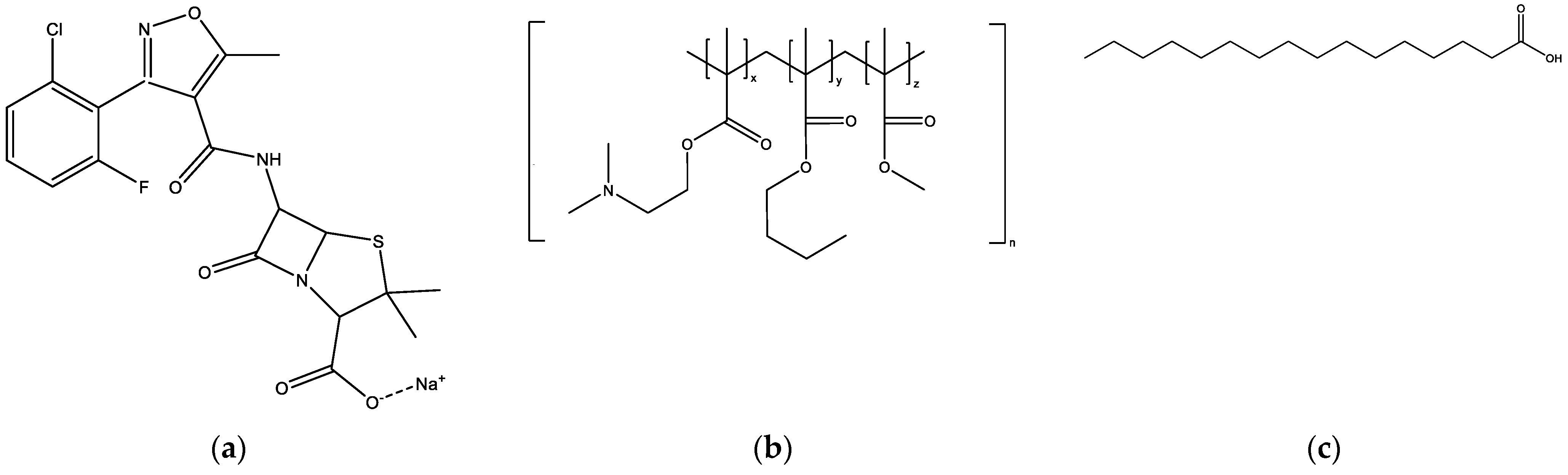
Figure 1.
Chemical structures of (a) flucloxacillin sodium; (b) Eudragit EPO, which consists of dimethylaminoethyl methacrylate (x), butyl methacrylate (y), and methyl methacrylate (z), at a ratio of 2:1:1; (c) palmitic acid.
Figure 1.
Chemical structures of (a) flucloxacillin sodium; (b) Eudragit EPO, which consists of dimethylaminoethyl methacrylate (x), butyl methacrylate (y), and methyl methacrylate (z), at a ratio of 2:1:1; (c) palmitic acid.
Figure 2.
Predicted model graph to determine optimal solvents for the two-phase systems in Option 1: (a) Predicted model graph of phase 1 showing opacity measured as a function of the proportion of ethanol to acetone. (b) Predicted model graph of phase 2 showing opacity measured as a function of the proportion of ethanol to acetone. A: proportion of ethanol; B: proportion of acetone volume. The blue line denotes the 95% confidence bands, while the red dots indicate design points.
Figure 2.
Predicted model graph to determine optimal solvents for the two-phase systems in Option 1: (a) Predicted model graph of phase 1 showing opacity measured as a function of the proportion of ethanol to acetone. (b) Predicted model graph of phase 2 showing opacity measured as a function of the proportion of ethanol to acetone. A: proportion of ethanol; B: proportion of acetone volume. The blue line denotes the 95% confidence bands, while the red dots indicate design points.
Figure 3.
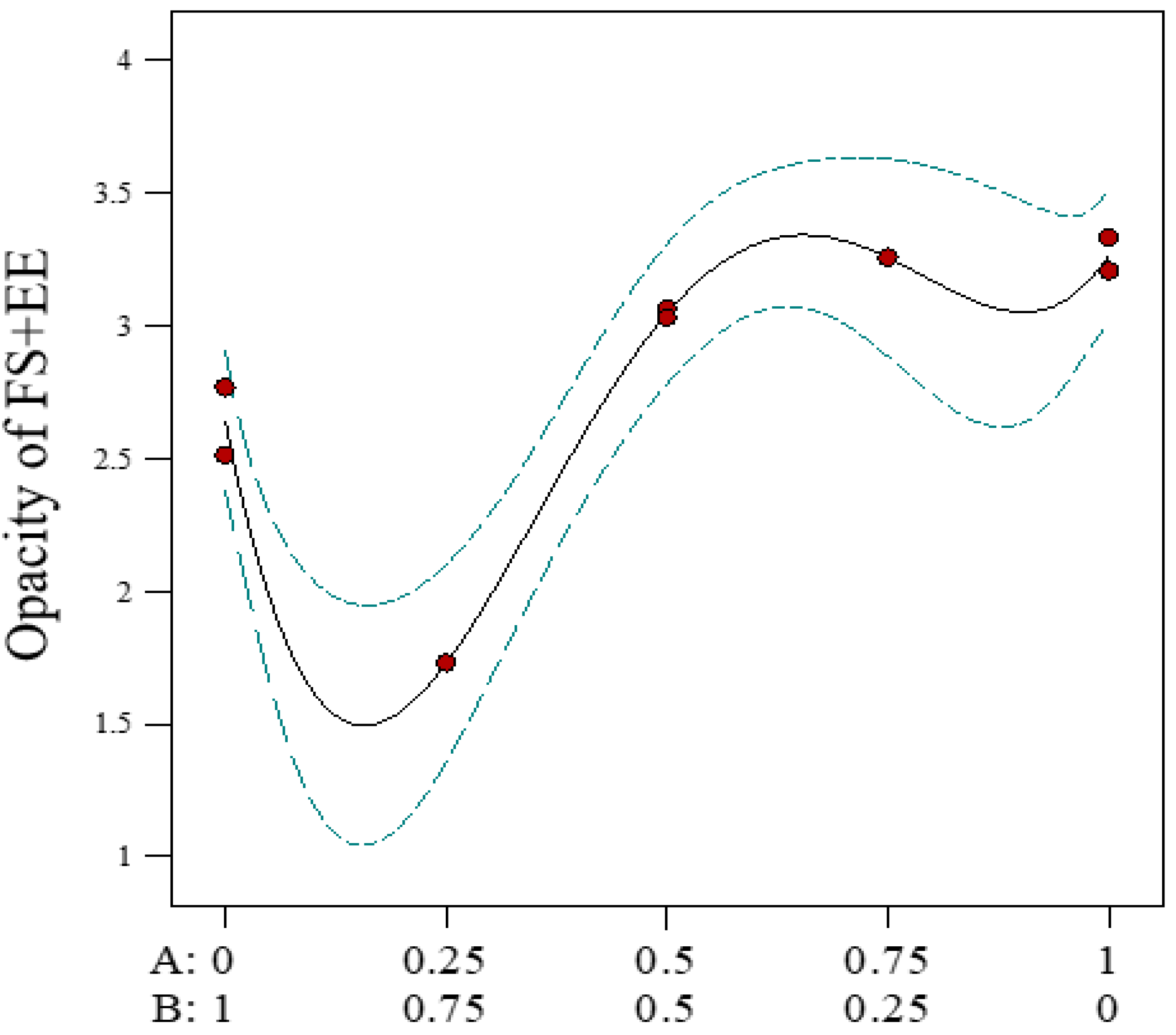
Predicted model graph to determine optimal solvents for phase 2 (FS-EE) of Option 3 of the two-phase systems, showing opacity measured as a function of the proportion of ethanol to acetone. A: proportion of ethanol; B: proportion of acetone volume. FS = flucloxacillin sodium, EE = Eudragit EPO. The blue line denotes the 95% confidence bands, while the red dots indicate design points.
Figure 3.
Predicted model graph to determine optimal solvents for phase 2 (FS-EE) of Option 3 of the two-phase systems, showing opacity measured as a function of the proportion of ethanol to acetone. A: proportion of ethanol; B: proportion of acetone volume. FS = flucloxacillin sodium, EE = Eudragit EPO. The blue line denotes the 95% confidence bands, while the red dots indicate design points.
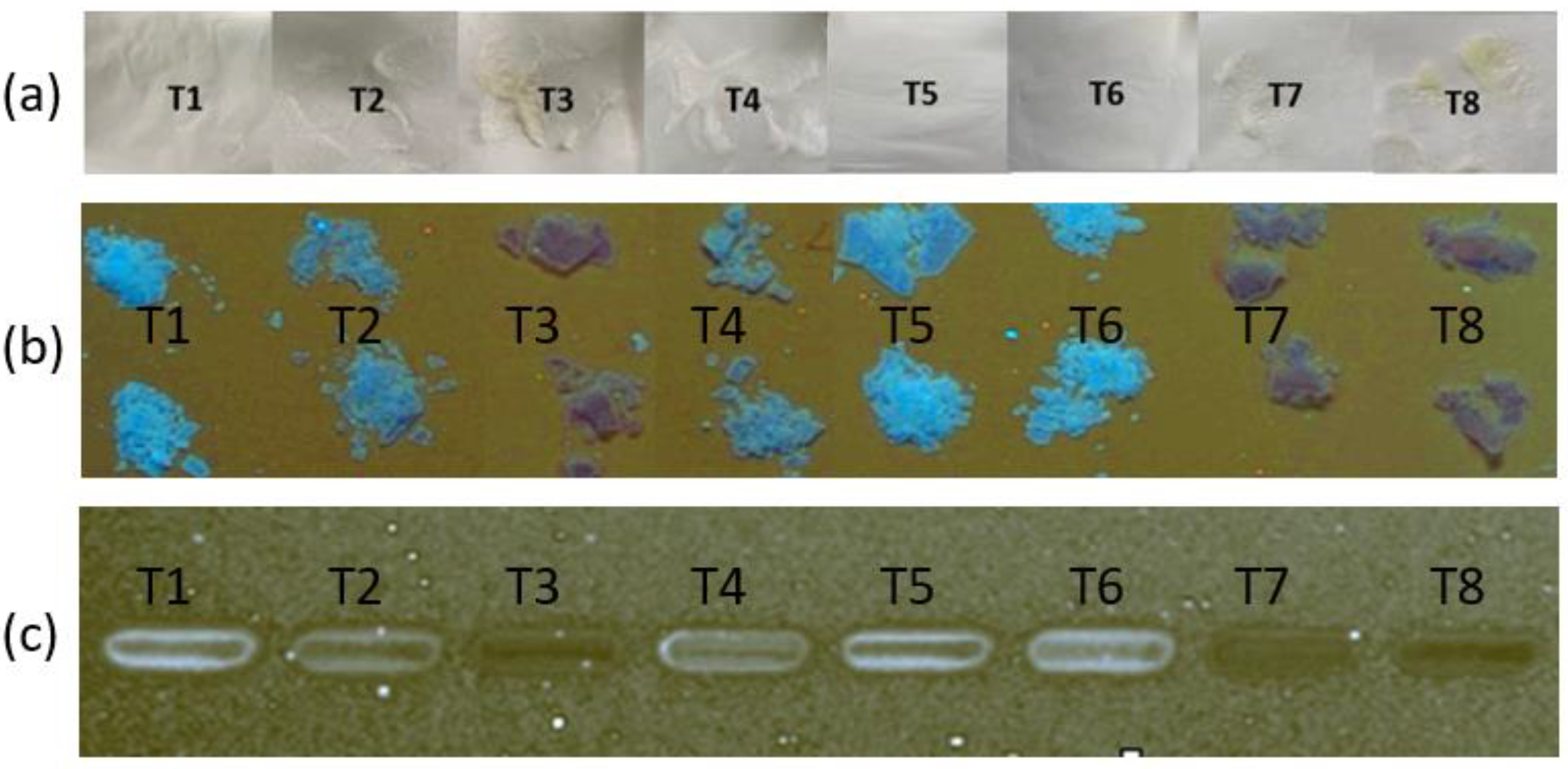
Figure 4.
Images of powders prepared by drying (at ambient temperature for 48 h) ternary suspensions prepared by triturating 0.2 g of FS, 0.16 g of EE, and 0.12 g of PA with 3 mL of solvent. Images were obtained of powder samples with (a) a camera, and with high-performance thin-layer chromatography (b) before and (c) after solubilisation in ethanol. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
Figure 4.
Images of powders prepared by drying (at ambient temperature for 48 h) ternary suspensions prepared by triturating 0.2 g of FS, 0.16 g of EE, and 0.12 g of PA with 3 mL of solvent. Images were obtained of powder samples with (a) a camera, and with high-performance thin-layer chromatography (b) before and (c) after solubilisation in ethanol. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
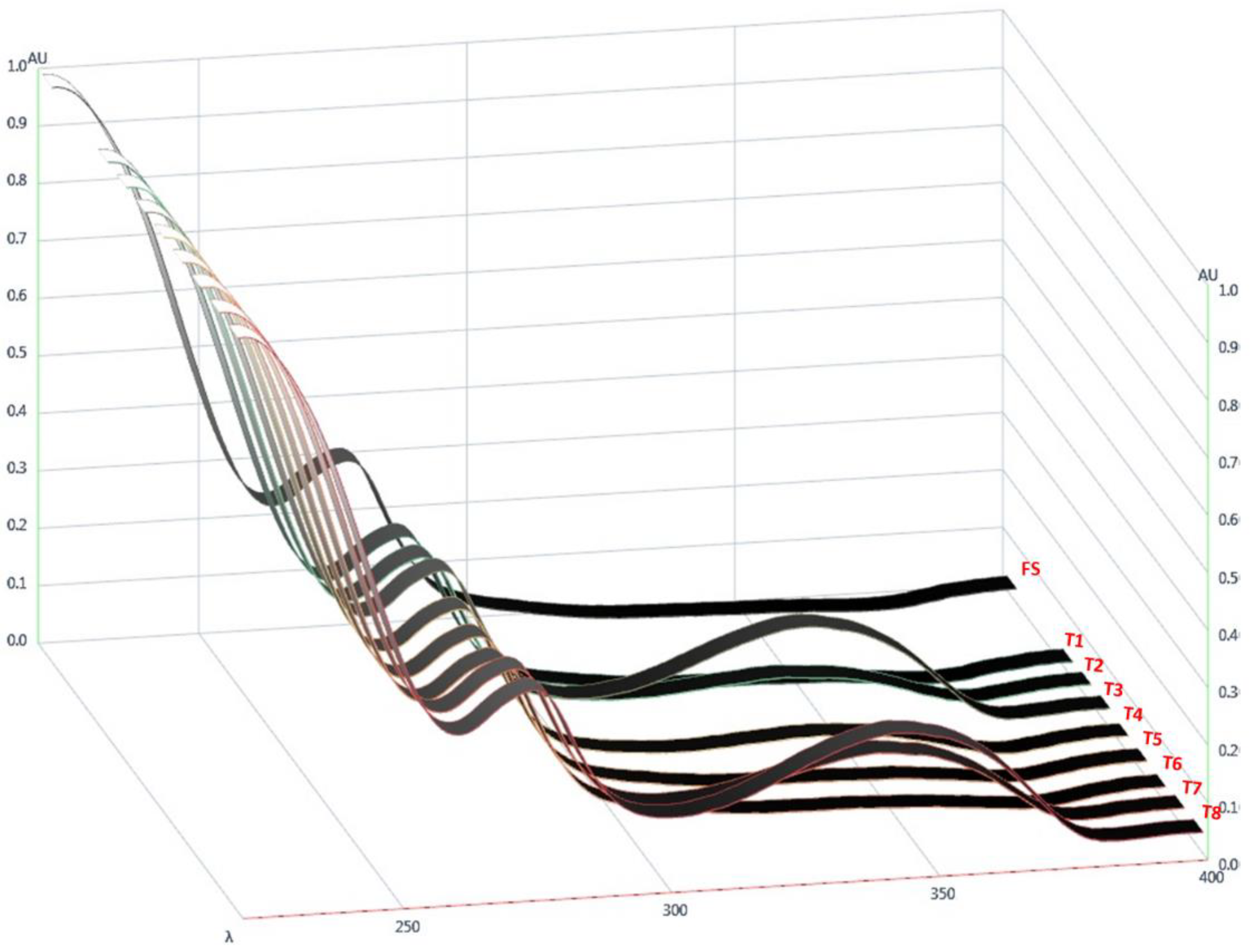
Figure 5.
UV spectra of flucloxacillin sodium (FS) and powders obtained by drying ternary systems of flucloxacillin sodium, Eudragit EPO, and palmitic acid prepared using different solvents (T1–T8).
Figure 5.
UV spectra of flucloxacillin sodium (FS) and powders obtained by drying ternary systems of flucloxacillin sodium, Eudragit EPO, and palmitic acid prepared using different solvents (T1–T8).
Figure 6.
Predicted model graphs of (a) AUC343, (b) FS loading efficiency (%), and (c) desirability, where AUC343 is minimised and FS loading efficiency (%) is maximised, as a function of the proportion of ethanol in the solvent system used to prepare the ternary samples of flucloxacillin sodium (FS), Eudragit EPO, and palmitic acid. A: proportion of ethanol; B: proportion of acetone volume. The blue line denotes the 95% confidence bands, while the red dots indicate design points.
Figure 6.
Predicted model graphs of (a) AUC343, (b) FS loading efficiency (%), and (c) desirability, where AUC343 is minimised and FS loading efficiency (%) is maximised, as a function of the proportion of ethanol in the solvent system used to prepare the ternary samples of flucloxacillin sodium (FS), Eudragit EPO, and palmitic acid. A: proportion of ethanol; B: proportion of acetone volume. The blue line denotes the 95% confidence bands, while the red dots indicate design points.
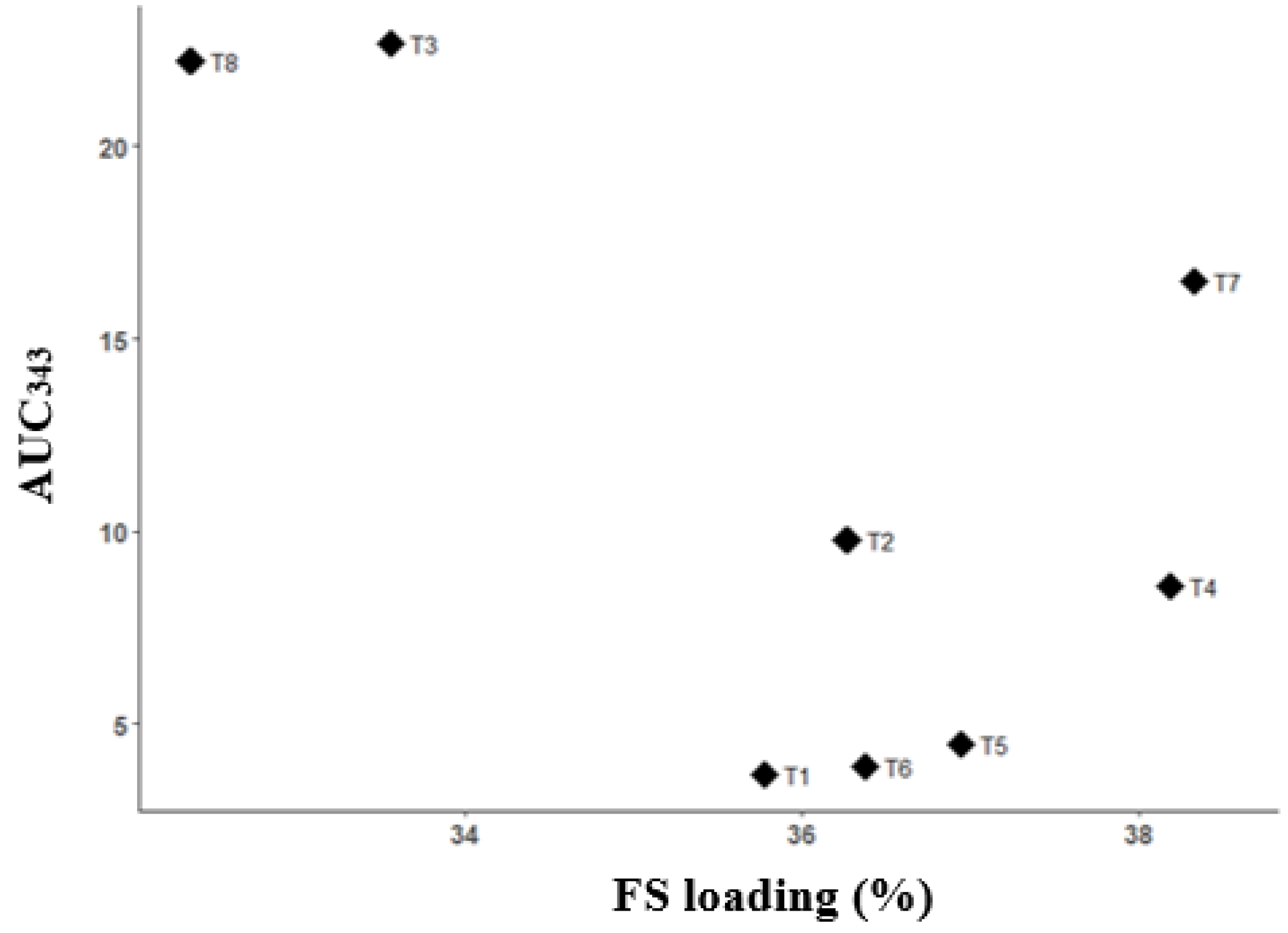
Figure 7.
Scatterplot of FS loading efficiency (%) vs. AUC343 for powders obtained by drying ternary systems of flucloxacillin sodium (FS), Eudragit EPO, and palmitic acid prepared using different solvents (T1–T8).
Figure 7.
Scatterplot of FS loading efficiency (%) vs. AUC343 for powders obtained by drying ternary systems of flucloxacillin sodium (FS), Eudragit EPO, and palmitic acid prepared using different solvents (T1–T8).
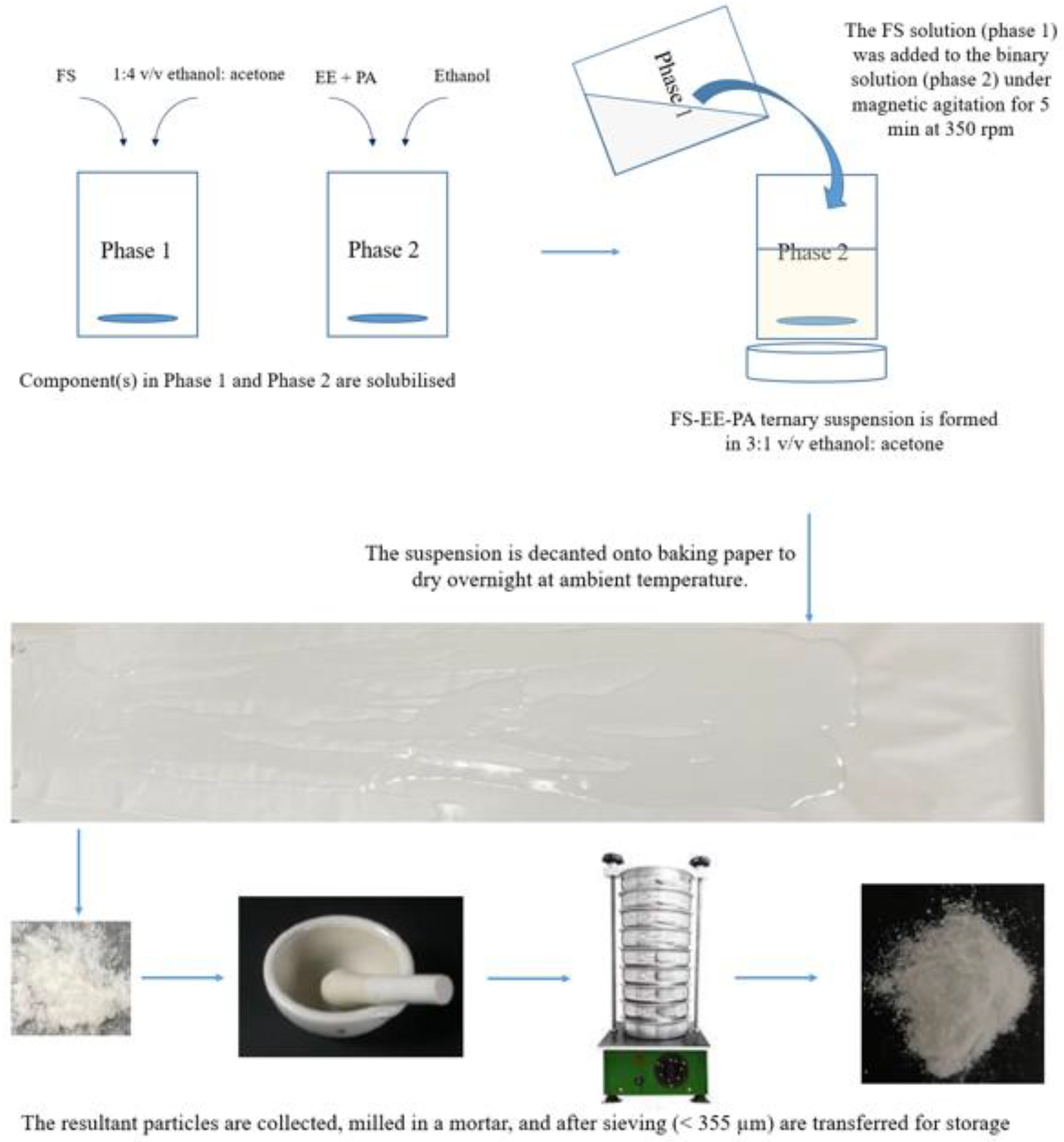
Figure 8.
Schematic diagram showing the optimised fabrication process for preparing a taste-masked powder from a ternary system comprising flucloxacillin sodium (FS), Eudragit EPO (EE), and palmitic acid (PA).
Figure 8.
Schematic diagram showing the optimised fabrication process for preparing a taste-masked powder from a ternary system comprising flucloxacillin sodium (FS), Eudragit EPO (EE), and palmitic acid (PA).
Figure 9.
FTM images: (a) produced via the optimised fabrication method at baseline; (b) after 6 months of storage at ambient temperature.
Figure 9.
FTM images: (a) produced via the optimised fabrication method at baseline; (b) after 6 months of storage at ambient temperature.
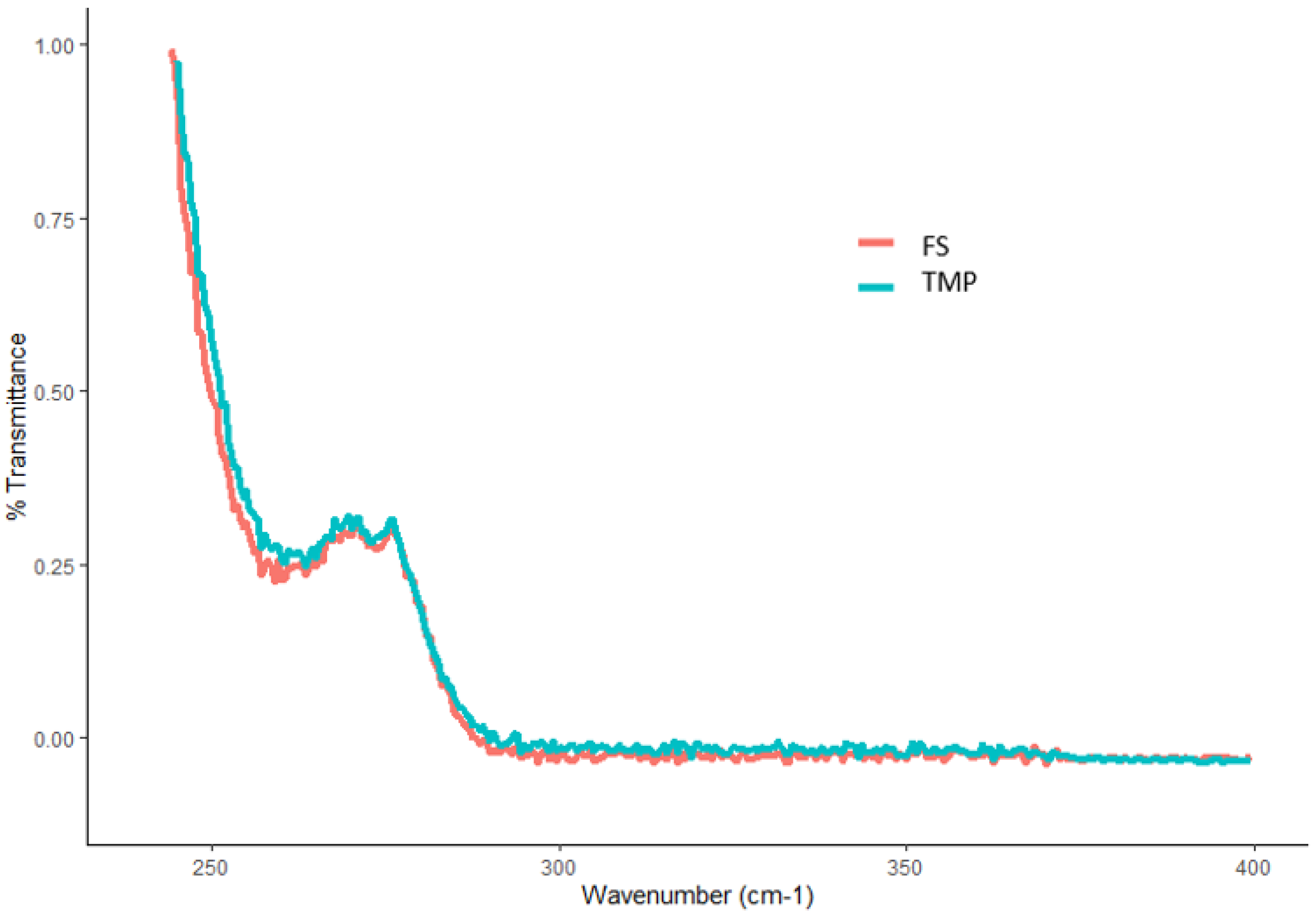
Figure 10.
UV spectra of an FTM sample produced by the optimal fabrication process (light blue) and flucloxacillin sodium (FS, pink).
Figure 10.
UV spectra of an FTM sample produced by the optimal fabrication process (light blue) and flucloxacillin sodium (FS, pink).
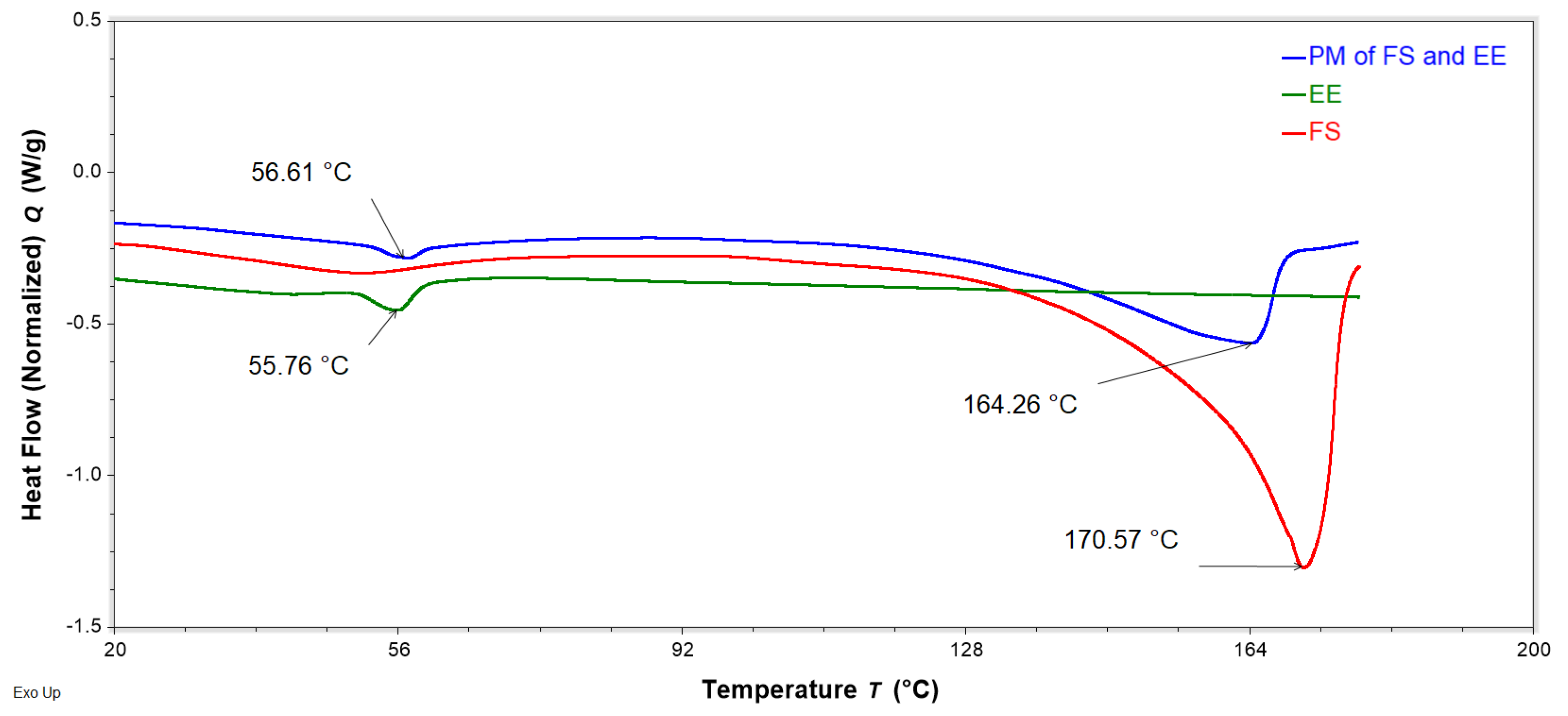
Figure 11.
DSC thermograms of neat flucloxacillin sodium (FS, red), neat Eudragit EPO (EE, green), and a binary physical mixture (PM) of FS and EE (1:0.8 w/w, blue) obtained with a heating rate of 10 °C/min, showing the peak temperature of the endotherms of FS, EE, and PM.
Figure 11.
DSC thermograms of neat flucloxacillin sodium (FS, red), neat Eudragit EPO (EE, green), and a binary physical mixture (PM) of FS and EE (1:0.8 w/w, blue) obtained with a heating rate of 10 °C/min, showing the peak temperature of the endotherms of FS, EE, and PM.
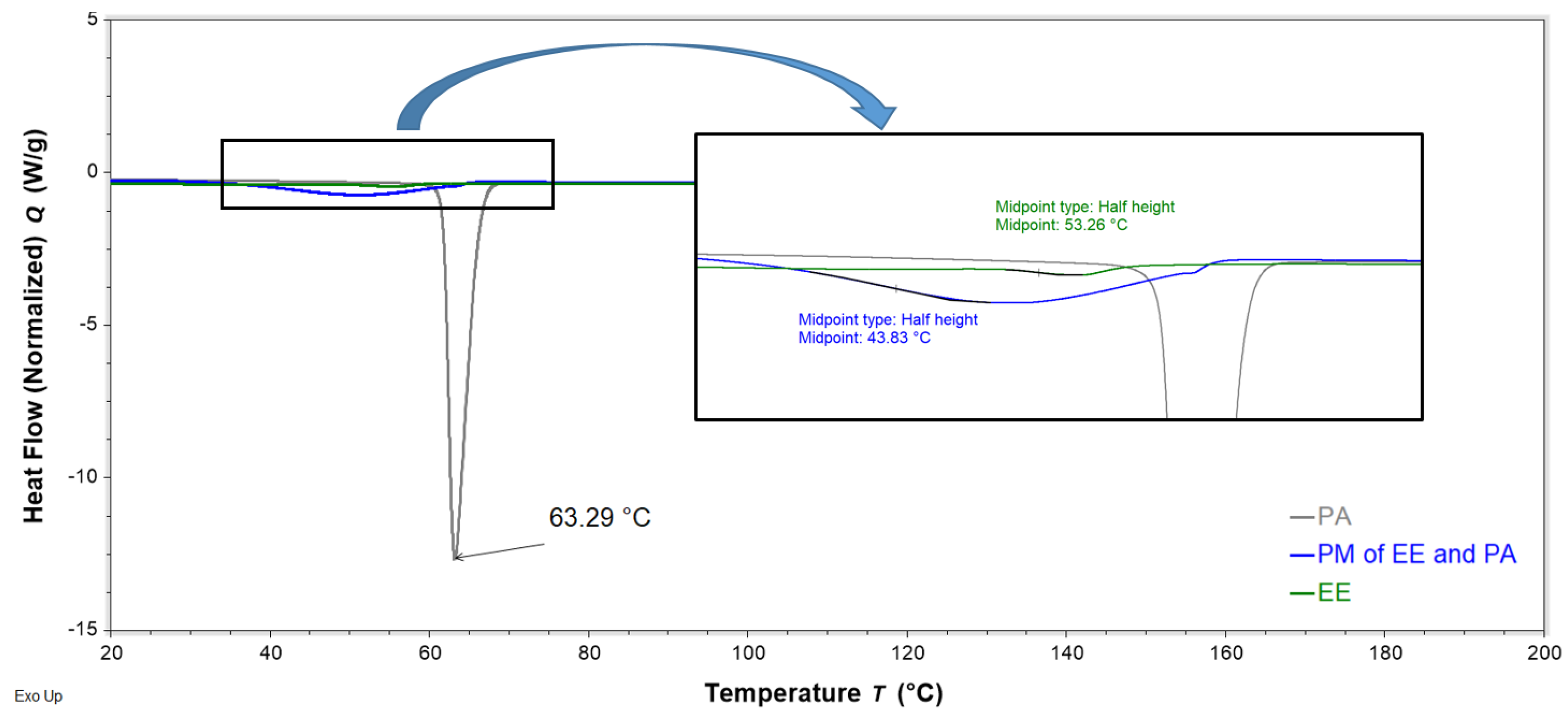
Figure 12.
DSC thermograms of neat flucloxacillin sodium (FS, red), neat palmitic acid (PA, grey), and a binary physical mixture (PM) of FS and PA (1:0.6 w/w, blue) obtained with a heating rate of 10 °C/min, showing the peak temperature of the endotherms of FS, PA, and PM.
Figure 12.
DSC thermograms of neat flucloxacillin sodium (FS, red), neat palmitic acid (PA, grey), and a binary physical mixture (PM) of FS and PA (1:0.6 w/w, blue) obtained with a heating rate of 10 °C/min, showing the peak temperature of the endotherms of FS, PA, and PM.
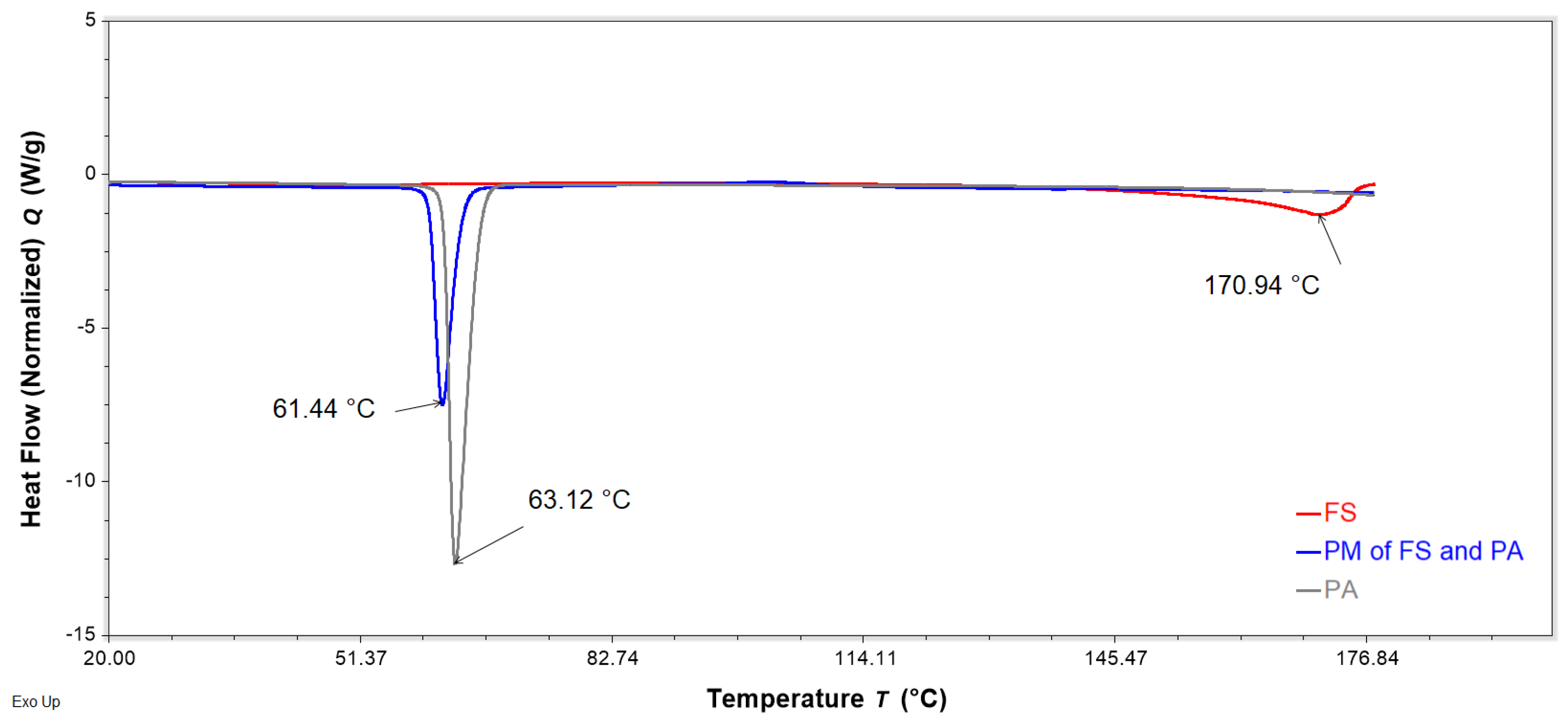
Figure 13.
DSC thermograms of neat Eudragit EPO (EE, green), neat palmitic acid (PA, grey), and a binary physical mixture (PM) of EE and PA (0.8:0.6 w/w, blue) obtained with a heating rate of 10 °C/min, showing the glass transition temperatures of EE and PM, as well as the peak temperature of the endotherm of PA. Phase transitions between 30 and 70 °C are amplified in the zoom box.
Figure 13.
DSC thermograms of neat Eudragit EPO (EE, green), neat palmitic acid (PA, grey), and a binary physical mixture (PM) of EE and PA (0.8:0.6 w/w, blue) obtained with a heating rate of 10 °C/min, showing the glass transition temperatures of EE and PM, as well as the peak temperature of the endotherm of PA. Phase transitions between 30 and 70 °C are amplified in the zoom box.
Figure 14.
DSC thermograms of FTMs (purple) fabricated using the optimised method and the ternary physical mixture (PM) (FS:EE:PA at 1:0.8:0.6 w/w/w, blue) obtained with a heating rate of 10 °C/min, showing the peak temperature of the endotherms of FTMs and PM. The analysis was not prolonged, due to the material degradation associated with temperature above 100 °C. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
Figure 14.
DSC thermograms of FTMs (purple) fabricated using the optimised method and the ternary physical mixture (PM) (FS:EE:PA at 1:0.8:0.6 w/w/w, blue) obtained with a heating rate of 10 °C/min, showing the peak temperature of the endotherms of FTMs and PM. The analysis was not prolonged, due to the material degradation associated with temperature above 100 °C. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
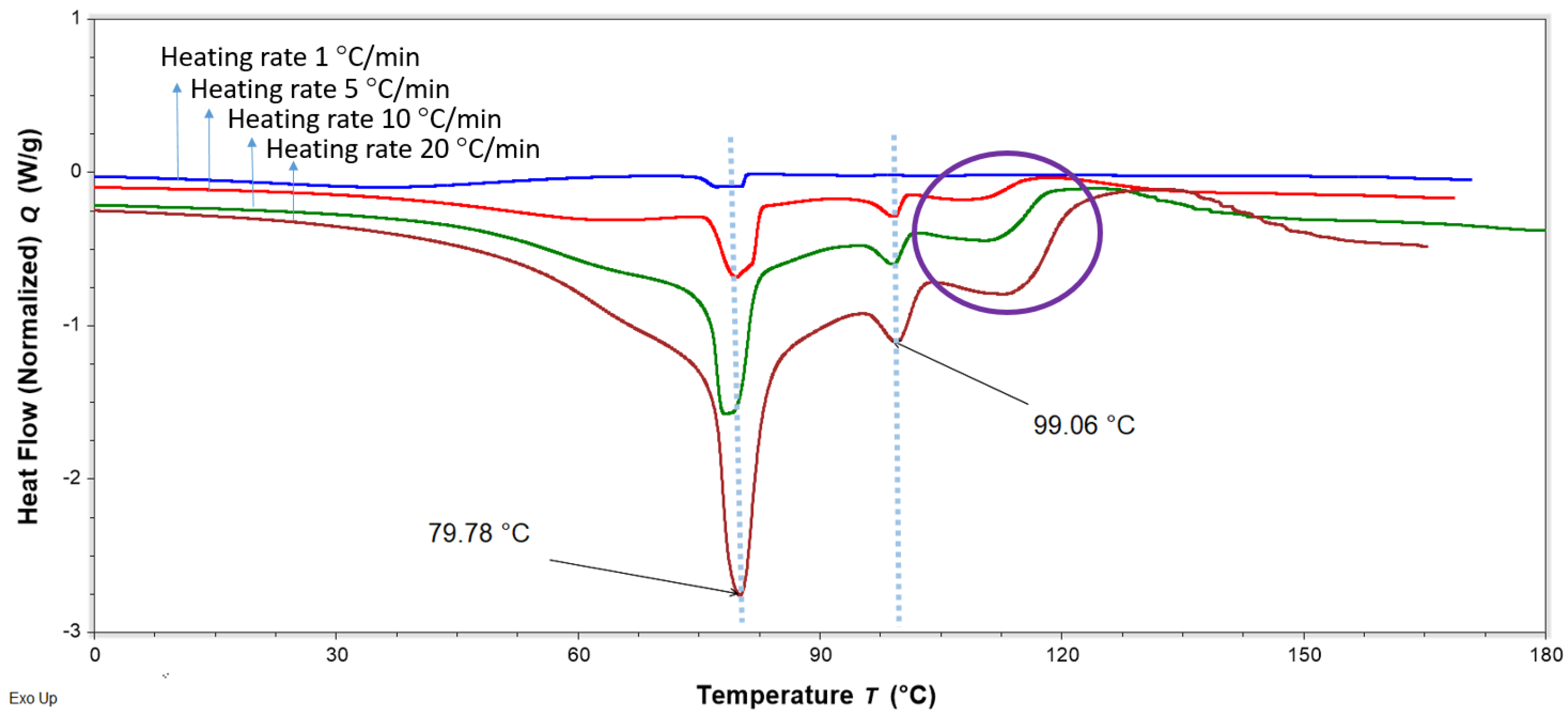
Figure 15.
DSC thermograms of FTMs fabricated using the optimised method (FS:EE:PA at 1:0.8:0.6 w/w/w), heated at different heating rates of 1 to 20 °C/min, showing variations in the phase transitions above 100 °C. The likely degradation events are circled in purple. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
Figure 15.
DSC thermograms of FTMs fabricated using the optimised method (FS:EE:PA at 1:0.8:0.6 w/w/w), heated at different heating rates of 1 to 20 °C/min, showing variations in the phase transitions above 100 °C. The likely degradation events are circled in purple. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
Figure 16.
FS-EE-PA microparticles that passed through a sieve with a pore diameter of 355 µm and were retained on sieves with pore diameters of 212 µm.
Figure 16.
FS-EE-PA microparticles that passed through a sieve with a pore diameter of 355 µm and were retained on sieves with pore diameters of 212 µm.
Table 1.
Opacity (UV absorbance at 550 nm) obtained for phase 1 and phase 2 for Option 1 of the two-phase systems when mixed with specified solvents. Phase 1 was mixed with 1 mL of solvent, while phase 2 was mixed with 2 mL of solvent. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
Table 1.
Opacity (UV absorbance at 550 nm) obtained for phase 1 and phase 2 for Option 1 of the two-phase systems when mixed with specified solvents. Phase 1 was mixed with 1 mL of solvent, while phase 2 was mixed with 2 mL of solvent. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
| Design Point | Proportion of Ethanol | Proportion of Acetone | Opacity for Phase 1 (FS) | Opacity for Phase 2 (EE + PA) |
|---|
| 1 | 1 | 0 | 2.96 | 0.06 |
| 2 | 0.5 | 0.5 | 2.43 | 0.03 |
| 3 | 0 | 1 | 2.32 | 0.55 |
| 4 | 0.5 | 0.5 | 2.28 | 0.05 |
| 5 | 0.75 | 0.25 | 2.86 | 0.03 |
| 6 | 1 | 0 | 2.41 | 0.05 |
| 7 | 0.25 | 0.75 | 0.14 | 0.41 |
| 8 | 0 | 1 | 2.68 | 0.46 |
Table 2.
Coefficients and confidence intervals (CIs) for the mixture model for phase 1 and 2, where the independent variables are the proportions of solvents and the respondents are opacity for phase 1 and phase 2 measured as visible spectral absorption at 550 nm on a microplate reader. A: proportion of ethanol; B: proportion of acetone volume.
Table 2.
Coefficients and confidence intervals (CIs) for the mixture model for phase 1 and 2, where the independent variables are the proportions of solvents and the respondents are opacity for phase 1 and phase 2 measured as visible spectral absorption at 550 nm on a microplate reader. A: proportion of ethanol; B: proportion of acetone volume.
| p-Value for Model for Phase 1: 0.02 |
|---|
| Model Terms for Phase 1 | Coefficient Estimate | 95% CI |
|---|
| A | 2.69 | 2.07–3.30 |
| B | 2.50 | 1.88–3.12 |
| AB | −0.94 | −3.97–2.09 |
| AB(A-B) | 13.99 | 6.99–20.99 |
| AB(A-B)2 | −19.52 | −36.18–−2.86 |
| Model Terms for Phase 2 | | |
| p-Value for Model for Phase 2: 0.025 |
| A | 0.05 | −0.094–0.19 |
| B | 0.53 | 0.39–0.68 |
| AB | −0.77 | −1.43–−0.11 |
Table 3.
AUC343 obtained for powders obtained by drying (at ambient temperature for 48 h) ternary suspensions prepared by triturating 0.2 g of FS, 0.16 g of EE, and 0.12 g of PA with 3 mL of the specified solvent. AUC343 is the UV absorption of all peaks measured at 343 nm using the HPLC assay developed for flucloxacillin sodium. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
Table 3.
AUC343 obtained for powders obtained by drying (at ambient temperature for 48 h) ternary suspensions prepared by triturating 0.2 g of FS, 0.16 g of EE, and 0.12 g of PA with 3 mL of the specified solvent. AUC343 is the UV absorption of all peaks measured at 343 nm using the HPLC assay developed for flucloxacillin sodium. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
| Sample Number | Proportion of Ethanol | Proportion of Acetone | AUC343 of Ternary Phase |
|---|
| T1 | 1 | 0 | 0.86 |
| T2 | 0.5 | 0.5 | 2.27 |
| T3 | 0 | 1 | 0.45 |
| T4 | 0.5 | 0.5 | 2.22 |
| T5 | 0.75 | 0.25 | 0.37 |
| T6 | 1 | 0 | 0.39 |
| T7 | 0.25 | 0.75 | 0.98 |
| T8 | 0 | 1 | 1.65 |
Table 4.
Coefficients and confidence intervals (CIs) for the mixture model, where the independent variables are the proportions of solvents and the respondents are the FS loading efficiency (%) and AUC343 for the ternary samples of flucloxacillin sodium (FS), Eudragit EPO, and palmitic acid (T1–T8). AUC343 is the UV absorption of all peaks measured at 343 nm using the HPLC assay developed for FS. A: proportion of ethanol; B: proportion of acetone volume.
Table 4.
Coefficients and confidence intervals (CIs) for the mixture model, where the independent variables are the proportions of solvents and the respondents are the FS loading efficiency (%) and AUC343 for the ternary samples of flucloxacillin sodium (FS), Eudragit EPO, and palmitic acid (T1–T8). AUC343 is the UV absorption of all peaks measured at 343 nm using the HPLC assay developed for FS. A: proportion of ethanol; B: proportion of acetone volume.
| p-Value for Model of % FS Loading Efficiency: <0.0359; p-Value for Lack of Fit: 0.1976 |
|---|
| Model Terms of % FS Loading Efficiency | Coefficient Estimate | 95% CI |
|---|
| A | 35.84 | 33.61–38.07 |
| B | 33.38 | 31.15–35.61 |
| AB | 12.52 | 2.19–22.85 |
| p-Value for Model of AUC343: <0.0001; p-Value for Lack of Fit: 0.4461 |
| Model Terms of AUC343 | Coefficient Estimate | 95% CI |
| A | 3.82 | −2.86–4.78 |
| B | 22.47 | 21.50–23.43 |
| AB | −15.29 | −19.63–−10.95 |
| AB(A-B) | −14.31 | −25.27–−3.35 |
Table 5.
Volumes of ethanol and acetone required to fulfil the optimal solvents predicted to solubilise phase 1 and phase 2, and to produce stable FTMs using 1 g of FS, 0.8 g of EE, and 0.6 g of PA as starting materials. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
Table 5.
Volumes of ethanol and acetone required to fulfil the optimal solvents predicted to solubilise phase 1 and phase 2, and to produce stable FTMs using 1 g of FS, 0.8 g of EE, and 0.6 g of PA as starting materials. FS = flucloxacillin sodium, EE = Eudragit EPO, PA = palmitic acid.
| Phase | Ethanol (mL) | Acetone (mL) | Ethanol:Acetone (v/v) | Weight of Component(s) (g) | Total Solvent (mL) (Equivalent Weight (g)) |
|---|
| FS (Phase 1) | 0.74 | 2.94 | 0.2:0.8 | 1 | 3.68 (2.88) |
| EE + PA (Phase 2) | 8.10 | 0 | 1:0 | 1.4 | 8.1 (6.40) |
| Final Ternary Mixture | 8.83 | 2.94 | 0.75:0.25 | 2.4 | 11.8 (9.27) |
Table 6.
Drug-loading efficiency in 3 independent batches of FTMs produced on the same day and over 2 different days. Triplicate samples were withdrawn from each batch of FTMs to determine the drug-loading efficiency via a validated HPLC assay.
Table 6.
Drug-loading efficiency in 3 independent batches of FTMs produced on the same day and over 2 different days. Triplicate samples were withdrawn from each batch of FTMs to determine the drug-loading efficiency via a validated HPLC assay.
| | Flucloxacillin Sodium Loading Efficiency in FTMs
(% w/w) (n = 3, Mean ± SD) |
|---|
| | Day 1 | Day 2 |
| Batch 1 | 39.6 ± 1.0 | 39.9 ± 0.5 |
| Batch 2 | 40.8 ± 1.0 | 39.3± 0.8 |
| Batch 3 | 39.6 ± 1.0 | 39.1 ± 0.9 |
Table 7.
Components present in phase 1 (single component) and phase 2 (two components) employed for the two-phase experiments.
Table 7.
Components present in phase 1 (single component) and phase 2 (two components) employed for the two-phase experiments.
| Option Number | Component in Phase 1 (Mass in g) | Components in Phase 2 (Mass in g) |
|---|
| 1 | FS (0.2) | EE (0.16) + PA (0.12) |
| 2 | EE (0.16) | FS (0.2) + PA (0.12) |
| 3 | PA (0.12) | FS (0.2) + EE (0.16) |
Table 8.
Simplex lattice mixture design with 8 experimental runs involving different proportions (v/v) of ethanol and acetone to determine optimal solvent systems to solubilise the components of each phase.
Table 8.
Simplex lattice mixture design with 8 experimental runs involving different proportions (v/v) of ethanol and acetone to determine optimal solvent systems to solubilise the components of each phase.
| Sample Number | Proportion of Ethanol | Proportion of Acetone |
|---|
| 1 | 1 | 0 |
| 2 | 0.5 | 0.5 |
| 3 | 0 | 1 |
| 4 | 0.5 | 0.5 |
| 5 | 0.75 | 0.25 |
| 6 | 1 | 0 |
| 7 | 0.25 | 0.75 |
| 8 | 0 | 1 |
Table 9.
Simplex lattice mixture design with 8 experimental runs involving different proportions of ethanol and acetone to determine the optimal solvent for a ternary system to generate stable FTMs.
Table 9.
Simplex lattice mixture design with 8 experimental runs involving different proportions of ethanol and acetone to determine the optimal solvent for a ternary system to generate stable FTMs.
| Sample Number | Proportion of Ethanol | Proportion of Acetone |
|---|
| T1 | 1 | 0 |
| T2 | 0.5 | 0.5 |
| T3 | 0 | 1 |
| T4 | 0.5 | 0.5 |
| T5 | 0.75 | 0.25 |
| T6 | 1 | 0 |
| T7 | 0.25 | 0.75 |
| T8 | 0 | 1 |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}