2270–2274. [Google Scholar] [CrossRef]
- Hitt, R.; López-Pousa, A.; Martínez-Trufero, J.; Escrig, V.; Carles, J.; Rizo, A.; Isla, D.; Vega, M.E.; Martí, J.L.; Lobo, F.; et al. Phase III Study Comparing Cisplatin Plus Fluorouracil to Paclitaxel, Cisplatin, and Fluorouracil Induction Chemotherapy Followed by Chemoradiotherapy in Locally Advanced Head and Neck Cancer. J. Clin. Oncol. 2005, 23, 8636–8645. [Google Scholar] [CrossRef]
- Manfredi, J.J.; Horwitz, S.B. Taxol: An antimitotic agent with a new mechanism of action. Pharmacol. Ther. 1984, 25, 83–125. [Google Scholar] [CrossRef]
- Huizing, M.T.; Giaccone, G.; van Warmerdam, L.J.; Rosing, H.; Bakker, P.J.; Vermorken, J.B.; Postmus, P.E.; van Zandwijk, N.; Koolen, M.G.; Huinink, W.W.T.B.; et al. Pharmacokinetics of paclitaxel and carboplatin in a dose-escalating and dose-sequencing study in patients with non-small-cell lung cancer. The European Cancer Centre. J. Clin. Oncol. 1997, 15, 317–329. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Joerger, M.; Huitema, A.D.R.; Huizing, M.T.; Willemse, P.H.B.; De Graeff, A.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H.; Vermorken, J.B. Safety and pharmacology of paclitaxel in patients with impaired liver function: A population pharmacokinetic-pharmacodynamic study. Br. J. Clin. Pharmacol. 2007, 64, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.O.; Molnar, V.; Freijs, A.; Nygren, P.; Bergh, J.; Larsson, R. Pharmacokinetic models for the saturable distribution of paclitaxel. Drug Metab. Dispos. 1999, 27, 1220–1223. [Google Scholar] [PubMed]
- Sonnichsen, D.S.; A Hurwitz, C.; Pratt, C.B.; Shuster, J.J.; Relling, M.V. Saturable pharmacokinetics and paclitaxel pharmacodynamics in children with solid tumors. J. Clin. Oncol. 1994, 12, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Huizing, M.T.; Panday, V.R.N.; Schellens, J.H.M.; Nooijen, W.J.; Beijnen, J.H. Cremophor EL causes (pseudo-) non-linear pharmacokinetics of paclitaxel in patients. Br. J. Cancer 1999, 81, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Abraxane (Nab-Paclitaxel, Package Insert); Celgene Corporation: Summit, NJ, USA, 2020.
- Joerger, M.; Huitema, A.D.; Richel, D.J.; Dittrich, C.; Pavlidis, N.; Briasoulis, E.; Vermorken, J.B.; Strocchi, E.; Martoni, A.; Sorio, R.; et al. Population Pharmacokinetics and Pharmacodynamics of Paclitaxel and Carboplatin in Ovarian Cancer Patients: A Study by the European Organization for Research and Treatment of Cancer-Pharmacology and Molecular Mechanisms Group and New Drug Development Group. Clin. Cancer Res. 2007, 13, 6410–6418. [Google Scholar] [CrossRef]
- Mielke, S.; Sparreboom, A.; Steinberg, S.M.; Gelderblom, H.; Unger, C.; Behringer, D.; Mross, K. Association of Paclitaxel Pharmacokinetics with the Development of Peripheral Neuropathy in Patients with Advanced Cancer. Clin. Cancer Res. 2005, 11, 4843–4850. [Google Scholar] [CrossRef]
- Augusto, C.; Pietro, M.; Cinzia, M.; Sergio, C.; Sara, C.; Luca, G.; Scaioli, V. Peripheral neuropathy due to paclitaxel: Study of the temporal relationships between the therapeutic schedule and the clinical quantitative score (QST) and comparison with neurophysiological findings. J. Neuro-Oncol. 2008, 86, 89–99. [Google Scholar] [CrossRef]
- Joerger, M.; Von Pawel, J.; Kraff, S.; Fischer, J.R.; Eberhardt, W.; Gauler, T.C.; Mueller, L.; Reinmuth, N.; Reck, M.; Kimmich, M.; et al. Open-label, randomized study of individualized, pharmacokinetically (PK)-guided dosing of paclitaxel combined with carboplatin or cisplatin in patients with advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2016, 27, 1895–1902. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, F.; Qi, H.; Ni, H.; Hu, Q.; Zhou, C.; Li, Y.; Baburina, I.; Courtney, J.; Salamone, S.J. Randomized study of individualized pharmacokinetically-guided dosing of paclitaxel compared with body-surface area dosing in Chinese patients with advanced non-small cell lung cancer. Br. J. Clin. Pharmacol. 2019, 85, 2292–2301. [Google Scholar] [CrossRef]
- Woo, M.H.; Relling, M.V.; Sonnichsen, D.S.; Rivera, G.K.; Pratt, C.B.; Pui, C.H.; Evans, W.E.; Pappo, A.S. Phase I targeted systemic exposure study of paclitaxel in children with refractory acute leukemias. Clin. Cancer Res. 1999, 5, 543–549. [Google Scholar] [PubMed]
- de Jonge, M.E.; van den Bongard, H.D.; Huitema, A.D.; Mathôt, R.A.; Rosing, H.; Baas, P.; van Zandwijk, N.; Beijnen, J.H.; Schellens, J.H.M. Bayesian pharmacokinetically guided dosing of paclitaxel in patients with non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Mielke, S.; Sparreboom, A.; Behringer, D.; Mross, K. Paclitaxel pharmacokinetics and response to chemotherapy in patients with advanced cancer treated with a weekly regimen. Anticancer. Res. 2005, 25, 4423–4427. [Google Scholar]
- Joerger, M.; Kraff, S.; Huitema, A.D.; Feiss, G.; Moritz, B.; Schellens, J.H.; Beijnen, J.H.; Jaehde, U. Evaluation of a pharmacology-driven dosing algorithm of 3-weekly paclitaxel using therapeutic drug moni-toring: A pharmacokinetic-pharmacodynamic simulation study. Clin. Pharmacokinet 2012, 51, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Kraff, S.; Nieuweboer, A.J.M.; Mathijssen, R.H.J.; Baty, F.; De Graan, A.-J.; Van Schaik, R.H.N.; Jaehde, U.; Joerger, M. Pharmacokinetically based dosing of weekly paclitaxel to reduce drug-related neurotoxicity based on a single sample strategy. Cancer Chemother. Pharmacol. 2015, 75, 975–983. [Google Scholar] [CrossRef]
- van Zuylen, L.; Karlsson, M.O.; Verweij, J.; Brouwer, E.; de Bruijn, P.; Nooter, K.; Stoter, G.; Sparreboom, A. Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemother. Pharmacol. 2001, 47, 309–318. [Google Scholar] [CrossRef]
- ten Tije, A.J.; Verweij, J.; Loos, W.J.; Sparreboom, A. Pharmacological effects of formulation vehicles: Implications for cancer chemotherapy. Clin. Pharmacokinet 2003, 42, 665–685. [Google Scholar] [CrossRef]
- Andriguetti, N.B.; Hahn, R.Z.; Lizot, L.F.; Raymundo, S.; Costa, J.L.; da Cunha, K.F.; Vilela, R.M.; Kluck, H.M.; Schwartsmann, G.; Antunes, M.V.; et al. Analytical and clinical validation of a dried blood spot assay for the determination of paclitaxel using high-performance liquid chromatography-tandem mass spectrometry. Clin. Biochem. 2018, 54, 123–130. [Google Scholar] [CrossRef]
- Spooner, N.; Denniff, P.; Michielsen, L.; De Vries, R.; Ji, Q.C.; Arnold, M.E.; Woods, K.; Woolf, E.J.; Xu, Y.; Boutet, V.; et al. A device for dried blood microsampling in quantitative bioanalysis: Overcoming the issues associated blood hematocrit. Bioanalysis 2015, 7, 653–659. [Google Scholar] [CrossRef]
- Capiau, S.; Veenhof, H.; Koster, R.A.; Bergqvist, Y.; Boettcher, M.; Halmingh, O.; Keevil, B.G.; Koch, B.C.; Linden, R.; Pistos, C.; et al. Official International Association for Therapeutic Drug Monitoring and Clinical Toxicology Guideline: Devel-opment and Validation of Dried Blood Spot-Based Methods for Therapeutic Drug Monitoring. Ther. Drug Monit. 2019, 41, 409–430. [Google Scholar] [CrossRef]
- ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis; European Medicines Agency: Amsterdam, The Netherlands, 2023.
- Vethe, N.T.; Gustavsen, M.T.; Midtvedt, K.; Lauritsen, M.E.; Andersen, A.M.; Åsberg, A.; Bergan, S. Tacrolimus Can Be Reliably Measured with Volumetric Absorptive Capillary Microsampling throughout the Dose Interval in Renal Transplant Recipients. Ther. Drug Monit. 2019, 41, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Martial, L.C.; Aarnoutse, R.E.; Schreuder, M.F.; Henriet, S.S.; Brüggemann, R.J.M.; Joore, M.A. Cost Evaluation of Dried Blood Spot Home Sampling as Compared to Conventional Sampling for Therapeutic Drug Monitoring in Children. PLoS ONE 2016, 11, e0167433. [Google Scholar] [CrossRef] [PubMed]
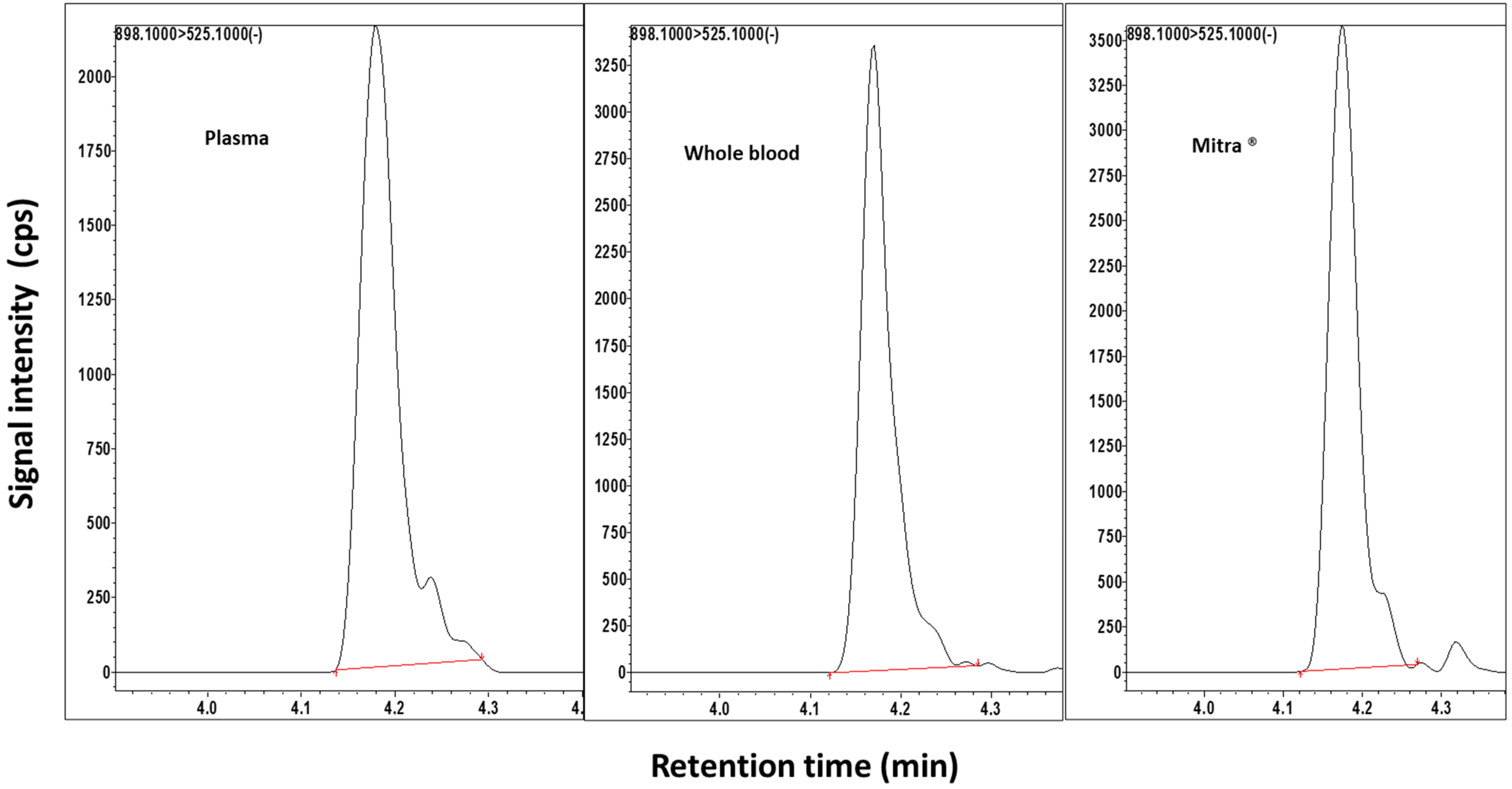

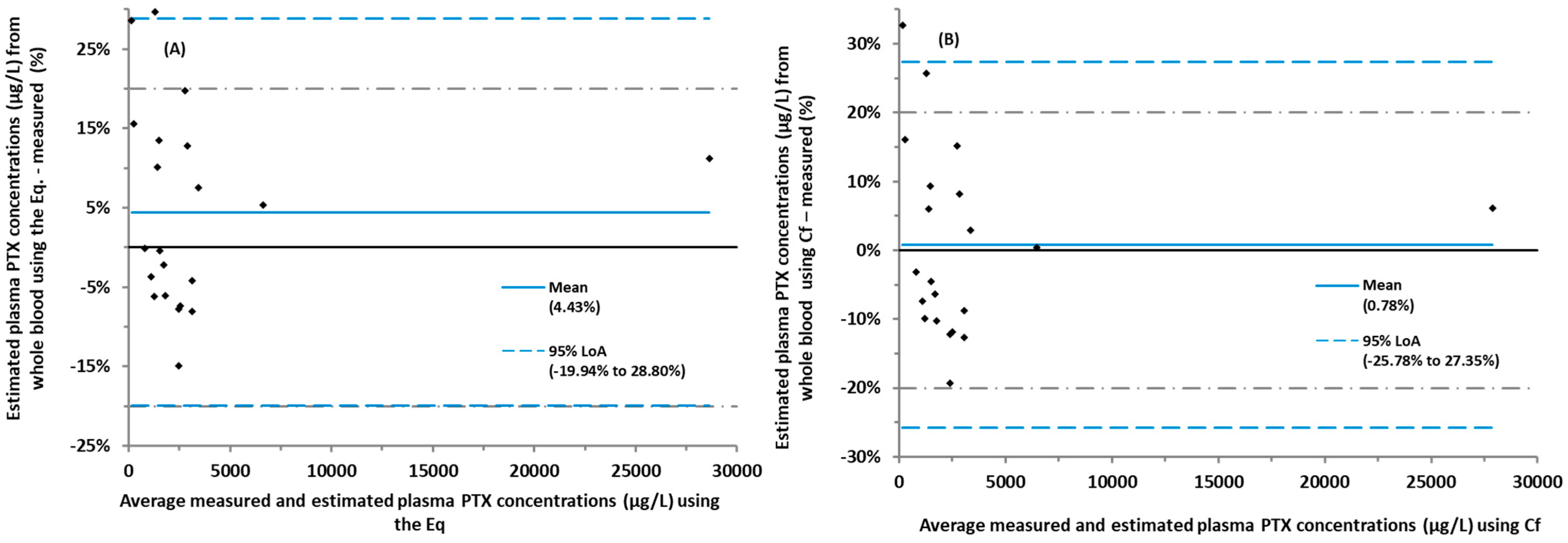


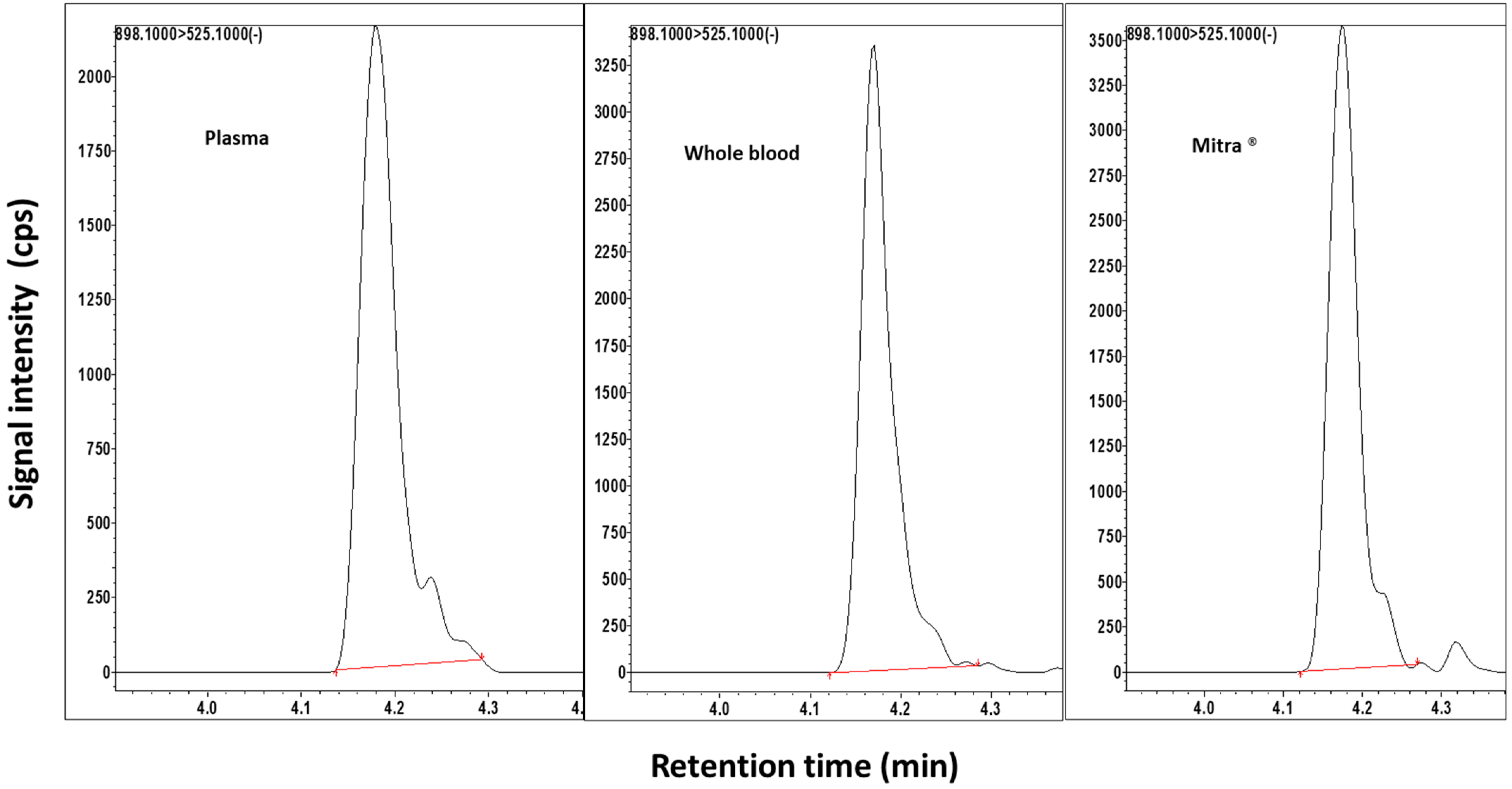{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasma | Whole Blood | Mitra® | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Concentration (µg/L) | Mean Concentration ± SD (n = 13) | CV (%) | Accuracy (%) | Mean Concentration ± SD (n = 13) | CV (%) | Accuracy (%) | Mean Concentration ± SD (n = 13) | CV (%) | Accuracy (%) |
| 5 # | 6.13 ± 0.36 | 5.8 | 123 | 5.9 ± 1.14 | 19 | 118 | 5.50 ± 0.32 | 5.7 | 110 |
| 8 | 8.22 ± 0.79 | 9.7 | 103 | 8.21 ± 1.47 | 17.8 | 103 | 7.96 ± 0.51 | 6.4 | 99.5 |
| 80 | 90.1 ± 6.5 | 7.2 | 113 | 84.5 ± 10.3 | 12.2 | 106 | 79.8 ± 7.3 | 9.2 | 99.7 |
| 400 | 420 ± 30 | 7.1 | 105 | 387 ± 69 | 17.8 | 97 | 412 ± 45 | 11.0 | 103 |
| 4000 * | 4009 ± 353 | 8.8 | 100 | 3625 ± 640 | 17.7 | 91 | 4456 ± 389 | 9 | 111 |
| E % Plasma Recovery Using an Eq | E % Plasma Recovery Using a Cf | ||||
|---|---|---|---|---|---|
| Whole Blood | Whole Blood Mitra® | Fingerprick Mitra® | Whole Blood | Whole Blood Mitra® | Fingerprick Mitra® |
| 111 | 101 | 119 | 106 | 111 | 120 |
| 114 | 143 | 93 | 109 | 158 | 105 |
| 122 | 133 | 147 | 116 | 147 | 175 |
| 135 | 105 | n/A | 130 | 115 | n/A |
| 96 | 69 | 60 | 92 | 76 | 64 |
| 94 | 51 | 76 | 90 | 56 | 73 |
| 114 | n/a | 84 | 110 | n/A | 74 |
| 94 | 66 | 147 | 91 | 72 | 156 |
| 93 | 74 | n/A | 88 | 81 | n/A |
| 86 | 72 | 71 | 82 | 80 | 74 |
| 112 | 92 | n/A | 106 | 102 | n/A |
| 100 | 80 | n/A | 96 | 88 | n/A |
| 100 | 98 | 100 | 97 | 107 | 69 |
| 133 | 122 | n/A | 139 | 127 | n/A |
| 93 | 104 | n/A | 89 | 115 | n/A |
| 98 | 86 | n/A | 94 | 95 | n/A |
| 117 | 131 | 287 | 118 | 141 | 176 |
| 92 | 80 | 92 | 88 | 88 | 106 |
| 96 | 130 | 124 | 93 | 143 | 120 |
| 106 | 106 | n/A | 100 | 118 | n/A |
| 108 | 117 | 141 | 103 | 129 | 172 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radovanovic, M.; Galettis, P.; Flynn, A.; Martin, J.H.; Schneider, J.J. Paclitaxel and Therapeutic Drug Monitoring with Microsampling in Clinical Practice. Pharmaceuticals 2024, 17, 63. https://doi.org/10.3390/ph17010063
Radovanovic M, Galettis P, Flynn A, Martin JH, Schneider JJ. Paclitaxel and Therapeutic Drug Monitoring with Microsampling in Clinical Practice. Pharmaceuticals. 2024; 17(1):63. https://doi.org/10.3390/ph17010063
Chicago/Turabian StyleRadovanovic, Mirjana, Peter Galettis, Alex Flynn, Jennifer H. Martin, and Jennifer J. Schneider. 2024. "Paclitaxel and Therapeutic Drug Monitoring with Microsampling in Clinical Practice" Pharmaceuticals 17, no. 1: 63. https://doi.org/10.3390/ph17010063
APA StyleRadovanovic, M., Galettis, P., Flynn, A., Martin, J. H., & Schneider, J. J. (2024). Paclitaxel and Therapeutic Drug Monitoring with Microsampling in Clinical Practice. Pharmaceuticals, 17(1), 63. https://doi.org/10.3390/ph17010063