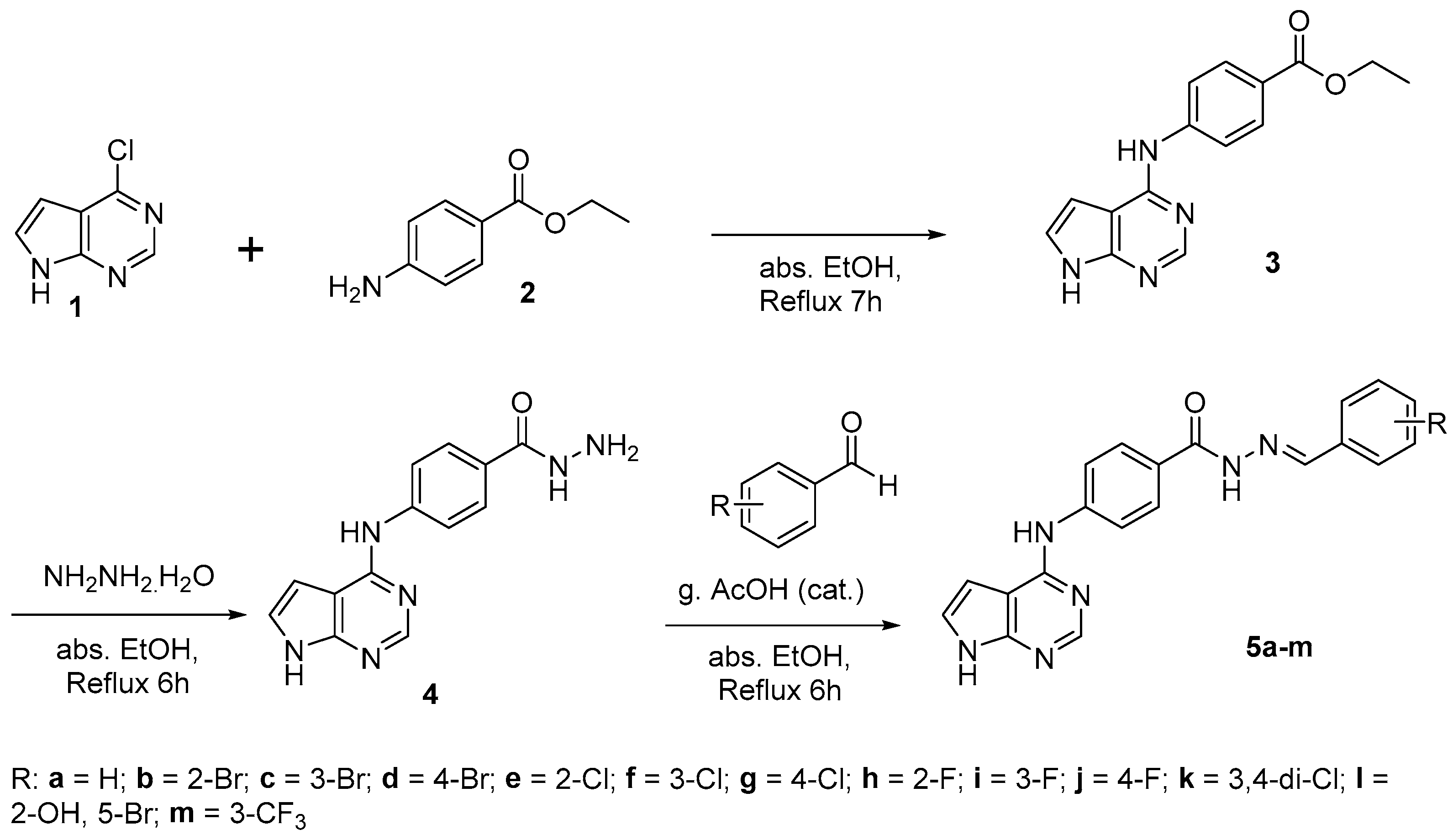
3.2.3. General Synthetic Method for the Synthesis of 5a–m
Compounds
5a–
m were synthesized according to our previously reported method [
31]. In brief, compound
4 (100 mg, 0.373 mmol) and substituted-aldehyde (1.2 equiv.) was placed in a round bottom flask (RBF) with 10 mL of absolute ethanol. Then, 3–4 drops of (cat.) glacial acetic acid were added to the reaction mixture, which was then refluxed overnight. The resulting precipitate was filtered, washed with ethanol, and dried under vacuum.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-benzylidenebenzohydrazide (5a) [31]
White solid (120 mg, 0.343 mmol, 92%). Mp. 294 °C (Lit Mp. 294 °C) [
31].
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(2-bromobenzylidene)benzohydrazide (5b)
White solid (157 mg, 0.362 mmol, 97%). Mp. 294 °C. FT-IR (KBr): ν (cm−1) = 3452, 3424, 3193, 1638, 1611, 1574, 1521, 1465, 1357, 1293, 1184, 1023, 898, 759, 719 and 607 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 12.03 (s, 1H), 11.87 (s, 1H), 9.64 (s, 1H), 8.84 (s, 1H), 8.38 (s, 1H), 8.11 (d, J = 8.4 Hz, 2H), 8.03 (d, J = 7.8 Hz, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.0 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.31 (t, J = 2.9 Hz, 1H) and 6.87 (dd, J = 3.6, 1.9 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.24, 153.50, 151.57, 151.07, 145.73, 144.52, 133.73, 133.64, 132.08, 128.97, 128.59 (2C), 127.70, 126.16, 123.94, 123.28, 119.31 (2C), 104.66 and 99.20 ppm. Mass (ESI): m/z 435 [79(Br)M + H]+, 437 [81(Br)M + H]+; 457 [79(Br)M + Na]+, 473 [79(Br)M + K]+, 475 [81(Br)M + K]+; HRMS (ESI): Calc. for C20H15BrN6O [M]+ 434.0491, found 434.6068.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(3-bromobenzylidene)benzohydrazide (5c)
White solid (151 mg, 0.347 mmol, 93%). Mp. 296 °C. FT-IR (KBr): ν (cm−1) = 3417, 3214, 2988, 2850, 2711, 1639, 1613, 1585, 1572, 1518, 1471, 1421, 1348, 1279, 1240, 1190, 1144, 1070, 951, 913, 895, 790, 759, 721, 683 and 521 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.90 (s, 1H), 11.87 (s, 1H), 9.63 (s, 1H), 8.44 (s, 1H), 8.38 (s, 1H), 8.11 (d, J = 8.8 Hz, 2H), 7.94 (d, J = 8.1 Hz, 3H), 7.74 (d, J = 7.6 Hz, 1H), 7.64 (d, J = 7.4 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.31 (dd, J = 3.4, 2.2 Hz, 1H) and 6.87 (dd, J = 3.5, 1.8 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.31, 153.51, 151.56, 151.07, 145.68, 144.48, 137.48, 132.90, 131.51, 129.48 (2C), 128.95, 126.64, 126.24, 123.27, 122.66, 119.32 (2C), 104.65 and 99.20 ppm. Mass (ESI): m/z 435 [79(Br)M + H]+, 437 [81(Br)M + H]+; 457 [79(Br)M + Na]+, 459 [81(Br)M + Na]+, 473 [79(Br)M + K]+, 475 [81(Br)M + K]+.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(4-bromobenzylidene)benzohydrazide (5d)
White solid (156 mg, 0.358 mmol, 96%). Mp. 321 °C. FT-IR (KBr): ν (cm−1) = 3440, 3396, 3212, 3098, 3048, 2992, 2850, 2708, 1653, 1618, 1588, 1575, 1527, 1477, 1424, 1357, 1313, 1298, 1245, 1193, 1667, 1009, 897, 828, 719, 980 and 514 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.87 (s, 1H), 11.83 (s, 1H), 9.63 (s, 1H), 8.45 (s, 1H), 8.37 (s, 1H), 8.10 (d, J = 8.8 Hz, 2H), 7.94 (d, J = 8.3 Hz, 2H), 7.73–7.65 (m, 4H), 7.31 (dd, J = 3.4, 2.3 Hz, 1H) and 6.87 (dd, J = 3.5, 1.9 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.23, 153.51, 151.56, 151.07, 146.26, 144.42, 134.28, 132.33 (4C), 129.34 (2C), 128.90, 126.33, 123.59, 123.26, 119.33 (2C), 104.64 and 99.20 ppm. Mass (ESI): m/z 435 [79(Br)M + H]+, 437 [81(Br)M + H]+; 457 [79(Br)M + Na]+, 459 [81(Br)M + Na]+, 473 [79(Br)M + K]+, 475 [81(Br)M + K]+.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(2-chlorobenzylidene)benzohydrazide (5e)
White solid (143 mg, 0.365 mmol, 98%). Mp. 298 °C. FT-IR (KBr): ν (cm−1) = 3301, 3214, 3110, 3066, 3015, 2923, 2850, 1655, 1617, 1569, 1464, 1360, 1281, 1265, 1246, 1074, 855, 757, 727 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 12.00 (s, 1H), 11.87 (s, 1H), 9.64 (s, 1H), 8.89 (s, 1H), 8.38 (s, 1H), 8.11 (d, J = 8.5 Hz, 2H), 8.06 (s, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.59–7.52 (m, 1H), 7.46 (dd, J = 6.7, 3.3 Hz, 2H), 7.31 (dd, J = 3.4, 2.3 Hz, 1H) and 6.87 (dd, J = 3.4, 1.9 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.23, 153.50, 151.56, 151.07, 144.52, 143.39, 133.56, 132.22, 131.83, 130.41 (2C), 128.96, 128.11 (2C), 127.32, 126.15, 123.28, 119.32 (2C), 104.66 and 99.20 ppm. Mass (ESI): m/z 391 [35(Cl)M + H]+, 393 [37(Cl)M + H]+, 413 [35(Cl)M + Na]+, 415 [37(Cl)M + Na]+; HRMS (ESI): Calc. for C20H15ClN6O [M]+ 390.0996, found 389.9511.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(3-chlorobenzylidene)benzohydrazide (5f)
White solid (138 mg, 0.354 mmol, 95%). Mp. 281 °C. FT-IR (KBr): ν (cm−1) = 3520, 3448, 3215, 3094, 2988, 2850, 1626, 1612, 1575, 1466, 1430, 1354, 1292, 1250, 1187, 1128, 1059, 897, 827, 783, 734, 685 and 520 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.87 (s, 1H), 11.85 (s, 1H), 9.61 (s, 1H), 8.43 (s, 1H), 8.36 (s, 1H), 8.09 (dd, 2H), 7.92 (d, J = 8.3 Hz, 2H), 7.78 (s, 1H), 7.68 (s, 1H), 7.49 (d, J = 4.5 Hz, 2H), 7.29 (dd, J = 3.4, 2.3 Hz, 1H) and 6.85 (dd, J = 3.5, 1.8 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.31, 153.51, 151.57, 151.07, 145.79, 144.47, 137.25, 134.13, 131.24 (2C), 130.02, 128.95, 126.64, 126.22, 123.27, 119.32 (2C), 104.65 and 99.20 ppm. Mass (ESI): m/z 391 [35(Cl)M + H]+, 393 [37(Cl)M + H]+; HRMS (ESI): Calc. for C20H15ClN6O [M]+ 390.0996, found 389.9585.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(4-chlorobenzylidene)benzohydrazide (5g)
White solid (134 mg, 0.343 mmol, 92%). Mp. 314 °C. FT-IR (KBr): ν (cm−1) = 3418, 3350, 3086, 2964, 2848, 1648, 1609, 1573, 1474, 1419, 1349, 1281, 1236, 1185, 1046, 899, 824, 721, 633 and 604 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.87 (s, 1H), 11.83 (s, 1H), 9.63 (s, 1H), 8.47 (s, 1H), 8.38 (s, 1H), 8.11 (d, J = 8.4 Hz, 2H), 7.94 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.31 (dd, J = 8.1 Hz, 1H) and 6.87 (dd, J = 3.3, 1.7 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.24, 153.52, 151.57, 151.07, 146.19, 144.43, 134.81, 133.94, 129.42 (4C), 129.10, 128.91 (2C), 126.35, 123.26, 119.34 (2C), 104.65 and 99.20 ppm. Mass (ESI): m/z 391 [M + H]+, 413 [M + Na]+; HRMS (ESI): Calc. for C20H15ClN6O [M]+ 390.0996, found 389.9400.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(2-fluorobenzylidene)benzohydrazide (5h)
White solid (130 mg, 0.347 mmol, 93%). Mp. 307 °C. FT-IR (KBr): ν (cm−1) = 3419, 3365, 3094, 2966, 2849, 1648, 1609, 1572, 1475, 1420, 1350, 1282, 1238, 1188, 1047, 900, 826, 719, 632 and 506 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.89 (s, 1H), 11.87 (s, 1H), 9.64 (s, 1H), 8.72 (s, 1H), 8.38 (s, 1H), 8.11 (d, J = 8.5 Hz, 2H), 7.98 (s, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.51 (q, J = 7.1 Hz, 1H), 7.33 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 3.1 Hz, 1H), 7.31 (d, J = 2.2 Hz, 1H) and 6.87 (dd, J = 3.5, 1.7 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 162.62 (1JF = 237 Hz), 160.51, 153.51, 151.52, 151.06, 144.48, 140.26, 132.38, 128.92 (2C), 126.79 (3JF = 2.6 Hz), 126.26, 125.44 (3JF = 3.2 Hz), 123.30, 122.45 (2JF = 9.8 Hz), 119.37 (2C), 116.48 (2JF = 18.0 Hz), 104.65 and 99.21 ppm. Mass (ESI): m/z 375 [M + H]+, 397 [M + Na]+, 413 [M + K]+; HRMS (ESI): Calc. for C20H15FN6O [M]+ 374.1291, found 374.0985.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(3-fluorobenzylidene)benzohydrazide (5i)
White solid (134 mg, 0.358 mmol, 96%). Mp. 295 °C. FT-IR (KBr): ν (cm−1) = 3432, 3227, 3119, 3063, 2924, 1653, 1610, 1576, 1474, 1293, 1235, 1194, 1065, 958, 889, 785, 728, 688 and 521 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.87 (s, 1H), 11.83 (s, 1H), 9.64 (s, 1H), 8.44 (s, 1H), 8.34 (s, 1H), 8.08 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.3 Hz, 2H), 7.56 (t, J = 10.7 Hz, 2H), 7.50 (q, J = 7.4 Hz, 1H), 7.28 (dd, J = 3.4, 2.3 Hz, 1H), 7.26 (td, J = 8.6, 2.6 Hz, 1H) and 6.84 (dd, J = 3.5, 1.9 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.42, 162.90 (d, 1JCF = 244 Hz), 153.51, 151.50, 151.05, 146.26, 144.45, 137.50 (d, 3JCF = 8 Hz), 131.42 (d, 3JCF = 8 Hz), 128.93 (2C), 126.20, 123.94, 123.30, 119.38 (2C), 117.19 (d, 2JCF = 21 Hz), 113.44 (d, 2JCF = 21 Hz), 104.64 and 99.21 ppm. Mass (ESI): m/z 375 [M + H]+, 397 [M + Na]+; HRMS (ESI): Calc. for C20H15FN6O [M]+ 374.1291, found 374.2093.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(4-fluorobenzylidene)benzohydrazide (5j)
White solid (134 mg, 0.358 mmol, 96%). Mp. 309 °C. FT-IR (KBr): ν (cm−1) = 3430, 3232, 1651, 1606, 1576, 1504, 1473, 1287, 1236, 1218, 1191, 1059, 963, 892, 840, 729, 676 and 512 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.85 (s, 1H), 11.75 (s, 1H), 9.60 (s, 1H), 8.46 (s, 1H), 8.36 (s, 1H), 8.09 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 8.3 Hz, 2H), 7.78 (dd, J = 7.2 Hz, 2H), 7.34–7.26 (m, 3H) and 6.85 (dd, J = 3.5, 1.7 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 13C NMR (176 MHz, DMSO) δ 163.51 (d, 1JCF = 248 Hz), 163.21, 153.52, 151.56, 151.08, 146.41, 144.37, 131.59 (d, 4JCF = 2.9 Hz, 2C), 129.62 (d, 3JCF = 8.6 Hz), 128.87 (2C), 126.44, 123.25, 119.34 (2C), 116.38 (d, 2JCF = 21.8 Hz, 2C), 104.64 and 99.20 ppm. Mass (ESI): m/z 375 [M + H]+; HRMS (ESI): Calc. for C20H16FN6O [M + H]+ 375.1364, found 375.5109.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(3,4-dichlorobenzylidene)benzohydrazide (5k)
White solid (147 mg, 0.347 mmol, 93%). Mp. 294 °C. FT-IR (KBr): ν (cm−1) = 3349, 3299, 3202, 3111, 2967, 1629, 1606, 1557, 1503, 1460, 1405, 1286, 1238, 1190, 1121, 1080, 891, 751, 598 and 516 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.96 (s, 1H), 11.87 (s, 1H), 9.64 (s, 1H), 8.44 (s, 1H), 8.38 (s, 1H), 8.12 (d, J = 8.8 Hz, 2H), 8.01–7.89 (m, 3H), 7.74 (d, J = 5.4 Hz, 2H), 7.31 (dd, J = 3.4, 2.4 Hz, 1H) and 6.87 (dd, J = 3.5, 1.9 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.36, 153.50, 151.57, 151.06, 144.76, 144.53, 135.85, 132.53, 132.19, 131.57, 128.98 (2C), 128.86, 127.30, 126.15, 123.27, 119.32 (2C), 104.67 and 99.20 ppm. Mass (ESI): m/z 425 [M (35(Cl) + H]+, 427 [M (37(Cl) + H]+, 447 [M + Na]+; HRMS (ESI): Calc. for C20H14Cl2N6O [M]+ 424.0606, found 424.4530.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(5-bromo-2-hydroxybenzylidene)benzohydrazide (5l)
White solid (152 mg, 0.358 mmol, 96%). Mp. 334 °C. FT-IR (KBr): ν (cm−1) = 3456, 3350, 3316, 3226, 3116, 1666, 1612, 1470, 1464, 1420, 1339, 1373, 1204, 892, 738, 628 and 593 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 12.11 (s, 1H), 11.87 (s, 1H), 11.41 (s, 1H), 9.65 (s, 1H), 8.62 (s, 1H), 8.38 (s, 1H), 8.12 (d, J = 8.5 Hz, 2H), 7.96 (d, J = 8.4 Hz, 2H), 7.80 (s, 1H), 7.44 (dd, J = 8.7, 2.5 Hz, 1H), 7.32 (dd, J = 3.4, 2.3 Hz, 1H), 6.92 (d, J = 8.7 Hz, 1H) and 6.87 (dd, J = 3.5, 1.9 Hz, 1H) ppm. 13C-NMR (176 MHz, DMSO-d6) δ 163.02, 156.90, 153.49, 151.58, 151.06, 145.59, 144.65, 133.88, 131.03, 128.99 (2C), 125.69, 123.31, 121.85, 119.34, 119.16 (2C), 110.88, 104.68 and 99.20 ppm. Mass (ESI): m/z 451 [79(Br)M + H]+, 453 [81(Br)M + H]+, 473 [79(Br)M + Na]+, 475 [81(Br)M + Na]+.
(E)-4-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-N’-(3-(trifluoromethyl)benzylidene)benzohydrazide (5m)
White solid (147 mg, 0.347 mmol, 93%). Mp. 280 °C. FT-IR (KBr): ν (cm−1) = 3480, 3438, 3236, 3103, 2991, 2851, 1656, 1620, 1575, 1525, 1475, 1425, 1355, 1290, 1249, 1199, 1158, 1121, 1074, 895, 719 and 530 cm−1. 1H-NMR (700 MHz, DMSO-d6) δ 11.96 (s, 1H), 11.87 (s, 1H), 9.64 (s, 1H), 8.56 (s, 1H), 8.38 (s, 1H), 8.11 (d, J = 8.8 Hz, 2H), 8.09 (s, 1H), 8.04 (d, J = 7.7 Hz, 1H), 7.95 (d, J = 8.3 Hz, 2H), 7.81 (d, J = 7.7 Hz, 1H), 7.73 (t, J = 7.7 Hz, 1H), 7.31 (dd, J = 3.4, 2.3 Hz, 1H) and 6.87 (dd, J = 3.4, 1.9 Hz, 1H) ppm. 13C NMR (176 MHz, DMSO-d6) δ 13C-NMR (176 MHz, DMSO) δ 163.42, 153.51, 151.56, 151.06, 145.74, 144.51, 136.17, 131.50, 130.54, 130.10 (q, 1JCF = 64, 32 Hz), 128.97 (2C), 126.39 (d, 2JCF = 78 Hz), 125.31, 123.76, 123.36 (d, 3JCF = 3.8 Hz), 123.28, 119.34 (2C), 104.66 and 99.20 ppm. Mass (ESI), m/z 425 [M + H]+. HRMS (ESI): Calc. for C21H15F3N6O [M]+ 424.1259 found 424.0000.



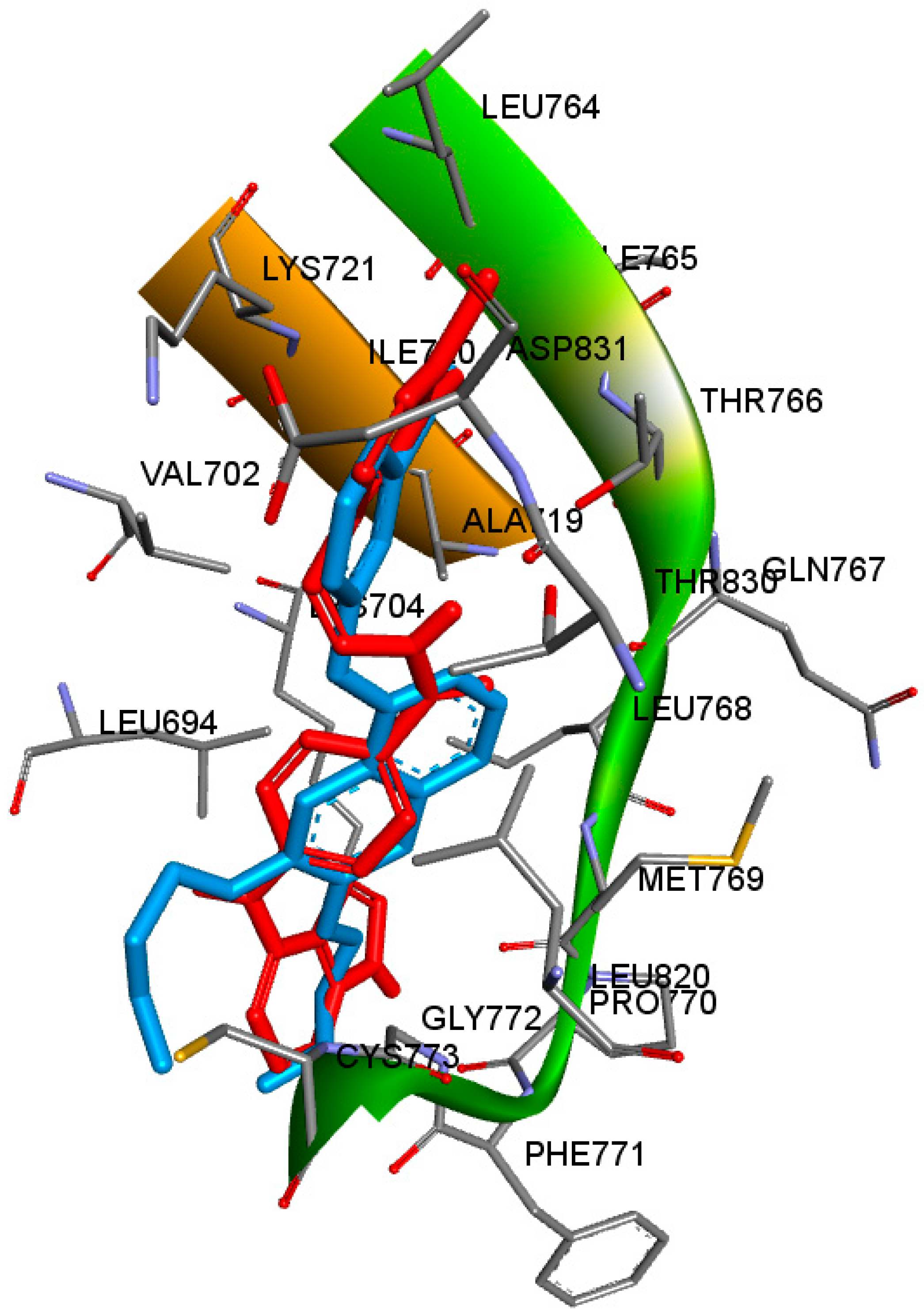
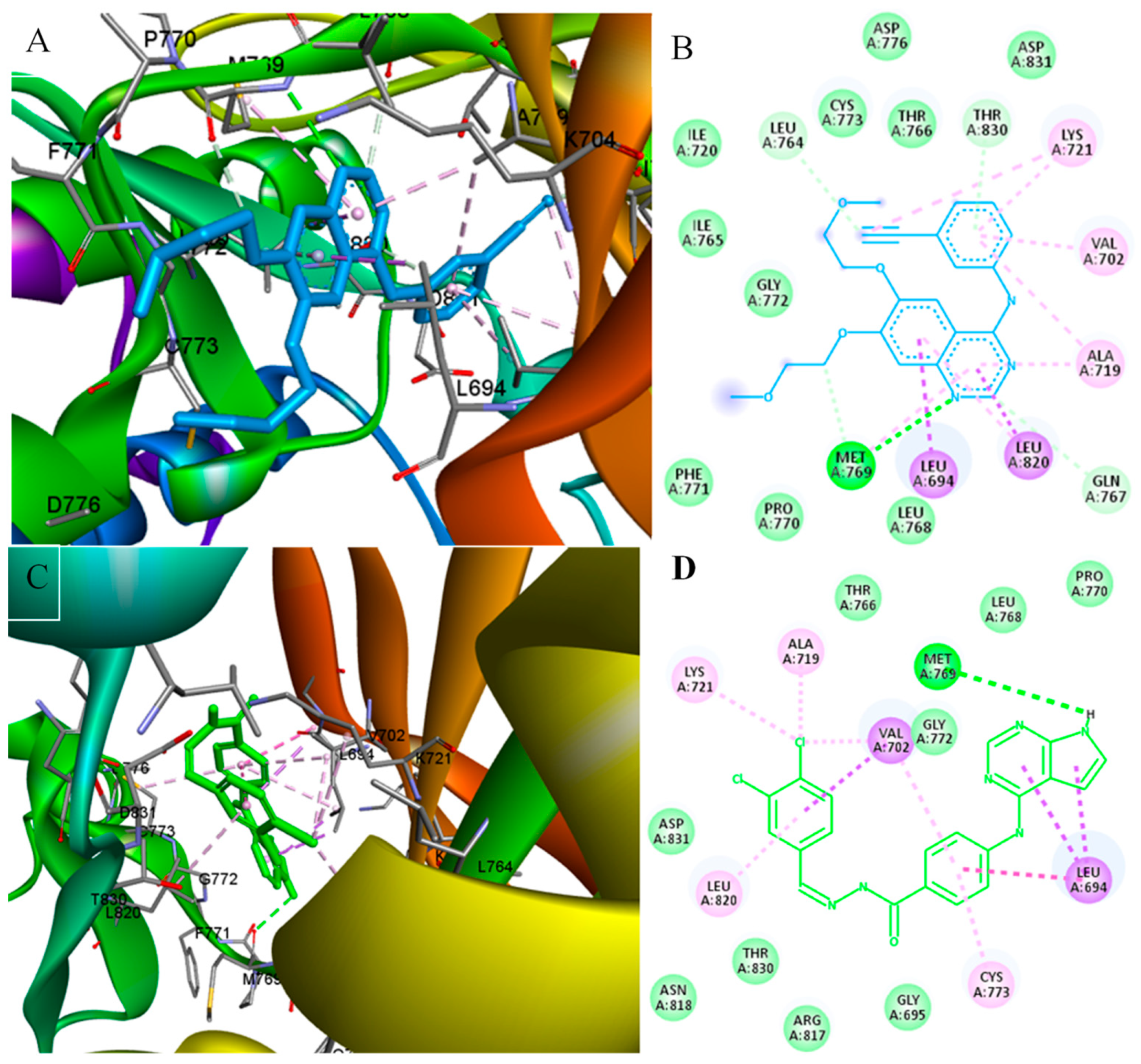
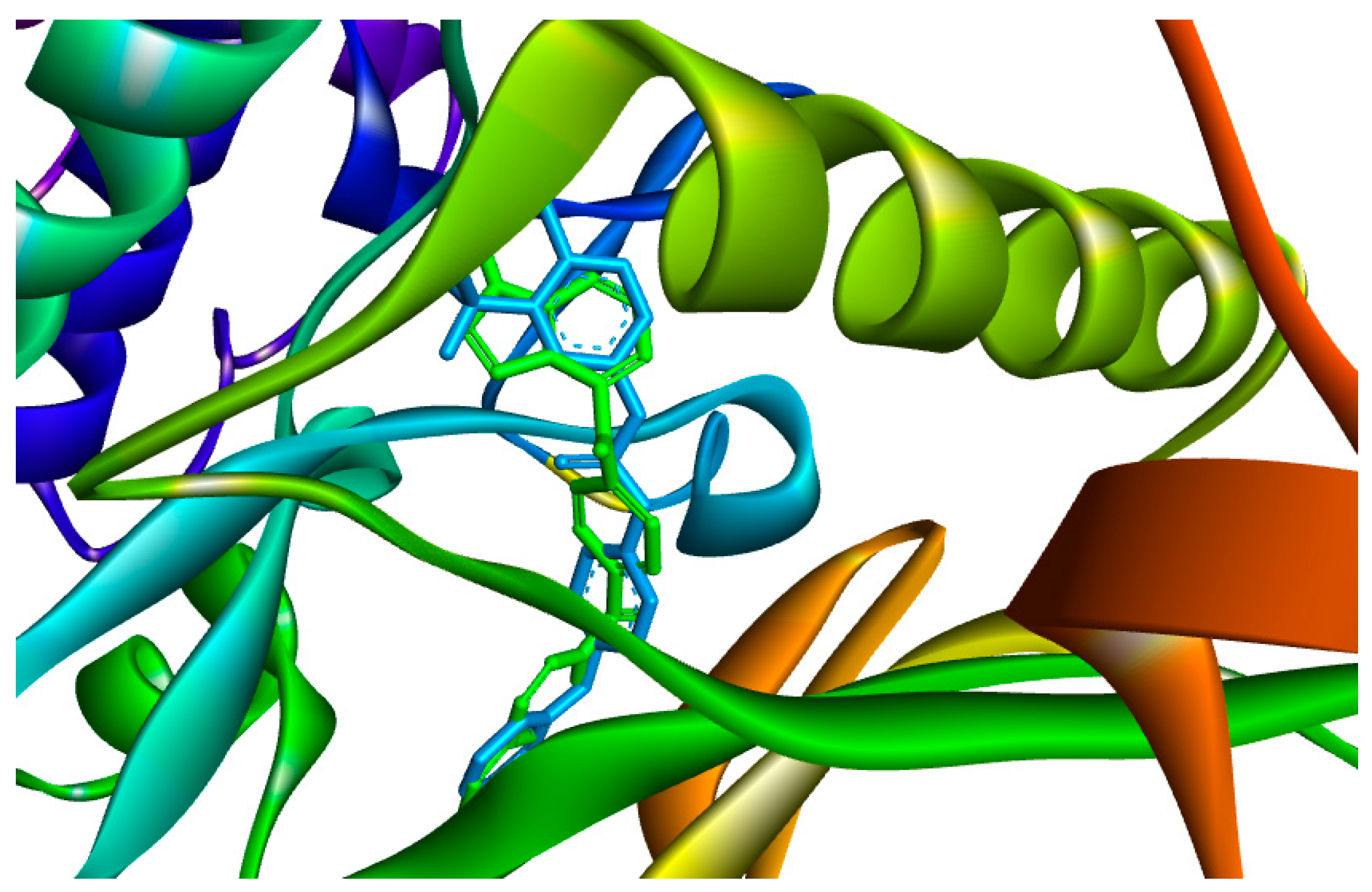
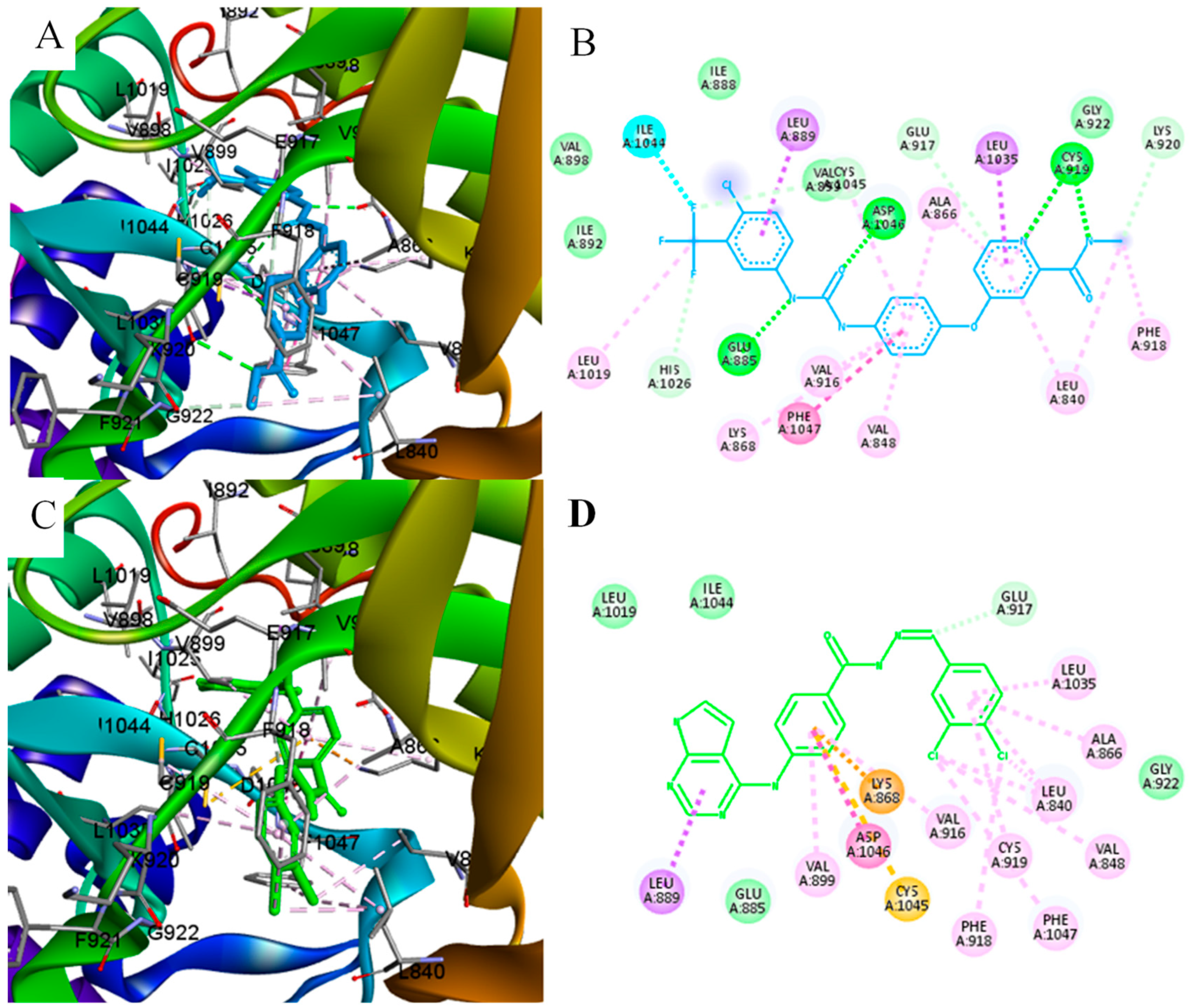

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}