

Quinoline- and Isoindoline-Integrated Polycyclic Compounds as Antioxidant, and Antidiabetic Agents Targeting the Dual Inhibition of α-Glycosidase and α-Amylase Enzymes



Abstract
:1. Introduction
2. Results and Discussion
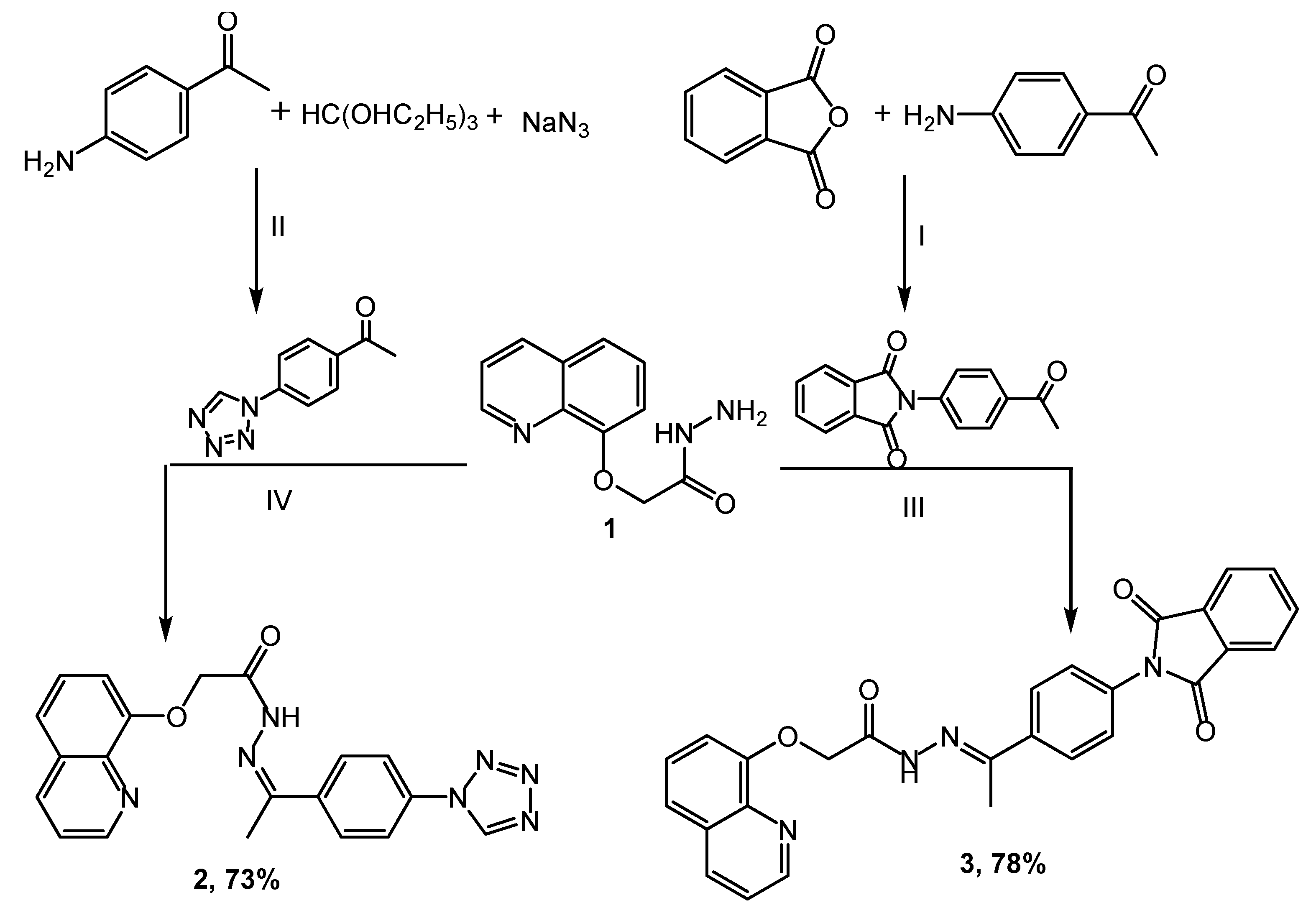
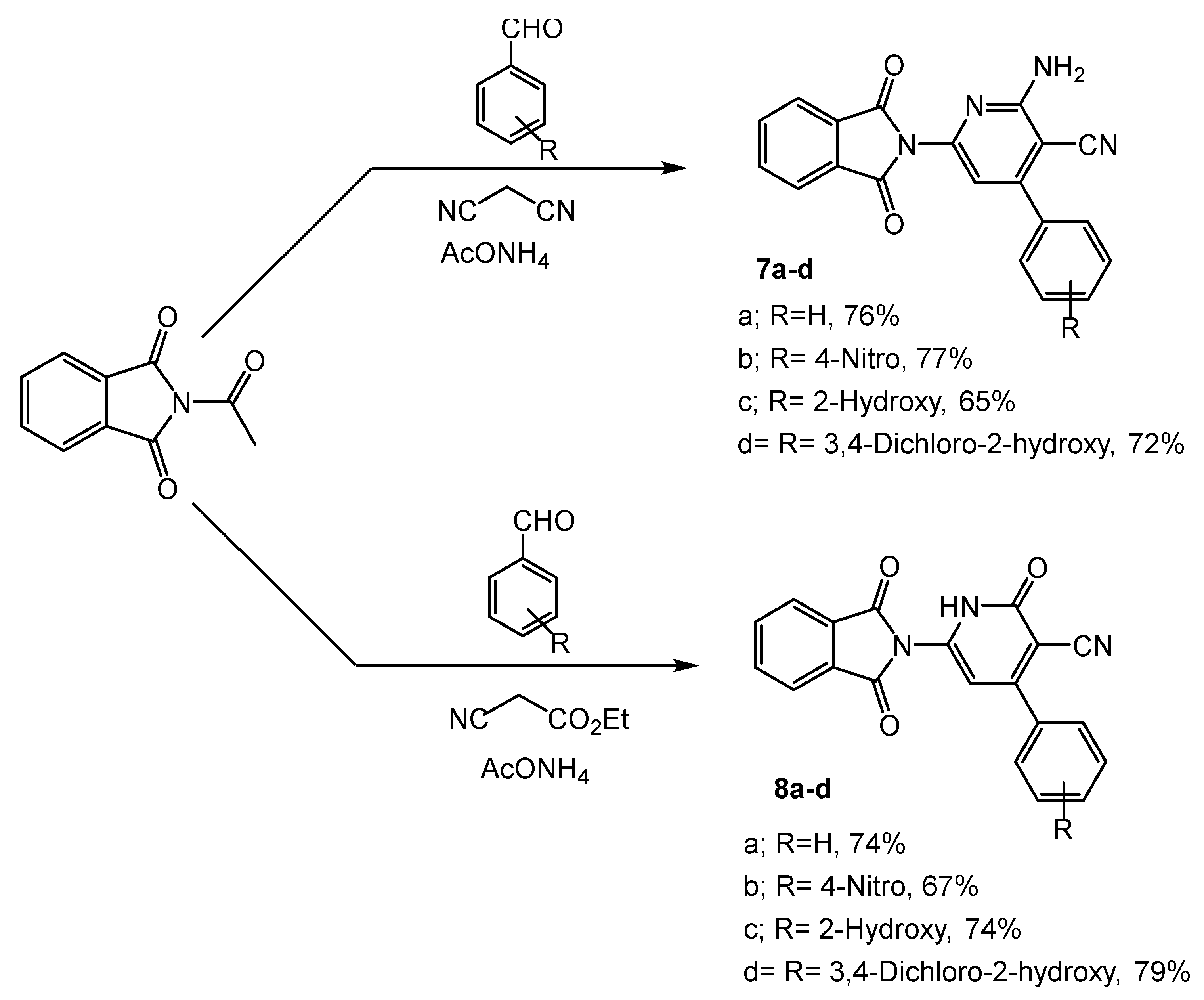
2.1. Chemistry
2.2. Biological Assays
2.2.1. Antioxidant Activity
2.2.2. Inhibitory Effects on Yeast α-Glycosidase and α-Amylase
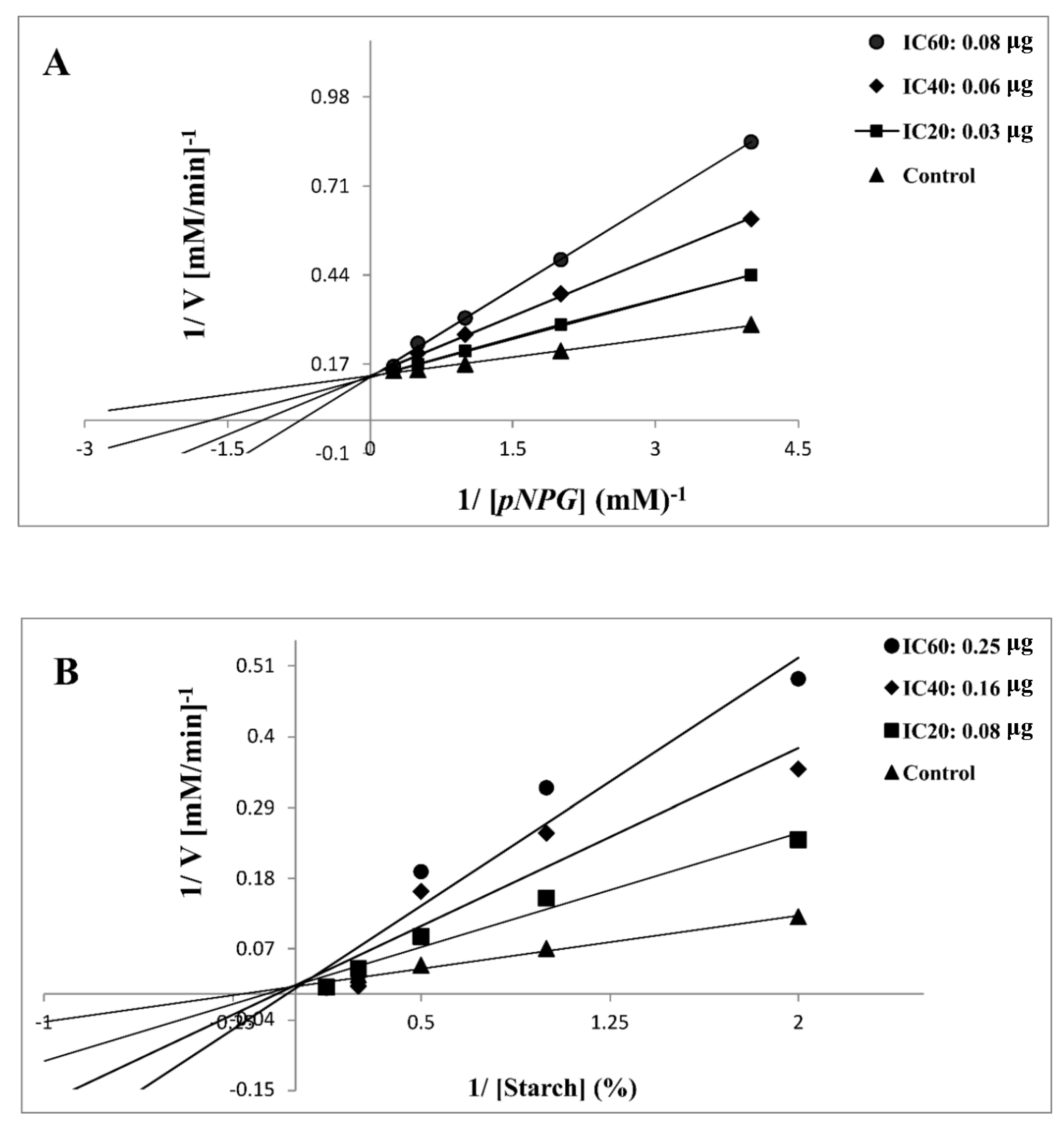
2.2.3. Kinetic Analysis of α-Glycosidase and α-Amylase Inhibition
2.3. Computational Assays
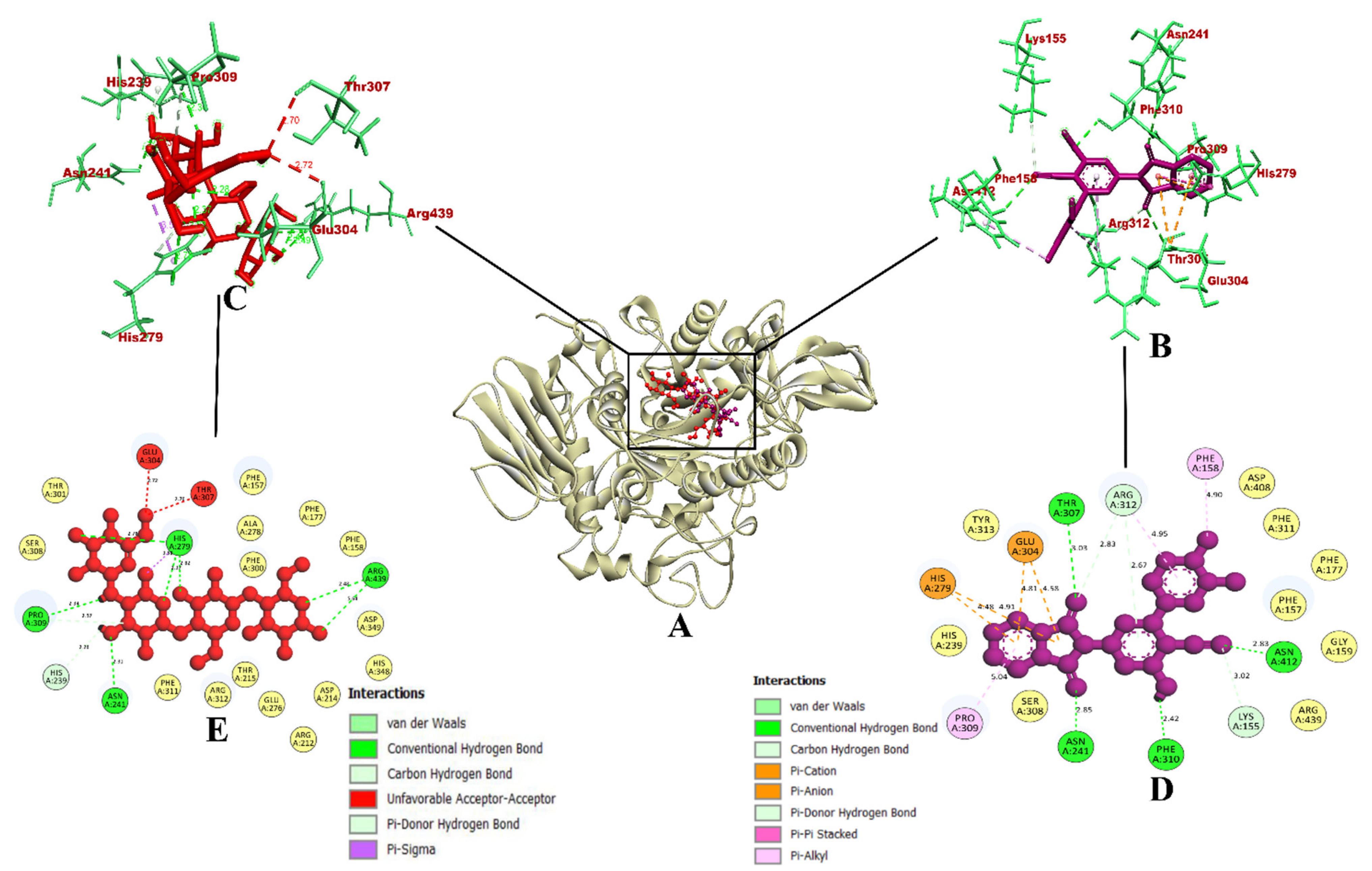
2.3.1. Molecular Docking Simulation
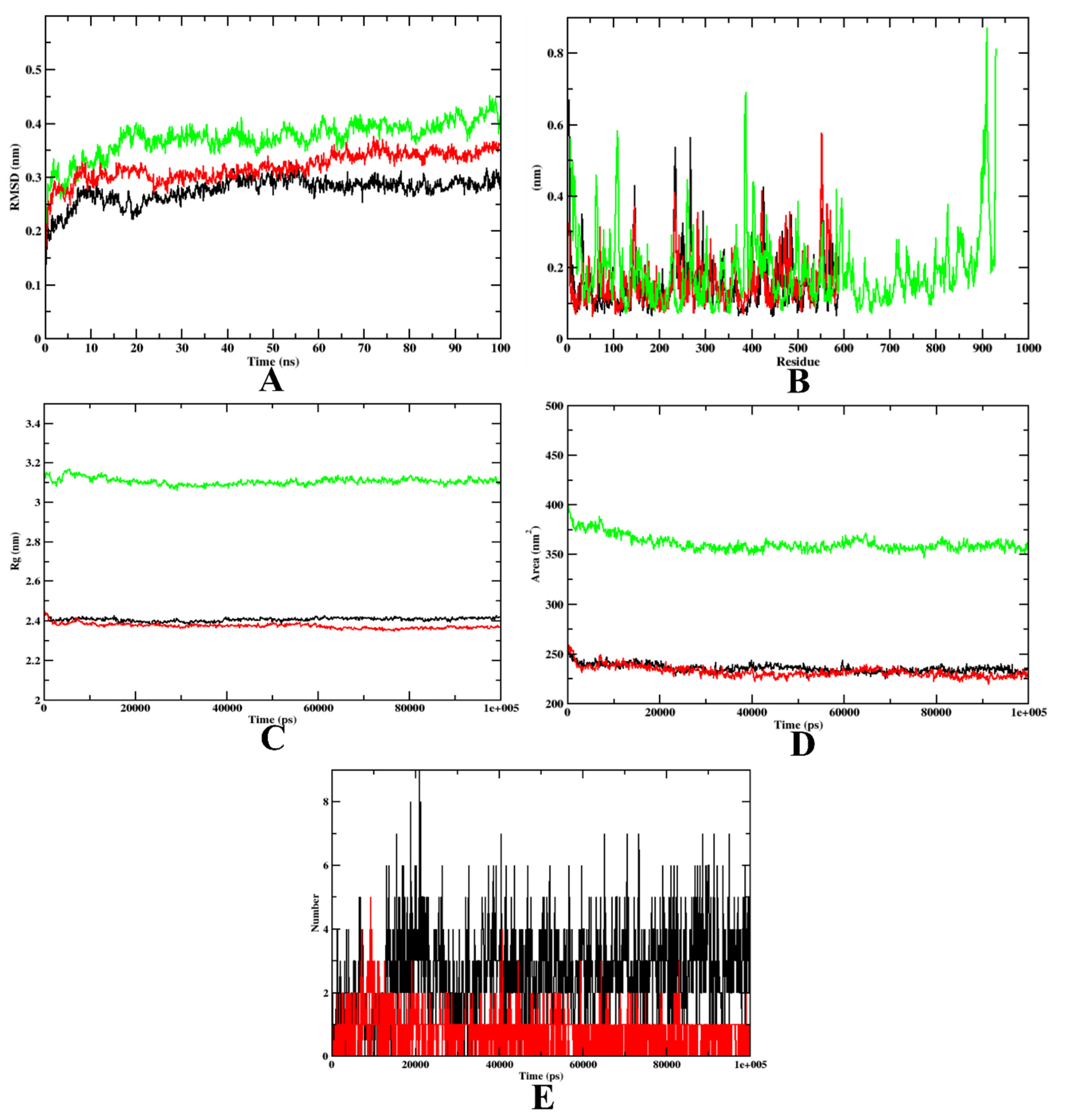
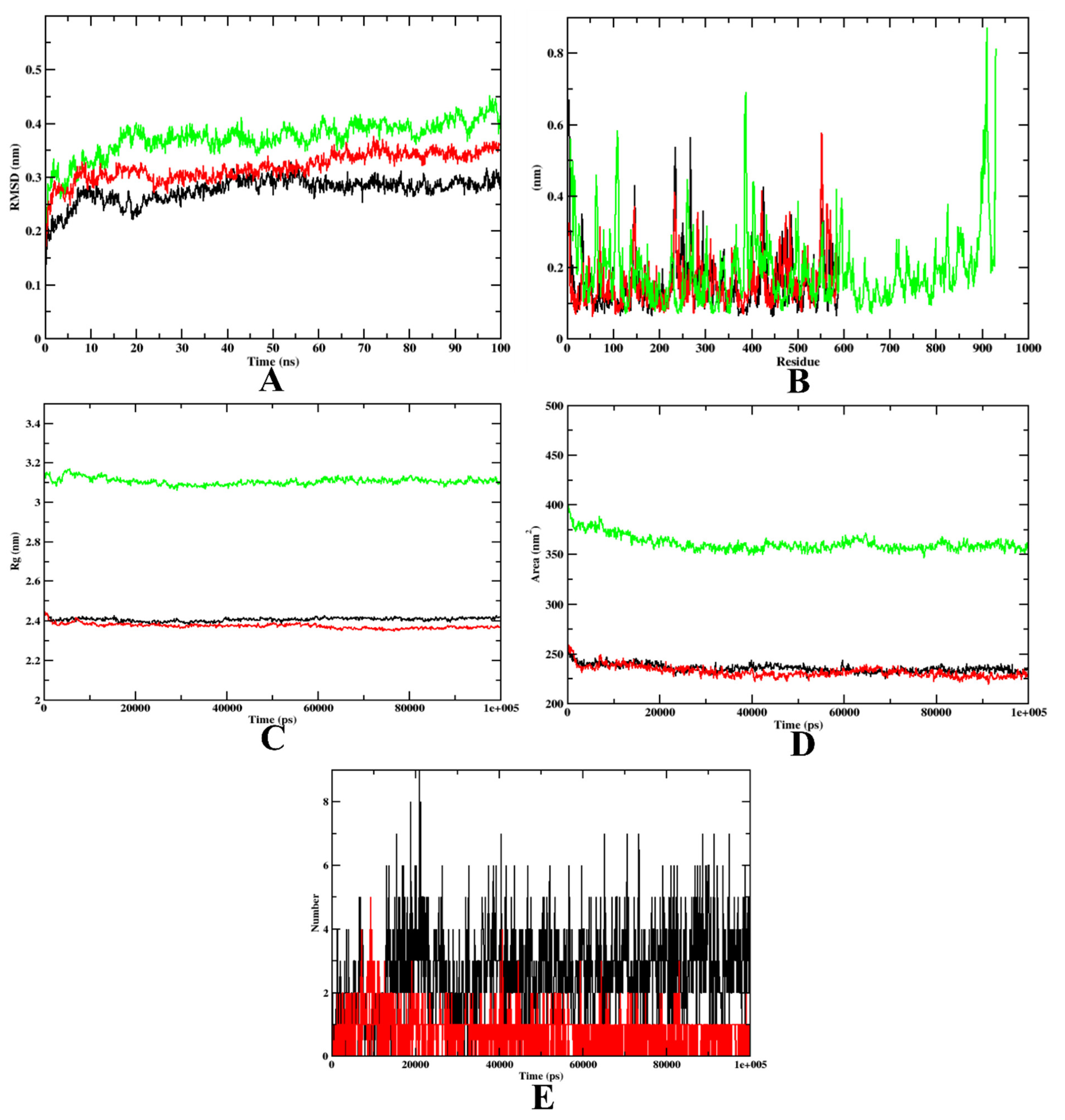
2.3.2. Molecular Dynamics Simulation
2.4. Binding Free Energy Calculations
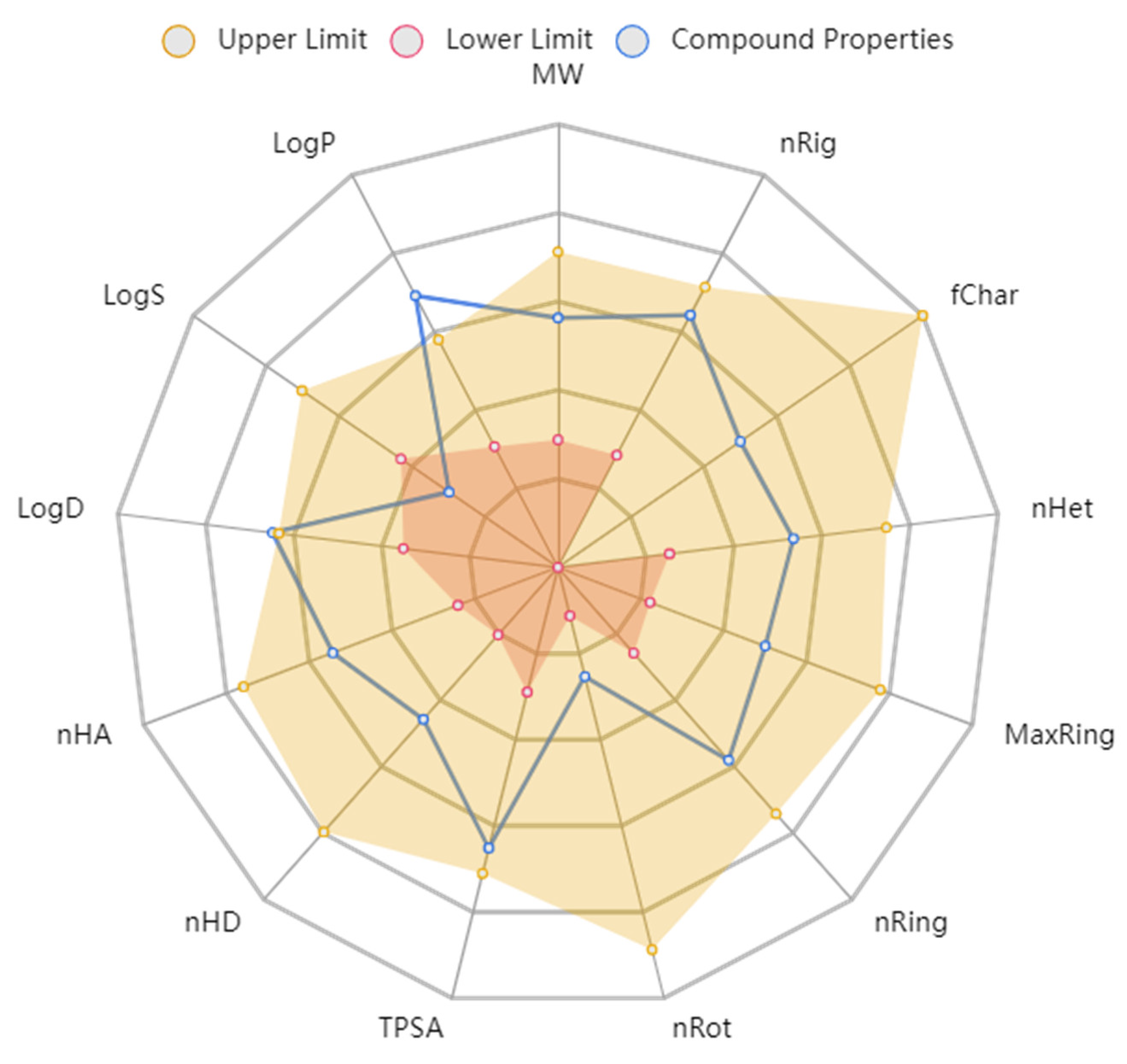
2.5. Physicochemical and ADMET Properties
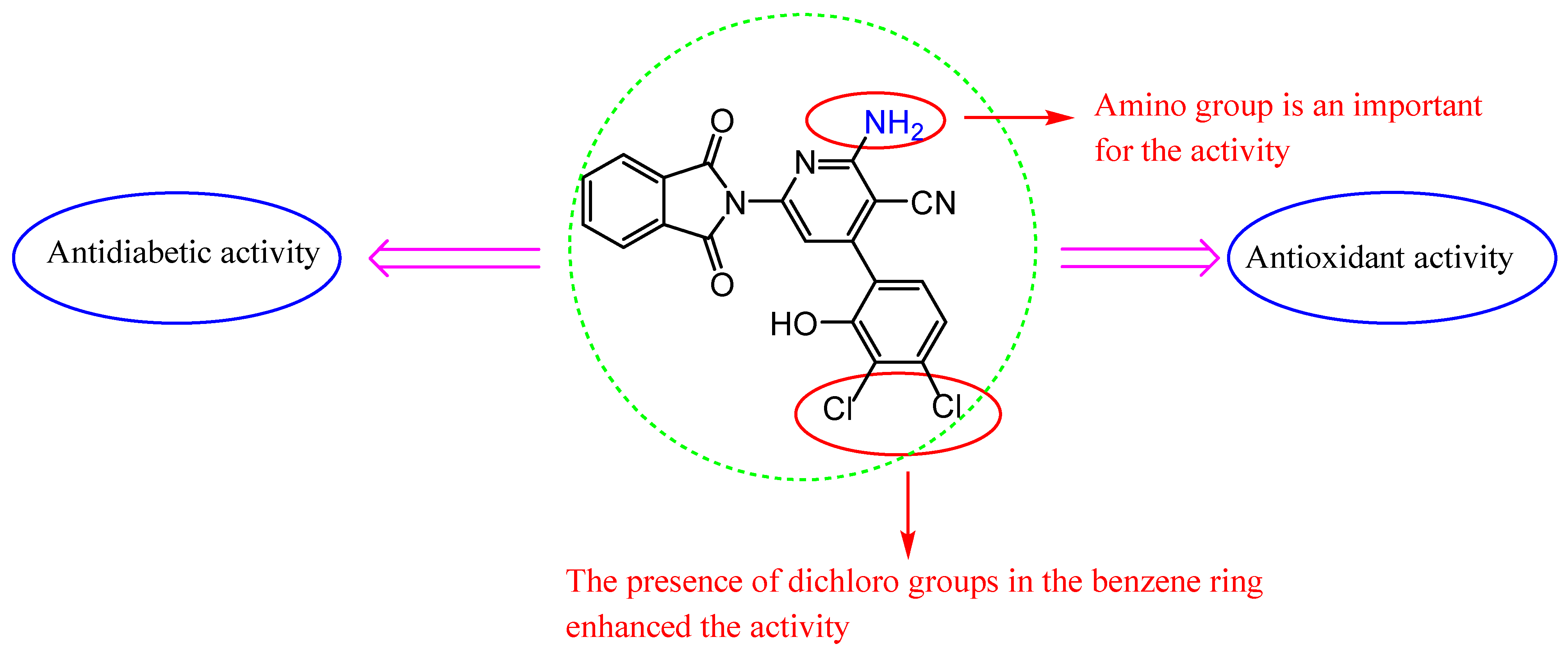
2.6. Structure–Activity Relationship (SAR)
3. Materials and Methods
3.1. Reagents and Instrumentation
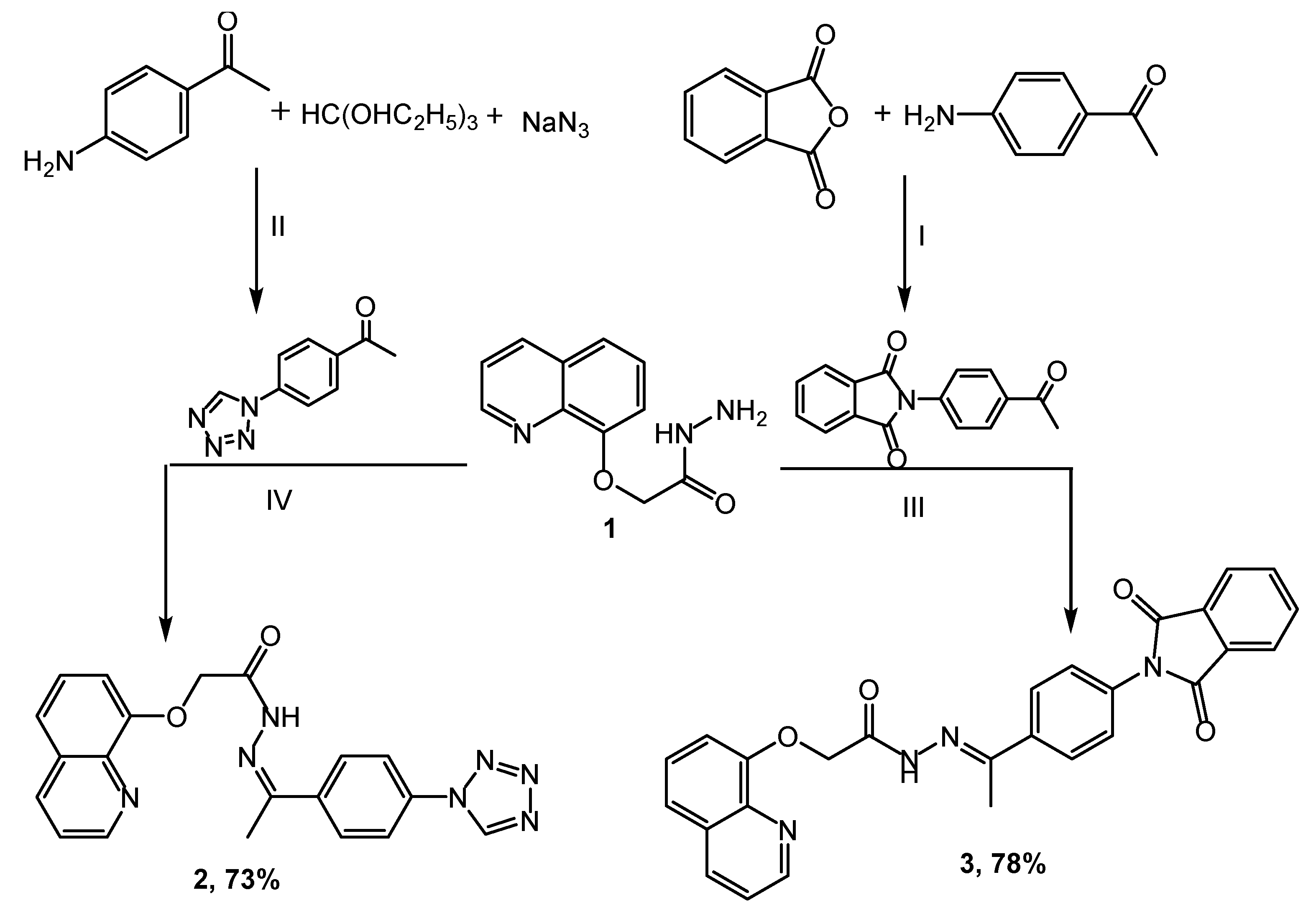
3.2. Synthesis
3.2.1. Synthesis of 2-(Quinolin-8-yloxy)acetohydrazide 1
3.2.2. Synthesis of 1-(4-(1H-Tetrazol-1-yl)phenyl)ethanone
3.2.3. Synthesis of 8-(2-((2-(1-(4-(1H-Tetrazol-1-yl)phenyl)ethylidene)hydrazinyl)oxy)-2-oxoethoxy)quinoline 2
3.2.4. Synthesis of 2-(4-Acetylphenyl)isoindoline-1,3-dione
3.2.5. Synthesis of N’-(1-(4-(1,3-Dioxoisoindolin-2-yl)phenyl)ethylidene)-2-(quinolin-8-yloxy)acetohydrazide 3
3.2.6. Synthesis of 5-Amino-3-(4-substitutedphenyl)-1-(2-(quinolin-8-yloxy)acetyl)-1H-pyrazole-4-carbonitrile 4a–c
3.2.7. Synthesis of 5-((Quinolin-8-yloxy)methyl)-3H-1,2,4-triazole-3-thione 6
3.2.8. Synthesis of 2-Amino-6-(1,3-dioxoisoindolin-2-yl)-4-subsstitued-phenyl-1,2-dihydropyridine-3-carbonitrile 7a–d
3.2.9. Synthesis of 6-(1,3-Dioxoisoindolin-2-yl)-2-oxo-4-substituted-phenyl-1,2-dihydropyridine-3-carbonitriles 8a–d
3.3. Biological Assays
3.3.1. Antioxidant Assays
3.3.2. Inhibition of α-Amylase and α-Glycosidase
3.3.3. Kinetics of α-Glycosidase and α-Amylase Inhibition
3.4. Computational Studies
3.4.1. Molecular Docking Simulation
3.4.2. Molecular Dynamics Simulation
3.4.3. Binding Free Energy Calculations
3.4.4. Physicochemical and ADMET Properties
3.5. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huseynova, M.; Taslimi, P.; Medjidov, A.; Farzaliyev, V.; Aliyeva, M.; Gondolova, G.; Şahin, O.; Yalçın, B.; Sujayev, A.; Orman, E.B.; et al. Synthesis, characterization, crystal structure, electrochemical studies and biological evaluation of metal complexes with thiosemicarbazone of glyoxylic acid. Polyhedron 2018, 155, 25–33. [Google Scholar] [CrossRef]
- Gülçin, I.; Scozzafava, A.; Supuran, C.T.; Koksal, Z.; Turkan, F.; Çetinkaya, S.; Bingöl, Z.; Huyut, Z.; Alwasel, S.H. Rosmarinic acid inhibits some metabolic enzymes including glutathione S-transferase, lactoperoxidase, acetylcholinesterase, butyrylcholinesterase and carbonic anhydrase isoenzymes. J. Enzym. Inhibit. Med. Chem. 2016, 31, 1698–1702. [Google Scholar] [CrossRef]
- Bursal, E.; Aras, A.; Kılıç, Ö.; Taslimi, P.; Gören, A.C.; Gülçin, İ. Phytochemical content, antioxidant activity, and enzyme inhibition effect of Salvia eriophora Boiss. & Kotschy against acetylcholinesterase, α-amylase, butyrylcholinesterase, and α-glycosidase enzymes’. J. Food Biochem. 2019, 43, e12776. [Google Scholar] [CrossRef]
- Patil, S.M.; Shirahatti, P.S.; Ramu, R. Azadirachta indica A. Juss (neem) against diabetes mellitus: A critical review on its phytochemistry, pharmacology, and toxicology. J. Pharm. Pharmacol. 2022, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Ramu, R.; Shirahatti, P.S.; Deepika, T.H.; Bajpe, S.N.; Sreepathi, N.; Patil, S.M.; Prasad, N. Investigating Musa paradisiaca (Var. Nanjangud rasa bale) pseudostem in preventing hyperglycemia along with improvement of diabetic complications. J. Appl. Biol. Biotechnol. 2022, 10, 56–65. [Google Scholar] [CrossRef]
- Maradesha, T.; Martiz, R.M.; Patil, S.M.; Prasad, A.; Babakr, A.T.; Silina, E.; Stupin, V.; Achar, R.R.; Ramu, R. Integrated network pharmacology and molecular modeling approach for the discovery of novel potential MAPK3 inhibitors from whole green jackfruit flour targeting obesity-linked diabetes mellitus. PLoS ONE 2023, 18, e0280847. [Google Scholar] [CrossRef]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Storr, S.J.; Royle, L.; Chapman, C.J.; Hamid, U.M.; Robertson, J.F.; Murray, A.; Dwek, R.A.; Rudd, P.M. The O-linked glycosylation of secretory/shed MUC1 from an advanced breast cancer patient’s serum. Glycobiology 2008, 18, 456–462. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Satish, A.M.; Shbeer, A.M.; Ageel, M.; Al-Ghorbani, M.; Parameswaran, S.; Ramu, R. Discovery of novel coumarin derivatives as potential dual inhibitors against α-glucosidase and α-amylase for the management of post-prandial hyperglycemia via molecular modelling approaches. Molecules 2022, 27, 3888. [Google Scholar] [CrossRef] [PubMed]
- Simsek, E.; Lu, X.; Ouzounov, S.; Block, T.M.; Mehta, A.S. α-Glucosidase inhibitors have a prolonged antiviral effect against hepatitis B virus through the sustained inhibition of the large and middle envelope glycoproteins. Antivir. Chem. Chemother. 2006, 17, 259–267. [Google Scholar] [CrossRef]
- Ramu, R.; Patil, S.M. A perspective on the effective conduction of functional-based coaching program on diabetic Indonesian communities. Oman Med. J. 2021, 36, e281. [Google Scholar] [CrossRef]
- Cabrele, C.; Reiser, O. The modern face of synthetic heterocyclic chemistry. J. Org. Chem. 2016, 81, 10109–10125. [Google Scholar] [CrossRef]
- Hosseini, H.; Bayat, M. Cyanoacetohydrazides in Synthesis of Heterocyclic Compounds. Top. Curr. Chem. 2018, 376, 40. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghorbani, M.; Pavankumar, G.S.; Naveen, P.; Shamanth Neralagundi, H.G.; Prabhakar, B.T.; Khanum, S.A. Synthesis and an angiolytic role of novel piperazine-benzothiozole analogues on neovascularisation, a chief tumoral parameter in neoplastic development. Bioorg. Chem. 2016, 65, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Sarshira, E.M.; Hamada, N.M.; Moghazi, Y.M.; Abdelrahman, M.M. Synthesis and biological evaluation of some heterocyclic compounds from salicylic acid hydrazide. J. Heter. Chem. 2016, 53, 1970–1982. [Google Scholar] [CrossRef]
- Grande, F.; Yamada, R.; Cao, X.; Aiello, F.; Garofalo, A.; Neamati, N. Synthesis and biological evaluation of novel hydrazide based cytotoxic agents. Exp. Opin. Investig. Drugs 2009, 18, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lei, L.; Liu, Z.; Wang, H.; Meng, Q. Design, Synthesis, and Biological Evaluation of Novel Nitrogen Heterocycle-Containing Ursolic Acid Analogs as Antitumor Agents. Molecules 2019, 24, 877. [Google Scholar] [CrossRef]
- Kella, C.R.; Balachandran, C.; Arun, Y.; Kaliyappan, E.; Mahalingam, S.M.; Ignacimuthu, S.; Arumugam, N.; Almansour, A.I.; Kumar, R.S.; Perumal, P.T. A novel class of 1,4-disubstituted 1,2,3-triazoles: Regioselective synthesis, antimicrobial activity and molecular docking studies. Arab. J. Chem. 2020, 13, 9047–9057. [Google Scholar] [CrossRef]
- Kalkhambkar, R.G.; Kulkarni, G.M.; Kamanavalli, C.M.; Premkumar, N.; Asdaq, S.M.B.; Sun, C.M. Synthesis and biological activities of some new fluorinated coumarins and 1-aza coumarins. Eur. J. Med. Chem. 2008, 10, 2178–2188. [Google Scholar] [CrossRef]
- Al-Ghorbani, M.; Gouda, M.A.; Baashen, M.; Alharbi, O.; Almalki, F.A.; Ranganatha, L.V. Piperazine heterocycles as potential anticancer agents: A review. Pharm. Chem. J. 2022, 56, 1. [Google Scholar] [CrossRef]
- Patil, B.S.; Krishnamurthy, G.; Shashikumar, N.D.; Lokesh, M.R.; Bhojya Naik, H.S. Synthesis and Antimicrobial Activity of Some [1,2,4]-Triazole Derivatives. J. Chem. 2012, 2013, e462594. [Google Scholar] [CrossRef]
- Sharshira, E.M.; Hamada, N.M.M. Synthesis and Antimicrobial Evaluation of Some Pyrazole Derivatives. Molecules 2012, 17, 4962–4971. [Google Scholar] [CrossRef]
- Vengurlekar, S.; Sharma, R.; Trivedi, P. Synthesis, Antifungal Evaluation and Molecular Docking Studies on 2-Thioxoimidazolidin-4-One Derivatives. Med. Chem. 2013, 9, 459–473. [Google Scholar] [CrossRef]
- Rashid, H.; Martines, M.A.U.; Duarte, A.P.; Jorge, J.; Rasool, S.; Muhammad, R.; Ahmad, N.; Umar, M.N. Research Developments in the Syntheses, Anti-Inflammatory Activities and Structure–Activity Relationships of Pyrimidines. RSC Adv. 2021, 11, 6060–6098. [Google Scholar] [CrossRef]
- De Oliveira, V.V.G.; de Souza, M.A.A.; Cavalcanti, R.R.M.; de Oliveira Cardoso, M.V.; Leite, A.C.L.; da Silva Junior, V.A.; de Figueiredo, R.C.B.Q. Study of in Vitro Biological Activity of Thiazoles on Leishmania (Leishmania) Infantum. J. Glob. Antimicrob. Resist. 2020, 22, 414–421. [Google Scholar] [CrossRef]
- Winyakul, C.; Phutdhawong, W.; Tamdee, P.; Sirirak, J.; Taechowisan, T.; Phutdhawong, W.S. 2,5-Diketopiperazine Derivatives as Potential Anti-Influenza (H5N2) Agents: Synthesis, Biological Evaluation, and Molecular Docking Study. Molecules 2022, 27, 4200. [Google Scholar] [CrossRef]
- Azzam, R.A.; Elsayed, R.E.; Elgemeie, G.H. Design and Synthesis of a New Class of Pyridine-Based N-Sulfonamides Exhibiting Antiviral, Antimicrobial, and Enzyme Inhibition Characteristics. ACS Omega 2020, 5, 26182–26194. [Google Scholar] [CrossRef]
- Riyadh, S.M.; Abdelhamid, I.A.; Al-Matar, H.M.; Hilmy, N.M.; Elnagdi, M.H. Enamines as Precursors to Polyfunctional Heteroaromatic Compounds; a Decade of Development. Heterocyclic 2008, 75, 1849–1905. [Google Scholar] [CrossRef]
- Abdel-Motaleb, R.M.; Makhloof, A.M.A.S.; Ibrahim, H.M.; Elnagdi, M.H. Studies with azoles and benzoazoles: A novel simple approach for synthesis of 3-functionally substituted 3-acylindoles. J. Heterocycl. Chem. 2007, 44, 109–114. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Makhseed, S.; Abdel-Motaleb, R.M.; Makhlouf, A.M.; Elnagdi, M.H. Utility of cyanoacetamides as precursors to pyrazolo[3,4-d] pyrimidin-4-ones,2-aryl-6-substituted 1,2,3-triazolo[4,5-d]pyrimidines and pyrazolo[1,5-a]pyrimidine-3-carboxamides. Heterocyclic 2007, 71, 1951–1966. [Google Scholar] [CrossRef]
- Salaheldin, A.M. Enaminonitriles in Heterocyclic Synthesis: Novel Synthesis of 3-Aminopyrroles and Pyrrolo[3,2-d]pyrimidine Derivatives. Zeitsch. Naturfor. B 2008, 63, 564–570. [Google Scholar] [CrossRef]
- Al-Ghorbani, M.; Thirusangu, P.; Gurupadaswamy, H.D.; Girish, V.; Neralagundi, H.S.; Prabhakar, B.T.; Khanum, S.A. Synthesis and antiproliferative activity of benzophenone tagged pyridine analogues towards activation of caspase activated DNase mediated nuclear fragmentation in Dalton’s lymphoma. Bioorg. Chem. 2016, 65, 73–81. [Google Scholar] [CrossRef]
- Al-Ghorbani, M.; Gouda, M.A. Synthesis and in vitro anticancer activity of some novel cyclohepta[b]thiophene-3-carboxamides bearing pyrazole moiety. J. Heter. Chem. 2020, 57, 3213–3221. [Google Scholar] [CrossRef]
- Al-Ghorbani, M.; Thirusangu, P.; Gurupadaswamy, H.D.; Vigneshwaran, V.; Mohammed, Y.H.; Prabhakar, B.T.; Khanum, S.A. Synthesis of novel morpholine conjugated benzophenone analogues and evaluation of antagonistic role against neoplastic development. Bioorg. Chem. 2017, 71, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghorbani, M. Design, Synthesis of Some Quinoline-Heterocyclic Derivatives. Russ. J. Heter. Chem. 2022, 58, 1272–1279. [Google Scholar] [CrossRef]
- Peng, Y.; Song, G. Simultaneous microwave and ultrasound irradiation: A rapid synthesis of hydrazides. Green Chem. 2001, 3, 302–304. [Google Scholar] [CrossRef]
- Sonawane, H.R.; Vibhute, B.T.; Aghav, B.D.; Deore, J.V.; Patil, S.K. Versatile Applications of Transition Metal Incorporating Quinoline Schiff Base Metal Complexes: An Overview. Eur. J. Med. Chem. 2023, 258, 115549. [Google Scholar] [CrossRef]
- Gorobets, N.Y.; Yousefi, B.H.; Belaj, F.; Kappe, C.O. Rapid microwave-assisted solution phase synthesis of substituted 2-pyridone libraries. Tetrahedron 2004, 60, 8633–8644. [Google Scholar] [CrossRef]
- Güven, L.; Erturk, A.; Miloğlu, F.D.; Alwasel, S.; Gulcin, İ. Screening of Antiglaucoma, Antidiabetic, Anti-Alzheimer, and Antioxidant Activities of Astragalus alopecurus Pall—Analysis of Phenolics Profiles by LC-MS/MS. Pharmaceuticals 2023, 16, 659. [Google Scholar] [CrossRef] [PubMed]
- Karagecili, H.; İzol, E.; Kirecci, E.; Gulcin, İ. Determination of Antioxidant, Anti-Alzheimer, Antidiabetic, Antiglaucoma and Antimicrobial Effects of Zivzik Pomegranate (Punica granatum)—A Chemical Profiling by LC-MS/MS. Life 2023, 13, 735. [Google Scholar] [CrossRef]
- Karagecili, H.; Yılmaz, M.A.; Ertürk, A.; Kiziltas, H.; Güven, L.; Alwasel, S.H.; Gulcin, İ. Comprehensive Metabolite Profiling of Berdav Propolis Using LC-MS/MS: Determination of Antioxidant, Anticholinergic, Antiglaucoma, and Antidiabetic Effects. Molecules 2023, 28, 1739. [Google Scholar] [CrossRef]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Ranganatha, L.V.; Prasad, M.N. Inhibitory effect of banana (Musa sp. var. Nanjangud rasa bale) flower extract and its constituents Umbelliferone and Lupeol on α-glucosidase, aldose reductase and glycation at multiple stages. S. Afr. J. Bot. 2014, 95, 54–63. [Google Scholar] [CrossRef]
- Maradesha, T.; Patil, S.M.; Al-Mutairi, K.A.; Ramu, R.; Madhunapantula, S.V.; Alqadi, T. Inhibitory effect of polyphenols from the whole green jackfruit flour against α-glucosidase, α-amylase, aldose reductase and glycation at multiple stages and their interaction: Inhibition kinetics and molecular simulations. Molecules 2022, 27, 1888. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, S.; Nivetha, N.; Patil, S.M.; Martiz, R.M.; Ramu, R.; Sreenivasa, S.; Velmathi, S. One-pot three-component synthesis of novel phenyl-pyrano-thiazol-2-one derivatives and their anti-diabetic activity studies. Results Chem. 2022, 4, 100439. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Kumar, B.P.; Kumar, N. Evaluation of flavonoids from banana pseudostem and flower (quercetin and catechin) as potent inhibitors of α-glucosidase: An in silico perspective. J. Biomol. Struct. Dyn. 2022, 40, 12491–12505. [Google Scholar] [CrossRef]
- Maradesha, T.; Patil, S.M.; Phanindra, B.; Achar, R.R.; Silina, E.; Stupin, V.; Ramu, R. Multiprotein Inhibitory Effect of Dietary Polyphenol Rutin from Whole Green Jackfruit Flour Targeting Different Stages of Diabetes Mellitus: Defining a Bio-Computational Stratagem. Separations 2022, 9, 262. [Google Scholar] [CrossRef]
- Banu, H.A.N.; Kalluraya, B.; Manju, N.; Ramu, R.; Patil, S.M.; Lokanatha Rai, K.M.; Kumar, N. Synthesis of Pyrazoline-Embedded 1,2,3-Triazole Derivatives via 1,3-Dipolar Cycloaddition Reactions with in vitro and in silico Studies. ChemistrySelect 2023, 8, e202203578. [Google Scholar] [CrossRef]
- Shivanna, C.; Patil, S.M.; Mallikarjunaswamy, M.; Ramu, R.; Akhileshwari, P.; Nagaraju, L.R.; Sridhar, M.A.; Khanum, S.A.; Ranganatha, L.; Silina, E.; et al. Synthesis, characterization, Hirschfeld surface analysis, crystal structure and molecular modeling studies of 1-(4-(Methoxy(phenyl)methyl)-2-methylphenoxy)butan-2-one derivative as a novel α-glucosidase inhibitor. Crystals 2022, 12, 960. [Google Scholar] [CrossRef]
- Casacchia, T.; Occhiuzzi, M.A.; Grande, F.; Rizzuti, B.; Granieri, M.C.; Rocca, C.; Gattuso, A.; Garofalo, A.; Angelone, T.; Statti, G. A pilot study on the nutraceutical properties of the Citrus hybrid Tacle® as a dietary source of polyphenols for supplementation in metabolic disorders. J. Funct. Foods 2019, 52, 370–381. [Google Scholar] [CrossRef]
- Kumar, V.; Ramu, R.; Shirahatti, P.S.; Kumari, V.C.; Sushma, P.; Mandal, S.P.; Patil, S.M. α-glucosidase; α-amylase inhibition; kinetics and docking studies of novel (2-chloro-6-(trifluoromethyl) benzyloxy) arylidene) based rhodanine and rhodanine acetic acid derivatives. ChemistrySelect 2021, 6, 9637–9644. [Google Scholar] [CrossRef]
- Kumar, V.; Shetty, P.; Arunodaya, H.S.; Chandra, K.S.; Ramu, R.; Patil, S.M.; Baliga, A.; Rai, V.M.; Shenoy, M.S.; Udupi, V.; et al. Potential fluorinated anti-MRSA thiazolidinone derivatives with antibacterial, antitubercular activity and molecular docking studies. Chem. Biodivers. 2022, 19, e202100532. [Google Scholar] [CrossRef] [PubMed]
- Simha, N.A.; Patil, S.M.; Chagalamari, A.; Satish, A.M.; Ramu, R. Protocol to identify multiple protein targets and therapeutic compounds using an in silico polypharmacological approach. STAR Protoc. 2023, 3, 102440. [Google Scholar] [CrossRef]
- Patil, S.M.; Phanindra, B.; Shirahatti, P.S.; Martiz, R.M.; Sajal, H.; Babakr, A.T.; Ramu, R. Computational approaches to define poncirin from Magnolia champaka leaves as a novel multi-target inhibitor of SARS-CoV-2. J. Biomol. Struct. Dyn. 2023, 10, 1–20. [Google Scholar] [CrossRef]
- Pushpa, V.H.; Jayanthi, M.K.; Rashmi, R.H.; Shivamurthy, V.K.N.; Patil, S.M.; Shirahatti, P.S.; Ramu, R. New insights on the phytochemical intervention for the treatment of neuropsychiatric disorders using the leaves of Michelia champaca: An in vivo and in silico approach. Pharm. Biol. 2022, 60, 1656–1668. [Google Scholar] [CrossRef]
- Pradeep, S.; Patil, S.M.; Dharmashekara, C.; Jain, A.; Ramu, R.; Shirahatti, P.S.; Mandal, S.P.; Reddy, P.; Srinivasa, C.; Patil, S.S.; et al. Molecular insights into the in silico discovery of corilagin from Terminalia chebula as a potential dual inhibitor of SARS-CoV-2 structural proteins. J. Biomol. Struct. Dyn. 2022, 10, 1–16. [Google Scholar] [CrossRef]
- Taha, M.; Salahuddin, M.; Rahim, F.; Imran, S.; Hussain, S.; Uddin, N.; Khan, K.M. New Quinoline Analogues: As Potential Diabetics Inhibitors and Molecular Docking Study. Polycycl. Arom. Comp. 2023, 1–23. [Google Scholar] [CrossRef]
- Rajendran, G.; Bhanu, D.; Aruchamy, B.; Ramani, P.; Pandurangan, N.; Bobba, K.N.; Oh, E.J.; Chung, H.Y.; Gangadaran, P.; Ahn, B.-C. Chalcone: A Promising Bioactive Scaffold in Medicinal Chemistry. Pharmaceuticals 2022, 15, 1250. [Google Scholar] [CrossRef]
- Aruchamy, B.; Drago, C.; Russo, V.; Pitari, G.M.; Ramani, P.; Aneesh, T.P.; Benny, S.; Vishnu, V. Imidazole-Pyridine Hybrids as Potent Anti-Cancer Agents. Eur. J. Pharm. Sci. 2023, 180, 106323. [Google Scholar] [CrossRef] [PubMed]
- Razali, N.; Razab, R.; Junit, S.M.; Aziz, A.A. Radical scavenging and reducing properties of extracts of cashew shoots (Anacardium occidentale). Food Chem. 2008, 111, 38–44. [Google Scholar] [CrossRef]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Lakkapa, D.B.; Nagendra, M. Evaluation of Banana (Musa sp. var. Nanjangud Rasa bale) flower and pseudostem extracts on antimicrobial, cytotoxicity and thrombolytic activities. Int. J. Pharm. Pharm. Sci. 2015, 7, 136–140. [Google Scholar]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Lakkappa Dhananjaya, B.; MN, N.P. Assessment of In Vivo Antidiabetic Properties of Umbelliferone and Lupeol Constituents of Banana (Musa sp. var. Nanjangud Rasa Bale) Flower in Hyperglycaemic Rodent Model. PLoS ONE 2016, 11, e0151135. [Google Scholar] [CrossRef]
- Ramu, R.; Shirahatti, P.S.; Nayakavadi, S.; Zameer, V.R.F.; Dhananjaya, B.L.; Nagendra Prasad, M.N. The effect of a plant extract enriched in stigmasterol and β-sitosterol on glycaemic status and glucose metabolism in alloxan-induced diabetic rats. Food Funct. 2016, 7, 3999–4011. [Google Scholar] [CrossRef] [PubMed]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Prasad, M.N.N. Investigation of antihyperglycaemic activity of banana (Musa sp. var. Nanjangud rasa bale) pseudostem in normal and diabetic rats. J. Sci. Food Agric. 2015, 95, 165–173. [Google Scholar] [CrossRef]
- Mallikarjunaswamy, C.; Lakshmi Ranganatha, V.; Ramu, R.; Nagaraju, G.U. Facile microwave-assisted green synthesis of ZnO nanoparticles: Application to photodegradation, antibacterial and antioxidant. J. Mater. Sci. Mater. Electron. 2020, 31, 1004–1021. [Google Scholar] [CrossRef]
- Köksal, E.; Tohma, H.; Kılıç, Ö.; Alan, Y.; Aras, A.; Gülçin, İ.; Bursal, E. Assessment of Antimicrobial and Antioxidant Activities of Nepeta trachonitica: Analysis of Its Phenolic Compounds Using HPLC-MS/MS. Sci. Pharm. 2017, 85, 24. [Google Scholar] [CrossRef] [PubMed]
- Eruygur, N.; Koçyiğit, U.M.; Taslimi, P.; Ataş, M.; Tekin, M.; Gülçin, İ. Screening the in vitro antioxidant, antimicrobial, anticholinesterase, antidiabetic activities of endemic Achillea cucullata (Asteraceae) ethanol extract. S. Afr. J. Bot. 2019, 120, 141–145. [Google Scholar] [CrossRef]
- Gülçin, I.; Bursal, E.; Sehitoğlu, M.H.; Bilsel, M.; Gören, A.C. Polyphenol contents and antioxidant activity of lyophilized aqueous extract of propolis from Erzurum, Turkey. Food Chem. Toxicol. 2010, 48, 2227–2238. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Martiz, R.M.; Patil, S.M.; Hombegowda, D.T.; Shbeer, A.M.; Alqadi, T.; Al-Ghorbani, M.; Ramu, R.; Prasad, A. Phyto-Computational Intervention of Diabetes Mellitus at Multiple Stages Using Isoeugenol from Ocimum tenuiflorum: A Combination of Pharmacokinetics and Molecular Modelling Approaches. Molecules 2022, 27, 6222. [Google Scholar] [CrossRef]
- Nivetha, N.; Martiz, R.M.; Patil, S.M.; Ramu, R.; Sreenivasa, S.; Velmathi, S. Benzodioxole grafted spirooxindole pyrrolidinyl derivatives: Synthesis, characterization, molecular docking and anti-diabetic activity. RSC Adv. 2022, 12, 24192–24207. [Google Scholar] [CrossRef] [PubMed]
- Shashank, P.M.; Prithvi, S.S.; Ramith, R. The pathogenicity of MERS-CoV, SARS-CoV and SARS-CoV-2: A comparative overview. Res. J. Biotech. 2021, 16, 182–192. [Google Scholar]
- Ammar, A.; Cavill, R.; Evelo, C.; Willighagen, E. PSnpBind: A database of mutated binding site protein–ligand complexes constructed using a multithreaded virtual screening workflow. J. Cheminform. 2022, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.P.; Singh, A.K.; Bansal, T.; Yadav, A.; Prajapati, K.S.; Shuaib, M.; Kumar, S. Identification of Natural Inhibitors Against SARS-CoV-2 Drugable Targets Using Molecular Docking, Molecular Dynamics Simulation, and MM-PBSA Approach. Front. Cell. Infect. Microbiol. 2021, 11, 730288. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Chandra, J.S.; Ranganatha, L.V. In silico identification of novel benzophenone-coumarin derivatives as SARS-CoV-2 RNAdependent RNA polymerase (RdRp) inhibitors. J. Biomol. Struct. Dyn. 2022, 40, 13032–13048. [Google Scholar] [CrossRef] [PubMed]
- Sajal, H.; Patil, S.M.; Raj, R.; Shbeer, A.M.; Ageel, M.; Ramu, R. Computer-Aided Screening of Phytoconstituents from Ocimum tenuiflorum against Diabetes Mellitus Targeting DPP4 Inhibition: A Combination of Molecular Docking, Molecular Dynamics, and Pharmacokinetics Approaches. Molecules 2022, 27, 5133. [Google Scholar] [CrossRef] [PubMed]
- Martiz, R.M.; Patil, S.M.; Ramu, R.; Jayanthi, M.K.; Ashwini, P.; Ranganatha, L.V.; Khanum, S.A.; Silina, E.; Stupin, V.; Achar, R.R. Discovery of novel benzophenone integrated derivatives as anti-Alzheimer’s agents targeting presenilin-1 and presenilin-2 inhibition: A computational approach. PLoS ONE 2022, 17, e0265022. [Google Scholar] [CrossRef]
- Gurupadaswamy, H.D.; Ranganatha, V.L.; Ramu, R.; Patil, S.M.; Khanum, S.A. Competent synthesis of biaryl analogs via asymmetric Suzuki–Miyaura cross-coupling for the development of anti-inflammatory and analgesic agents. J. Iran. Chem. Soc. 2022, 19, 2421–2436. [Google Scholar] [CrossRef]
- Martiz, R.M.; Patil, S.M.; Abdulaziz, M.; Babalghith, A.; Al-Areefi, M.; Al-Ghorbani, M.; Kumar, J.M.; Prasad, A.; Nagalingaswamy, N.P.M.; Ramu, R. Defining the role of isoeugenol from Ocimum tenuiflorum against diabetes mellitus-linked Alzheimer’s disease through network pharmacology and computational methods. Molecules 2022, 27, 2398. [Google Scholar] [CrossRef]
- Bagewadi, Z.K.; Khan, T.Y.; Gangadharappa, B.; Kamalapurkar, A.; Shamsudeen, S.M.; Yaraguppi, D.A. Molecular dynamics and simulation analysis against superoxide dismutase (SOD) target of Micrococcus luteus with secondary metabolites from Bacillus licheniformis recognized by genome mining approach. Saudi J. Biol. Sci. 2023, 30, 103753. [Google Scholar] [CrossRef]
- Patil, S.M.; Manu, G.; Shivachandra, J.C.; Anil Kumar, K.M.; Vigneswaran, J.; Ramu, R.; Shirahatti, P.S.; Ranganatha, V.L. Computational screening of benzophenone integrated derivatives (BIDs) targeting the NACHT domain of the potential target NLRP3 inflammasome. Adv. Cancer Bio. Metastasis 2022, 5, 100056. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compounds | EC50 *,# (mM) | ||
|---|---|---|---|
| Radical Scavenging Activities | |||
| DPPH | ABTS | Superoxide Anion Radicals | |
| 1 | 4.65 ± 2.25 f | 4.50 ± 1.14 d | 4.50 ± 1.11 c |
| 2 | 2.45 ± 0.88 d | 5.00 ± 0.01 d | 4.20 ± 1.33 c |
| 3 | 1.60 ± 1.02 c | 1.35 ± 1.10 b | 1.10 ± 0.55 a |
| 4a | 1.50 ± 0.77 c | 1.30 ± 1.00 b | 1.11 ± 0.27 a |
| 4b | 5.00 ± 0.25 g | 4.75 ± 0.42 d | 4.50 ± 2.00 c |
| 4c | 4.55 ± 1.57 f | 7.86 ± 0.55 f | 5.95 ± 1.05 d |
| 6 | 3.33 ± 0.86 e | 8.01 ± 0.33 f | 6.10 ± 1.54 d |
| 7a | 1.99 ± 0.09 c | 2.25 ± 0.22 c | 2.25 ± 0.66 b |
| 7b | 2.00 ± 0.15 c | 2.60 ± 0.77 c | 2.30 ± 1.99 b |
| 7c | 1.00 ± 0.85 b | 1.23 ± 0.48 b | 1.00 ± 0.00 a |
| 7d | 0.65 ± 0.03 a | 0.52 ± 0.25 a | 0.93 ± 0.12 a |
| 8a | 3.25 ± 1.23 e | 10.05 ± 1.17 e | 5.78 ± 3.01 d |
| 8b | 4.50 ± 0.79 f | 9.88 ± 2.25 e | 6.00 ± 2.45 d |
| 8c | 2.50 ± 1.11 d | 2.44 ± 0.67 c | 5.15 ± 0.77 c,d |
| 8d | 5.05 ± 2.22 g | 9.99 ± 1.89 e | 5.22 ± 0.85 c,d |
| BHA | 0.80 ± 0.32 b | 0.64 ± 0.31 a | 0.95 ± 0.17 a |
| Test Compounds | IC50 x,y (mM) | |
|---|---|---|
| Enzymes | ||
| α-Amylase | α-Glycosidase | |
| 1 | 0.90 ± 1.04 c | 0.50 ± 0.66 c |
| 2 | 0.75 ± 0.26 b | 0.25 ± 0.52 b |
| 3 | 0.91 ± 1.55 c | 0.47 ± 0.35 c |
| 4a | 0.72 ± 0.51 b | 0.24 ± 0.05 b |
| 4b | 1.55 ± 1.67 d | 0.99 ± 0.33 d |
| 4c | 1.60 ± 0.77 d | 1.11 ± 0.74 d |
| 6 | 3.65 ± 2.47 e | 1.50 ± 2.00 e |
| 7a | 3.50 ± 1.00 e | 1.78 ± 0.06 f |
| 7b | 1.50 ± 1.50 d | 1.05 ± 0.88 d |
| 7c | 0.26 ± 0.32 a | 0.11 ± 0.04 a |
| 7d | 0.21 ± 0.06 a | 0.07 ± 0.10 a |
| 8a | 0.88 ± 0.36 c | 1.00 ± 0.00 d |
| 8b | 1.60 ± 0.83 d | 1.01 ± 0.04 d |
| 8c | 3.55 ± 1.55 e | 1.80 ± 0.72 f |
| 8d | 5.00 ± 3.00 f | 2.22 ± 0.11 g |
| Acarbose | 0.25 ± 0.14 a | 0.09 ± 0.05 a |
| Compound | Enzyme | Treatment | Mode of Inhibition x | Km (mM) | Vmax 103 (µM/min)−1 | Ki (mg) y,z |
|---|---|---|---|---|---|---|
| 7d | α-glycosidase | Control | Competitive | 0.86 | 27.86 | 0.47 ± 0.35 |
| IC20 0.03 μg | 1.79 | 28.82 | ||||
| IC40 0.06 μg | 2.50 | 29.00 | ||||
| IC60 0.08 μg | 4.20 | 29.29 | ||||
| α-amylase | Control | Competitive | 0.44 | 13.00 | 0.75 ± 0.25 | |
| IC20 0.08 μg | 0.94 | 13.01 | ||||
| IC40 0.16 μg | 1.43 | 13.90 | ||||
| IC60 0.25 μg | 1.99 | 13.15 |
| Sl. No. | Compound | Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrogen Bonds | |||
|---|---|---|---|---|---|---|---|
| AG | AM | AG | AM | AG | AM | ||
| 1 | 1 | −7.6 | −6.6 | 7 | 11 | 7 | 4 |
| 2 | 2 | −9.5 | −7.9 | 17 | 9 | 6 | 2 |
| 3 | 3 | −10.2 | −10.1 | 6 | 11 | 2 | 2 |
| 4 | 4a | −10.4 | −8.8 | 12 | 5 | 2 | 1 |
| 5 | 4b | −9.7 | −8.7 | 13 | 10 | 5 | 3 |
| 6 | 4c | −10.1 | −8.5 | 11 | 13 | 2 | 0 |
| 7 | 6 | −7.2 | −6.9 | 6 | 9 | 2 | 1 |
| 8 | 7a | −9.2 | −8.5 | 12 | 7 | 4 | 1 |
| 9 | 7b | −9.1 | −7.8 | 15 | 9 | 7 | 4 |
| 10 | 7c | −9.1 | −8.3 | 6 | 8 | 1 | 3 |
| 11 | 7d | −10.9 | −9.0 | 17 | 10 | 7 | 3 |
| 12 | 7a | −9.8 | −8.8 | 13 | 9 | 5 | 3 |
| 13 | 8a | −10.3 | −8.6 | 14 | 11 | 4 | 3 |
| 14 | 8b | −10.5 | −8.8 | 7 | 11 | 3 | 2 |
| 15 | 8c | −10.0 | −8.7 | 14 | 11 | 6 | 4 |
| 16 | 8d | −9.9 | −8.8 | 9 | 8 | 3 | 3 |
| 17 | Acarbose | −8.6 | −6.0 | 10 | 6 | 9 | 6 |
| MD Trajectories | Apoprotein | Protein–7d Complex | Protein–Acarbose Complex |
|---|---|---|---|
| RMSD (nm) | 0.25–0.40 | 0.25–0.40 | 0.25–0.40 |
| Rg (nm) | 3.1–3.5 | 2.39–2.45 | 2.39–2.45 |
| SASA (nm2) | 380–400 | 240–250 | 240–250 |
| Ligand H-Bonds | - | 9 | 5 |
| MD Trajectories | Apoprotein | Protein–7d Complex | Protein–Acarbose Complex |
|---|---|---|---|
| RMSD (nm) | 0.3–0.4 | 0.3–0.4 | 0.3–0.4 |
| Rg (nm) | 2.25–2.30 | 2.25–2.30 | 2.25–2.30 |
| SASA (nm2) | 180–195 | 180–195 | 180–195 |
| Ligand H-Bonds | - | 10 | 7 |
| Types of Binding Free Energy | 7d–AG Complex | Acarbose–AG Complex | 7d–AM Complex | Acarbose–AM Complex |
|---|---|---|---|---|
| Values (kJ/mol) | Values (kJ/mol) |
Values (kJ/mol) |
Values (kJ/mol) | |
| Van der Waal energy | −228.101 | −218.605 | −129.410 | −150.112 |
| Electrostatic energy | −21.317 | −4.761 | −11.217 | −10.911 |
| Polar solvation energy | 122.686 | −103.307 | 59.185 | 60.951 |
| SASA energy | −19.612 | −17.835 | −26.185 | −15.929 |
| Binding energy | −186.186 | −137.894 | −159.136 | −112.119 |
| Physicochemical Parameters | Values | ADMET Parameters | Values |
|---|---|---|---|
| Molecular Weight (MW) | 424.010 | Absorption | |
| Volume | 386.756 | Caco-2 Permeability | −4.616 |
| Density | 1.096 | MDCK Permeability | 2.3 × 10−5 |
| nHA | 7 | Pgp inhibitor | - |
| nHD | 3 | Distribution | |
| nRot | 2 | VD | 0.309 |
| nRing | 4 | BBB | - |
| nHet | 9 | Metabolism | |
| fChar | 0 | CYP1A2 | - |
| nRig | 25 | CYP2C19 | - |
| Flexibility | 0.080 | CYP2C9 | - |
| TPSA | 120.310 | CYP2D6 | - |
| logS | −6.188 | Excretion | |
| logP | 4.233 | Clearance | 15 mL/kg/min |
| logD | 3.102 | Toxicity | |
| Stereo centers | 0 | hERG blockers | - |
| Lipinski Rule | Accepted | AMES toxicity | 0 alerts |
| Pfizer Rule | Accepted | Carcinogenicity | 0 alerts |
| PAINS | 0 alerts | Acute Toxicity Rule | 0 alerts |
| Protein Targets | Coordinates of the Grid Box | Size of the Grid Box | ||
|---|---|---|---|---|
| x | y | z | ||
| α-glycosidase | −17.48 Å | −8.62 Å | −19.65Å | 40 Å × 40 Å × 40 Å |
| α-amylase | 103.46 Å | 37.17 Å | 19.60 Å | 40 Å × 40 Å × 40 Å |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Ghorbani, M.; Alharbi, O.; Al-Odayni, A.-B.; Abduh, N.A.Y. Quinoline- and Isoindoline-Integrated Polycyclic Compounds as Antioxidant, and Antidiabetic Agents Targeting the Dual Inhibition of α-Glycosidase and α-Amylase Enzymes. Pharmaceuticals 2023, 16, 1222. https://doi.org/10.3390/ph16091222
Al-Ghorbani M, Alharbi O, Al-Odayni A-B, Abduh NAY. Quinoline- and Isoindoline-Integrated Polycyclic Compounds as Antioxidant, and Antidiabetic Agents Targeting the Dual Inhibition of α-Glycosidase and α-Amylase Enzymes. Pharmaceuticals. 2023; 16(9):1222. https://doi.org/10.3390/ph16091222
Chicago/Turabian StyleAl-Ghorbani, Mohammed, Osama Alharbi, Abdel-Basit Al-Odayni, and Naaser A. Y. Abduh. 2023. "Quinoline- and Isoindoline-Integrated Polycyclic Compounds as Antioxidant, and Antidiabetic Agents Targeting the Dual Inhibition of α-Glycosidase and α-Amylase Enzymes" Pharmaceuticals 16, no. 9: 1222. https://doi.org/10.3390/ph16091222
APA StyleAl-Ghorbani, M., Alharbi, O., Al-Odayni, A.-B., & Abduh, N. A. Y. (2023). Quinoline- and Isoindoline-Integrated Polycyclic Compounds as Antioxidant, and Antidiabetic Agents Targeting the Dual Inhibition of α-Glycosidase and α-Amylase Enzymes. Pharmaceuticals, 16(9), 1222. https://doi.org/10.3390/ph16091222