


Discovery of Potential Noncovalent Inhibitors of Dehydroquinate Dehydratase from Methicillin-Resistant Staphylococcus aureus through Computational-Driven Drug Design

 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
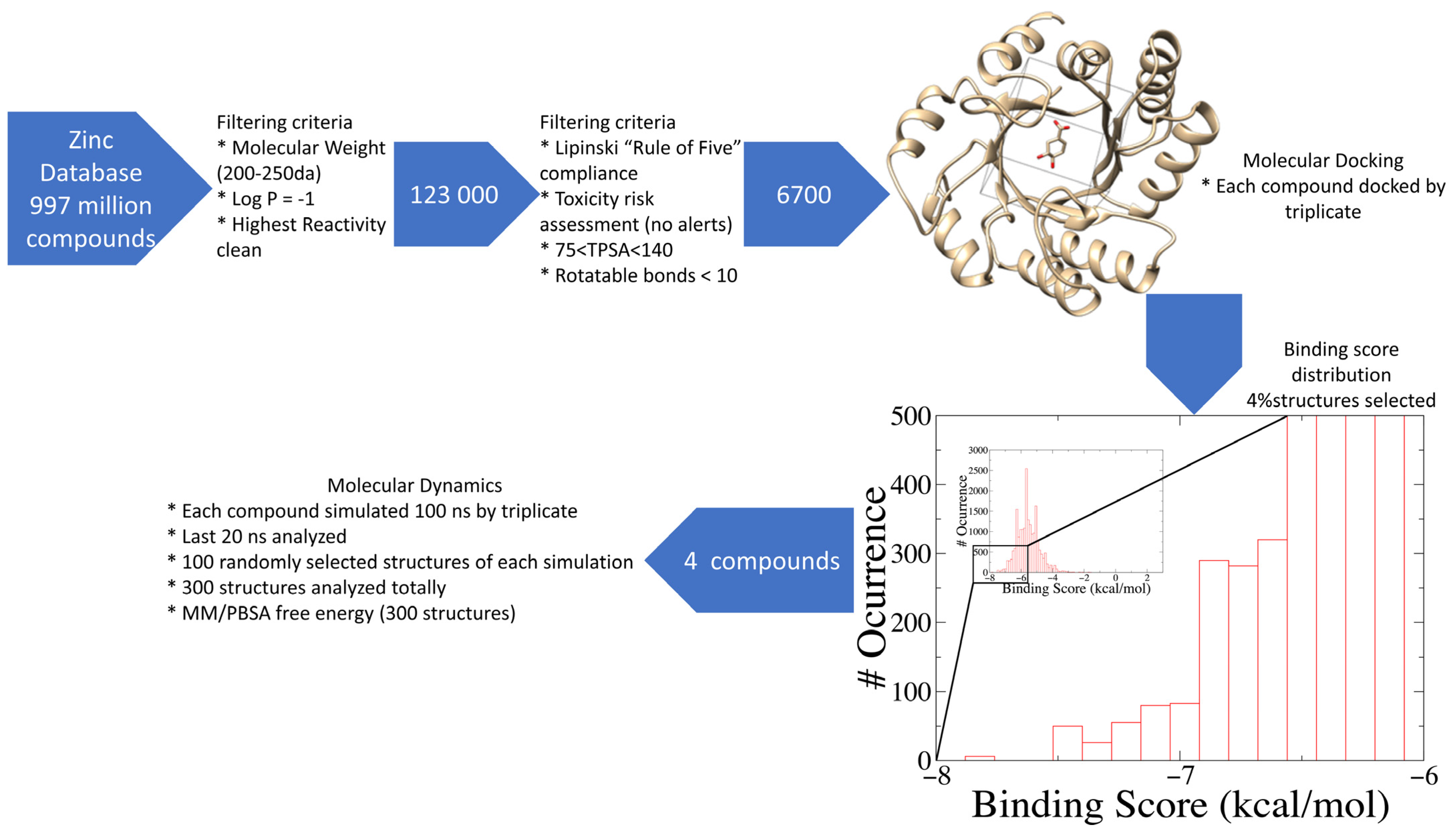
2.1. Database Filtering and Virtual Screening
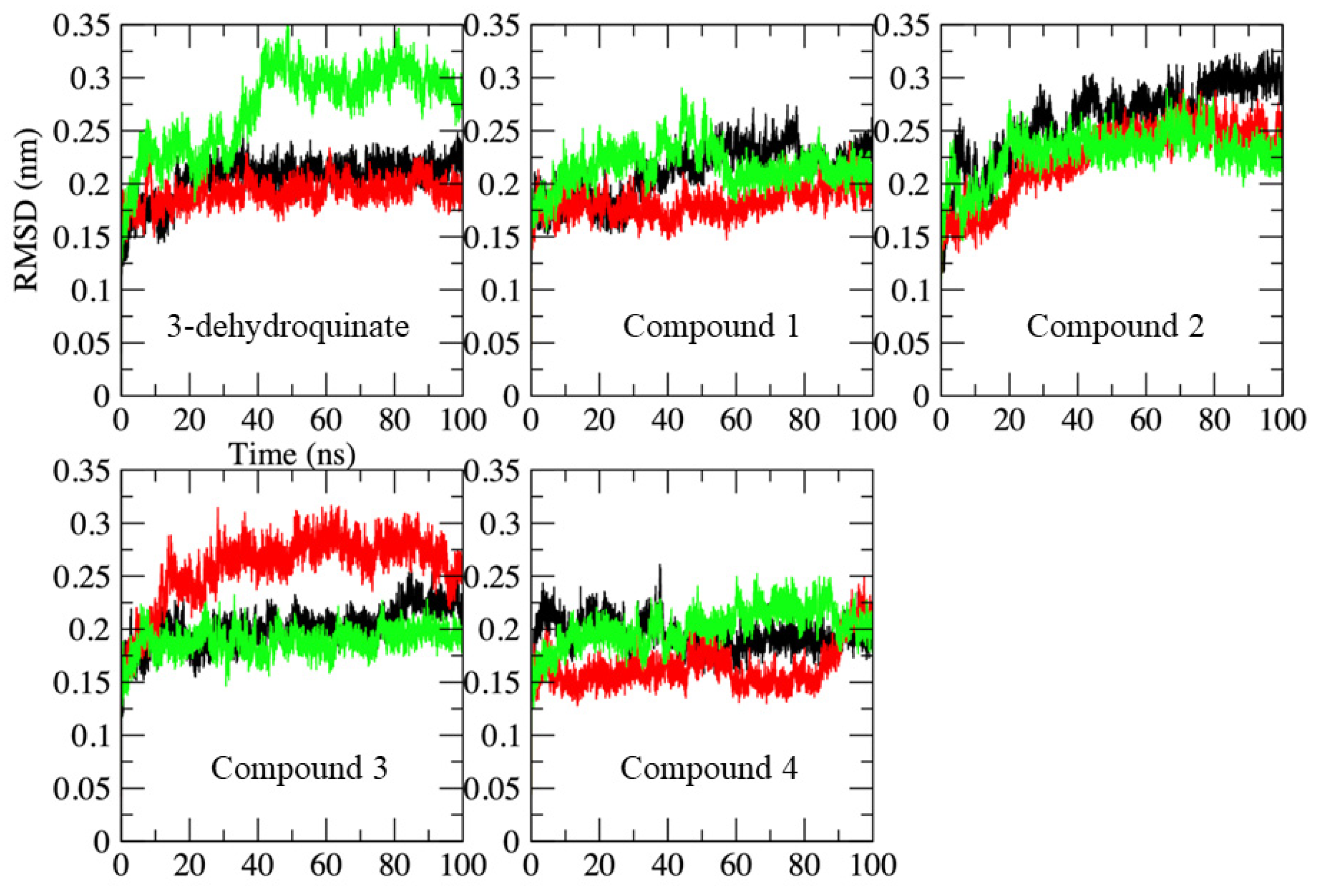
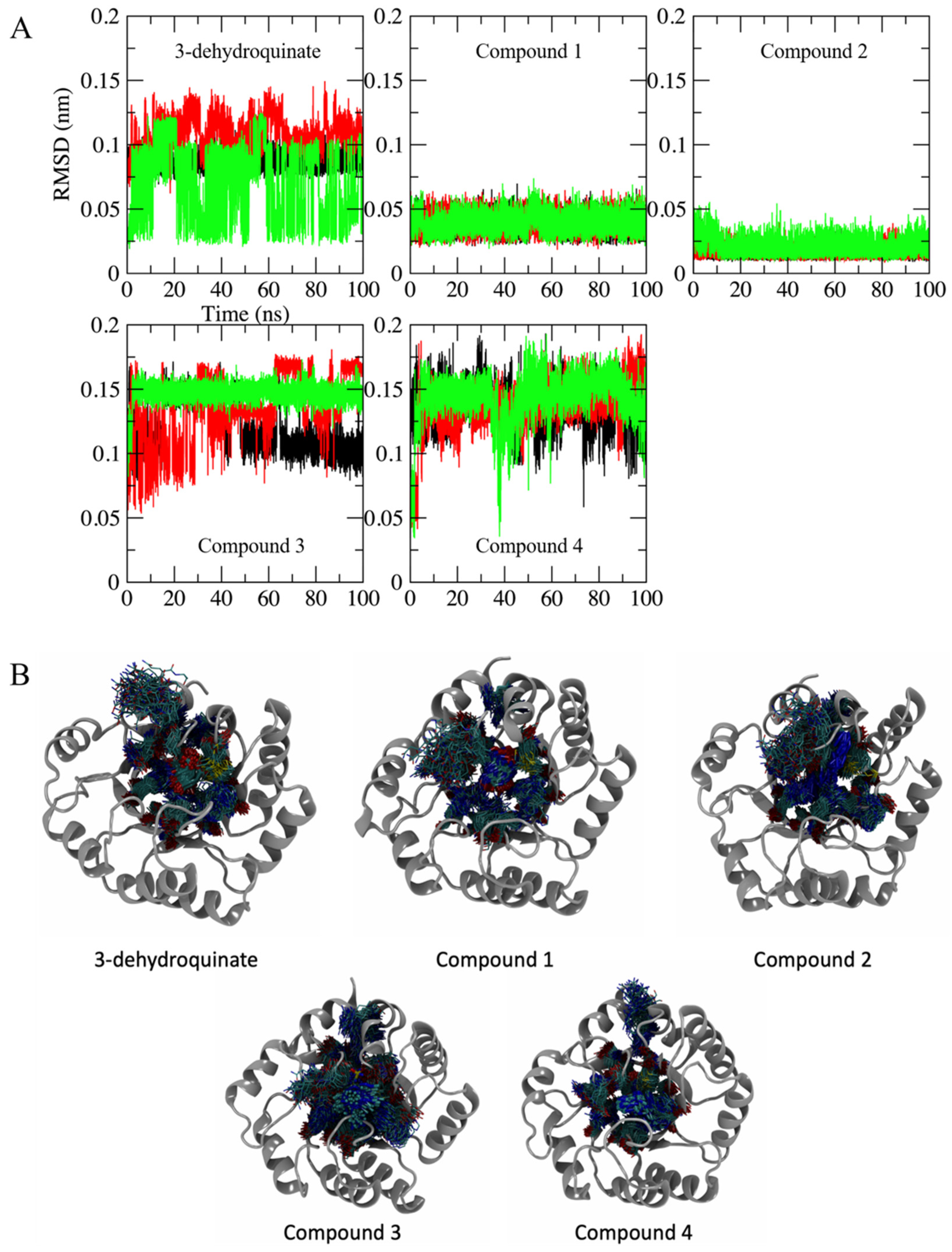
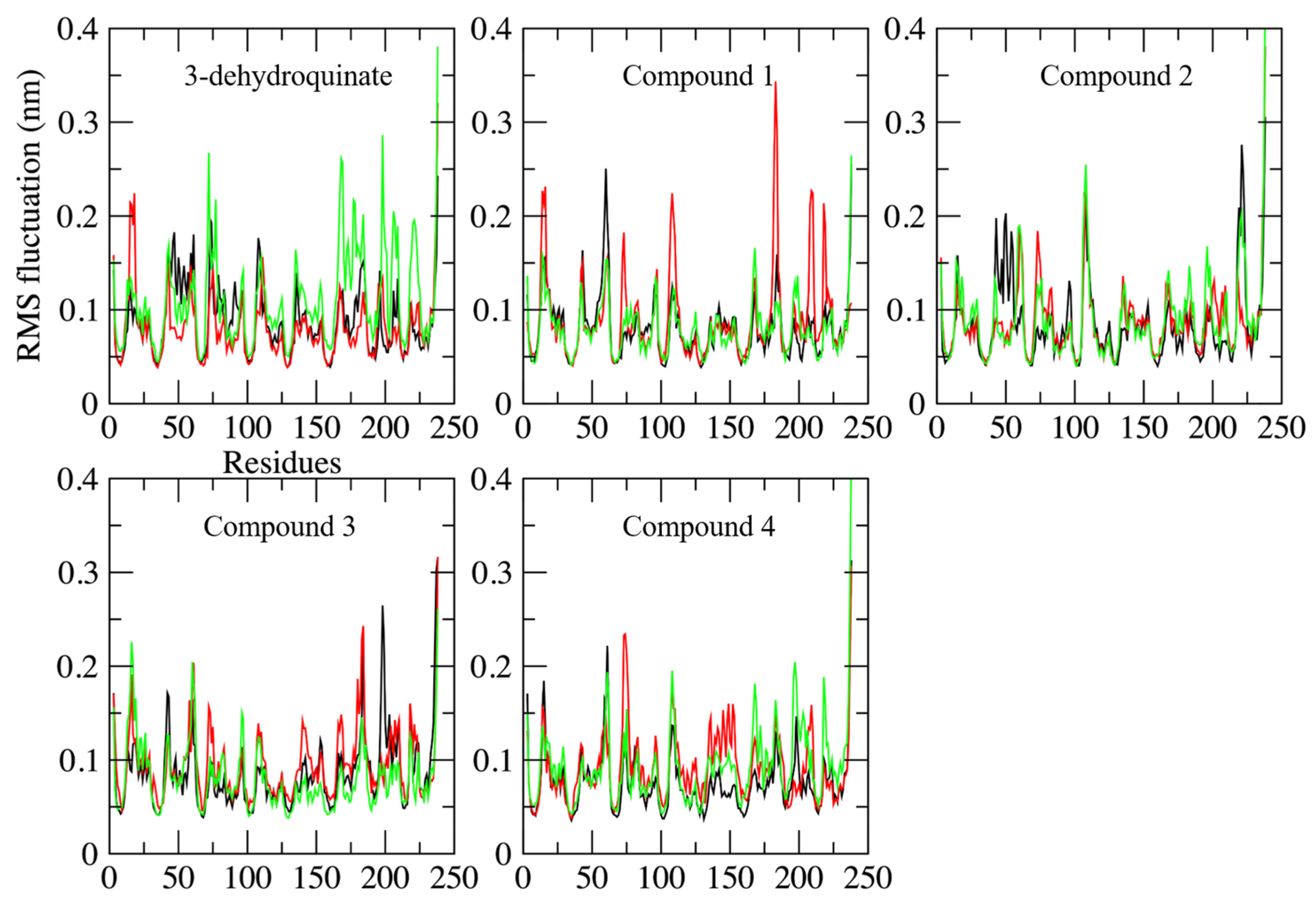
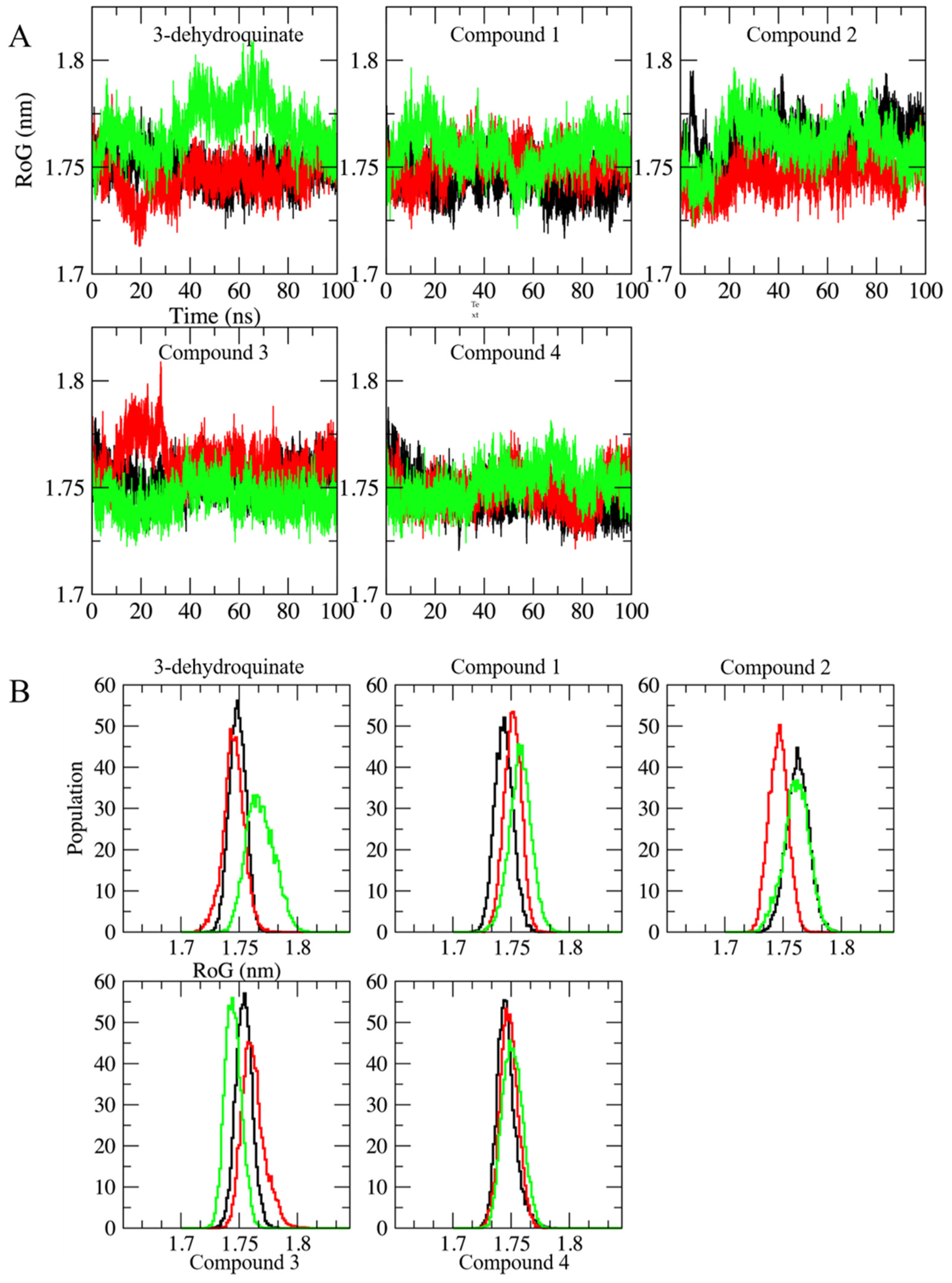
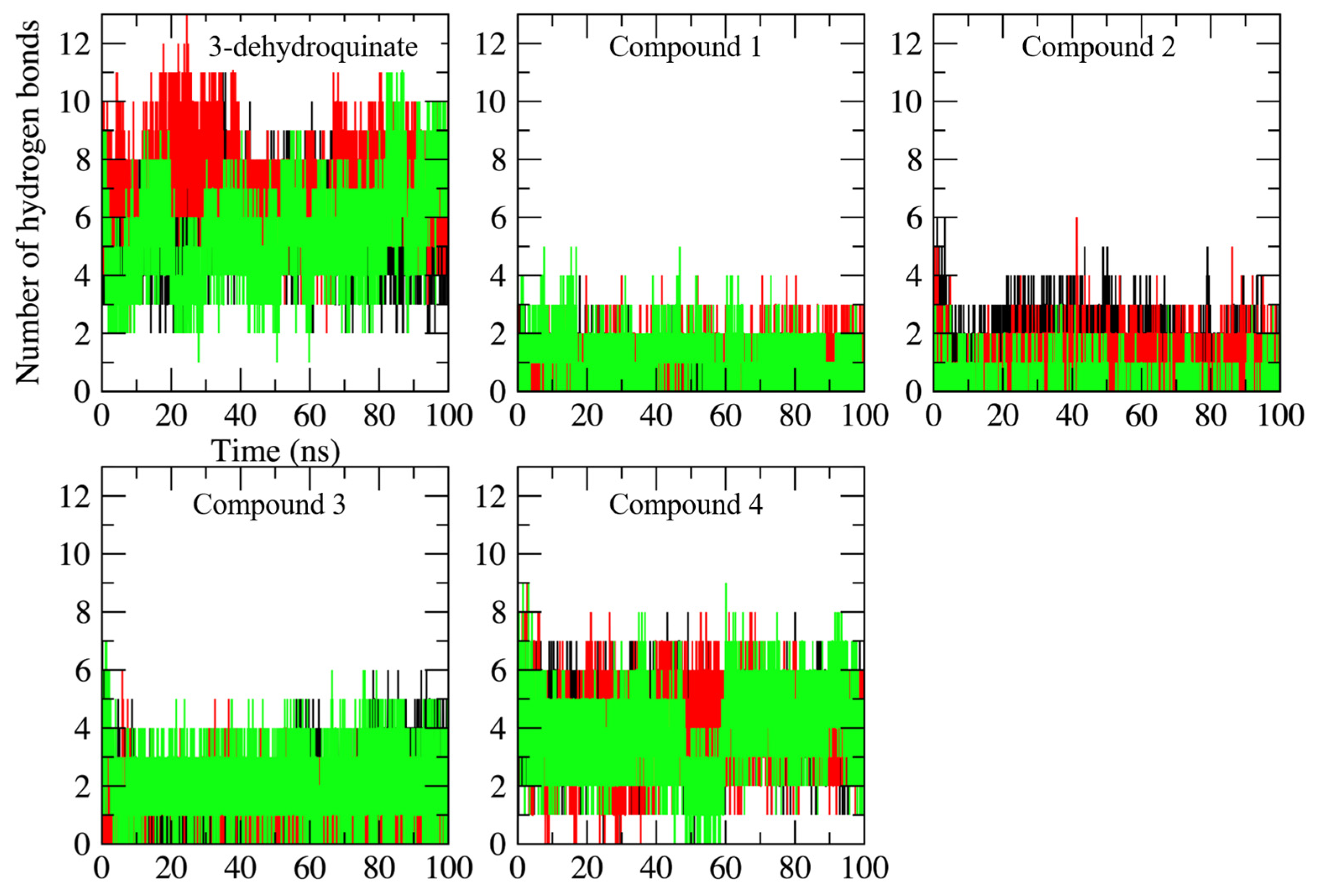
2.2. Molecular Dynamics Simulations
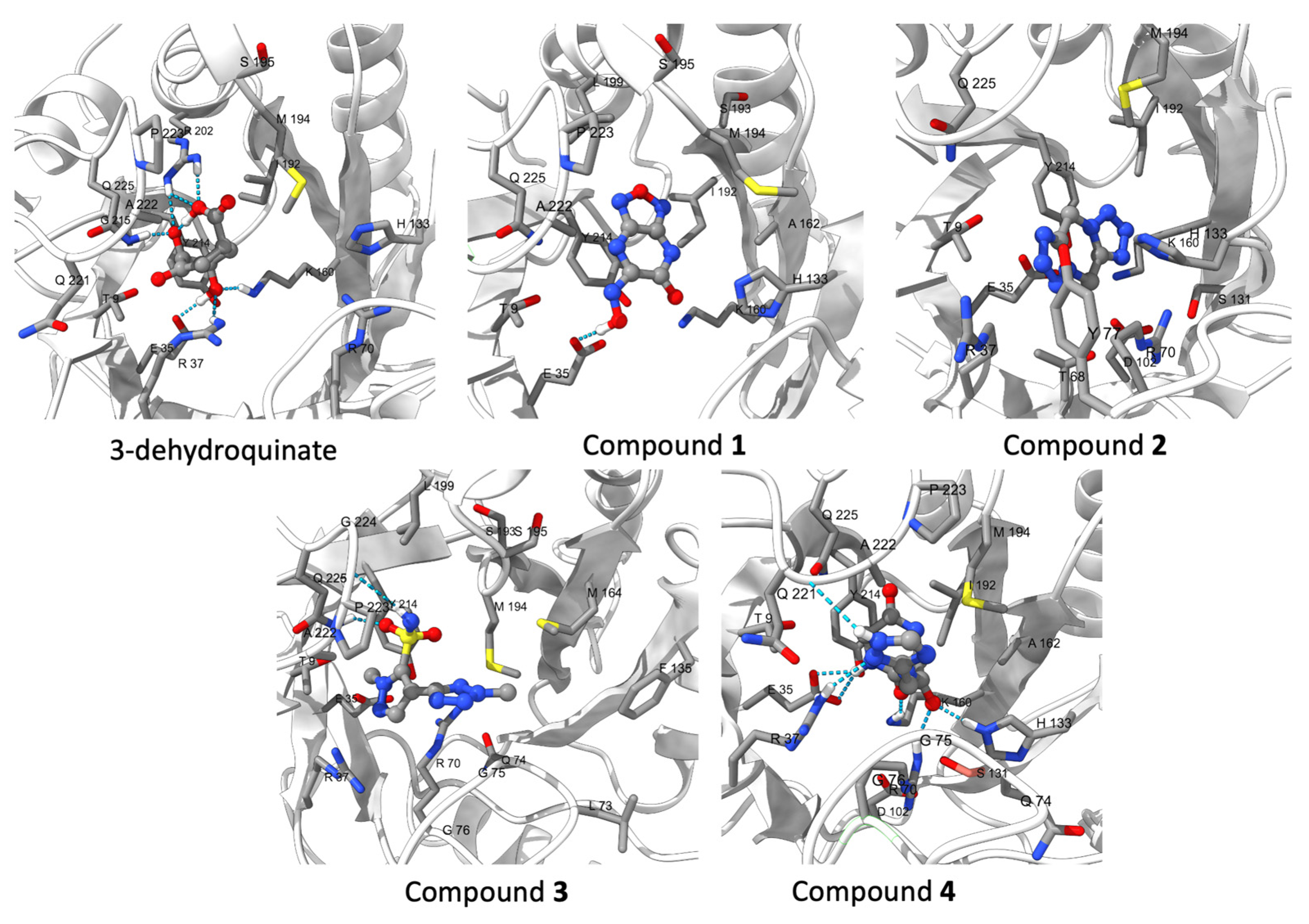
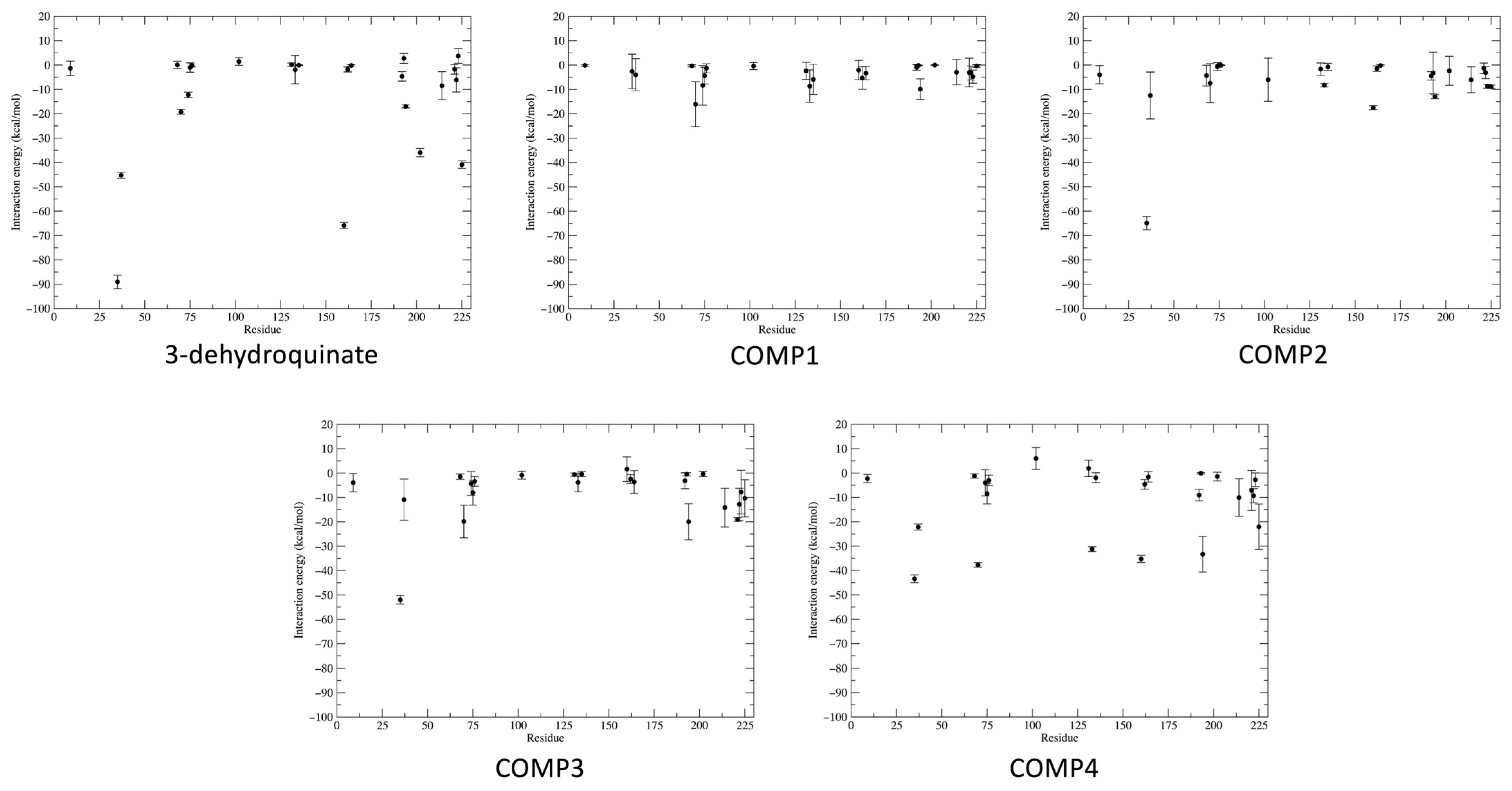
2.3. Binding Free and Interaction Energies
2.4. ADMETox Properties
3. Materials and Methods
3.1. Compound Selection and Filtering
3.2. Exhaustive Virtual Screening
3.3. Exhaustive Molecular Dynamics Simulations
3.4. Binding Free Energy
3.5. ADMETox Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, Government of the United Kingdom. Available online: https://apo.org.au/node/63983 (accessed on 23 December 2021).
- Pärnänen, K.M.M.; Narciso-da-Rocha, C.; Kneis, D.; Berendonk, T.U.; Cacace, D.; Do, T.T.; Elpers, C.; Fatta-Kassinos, D.; Henriques, I.; Jaeger, T.; et al. Antibiotic resistance in European wastewater treatment plants mirrors the pattern of clinical antibiotic resistance prevalence. Sci. Adv. 2019, 5, eaau9124. [Google Scholar] [CrossRef] [PubMed]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Frost, I.; Van Boeckel, T.P.; Pires, J.; Craig, J.; Laxminarayan, R. Global geographic trends in antimicrobial resistance: The role of international travel. J. Travel. Med. 2019, 26, taz036. [Google Scholar] [CrossRef]
- Abushaheen, M.A.; Muzaheed; Fatani, A.J.; Alosaimi, M.; Mansy, W.; George, M.; Acharya, S.; Rathod, S.; Divakar, D.D.; Jhugroo, C.; et al. Antimicrobial resistance, mechanisms and its clinical significance. Dis. Mon. 2020, 66, 100971. [Google Scholar] [CrossRef]
- Adebisi, Y.A.; Alaran, A.J.; Okereke, M.; Oke, G.I.; Amos, O.A.; Olaoye, O.C.; Oladunjoye, I.; Olanrewaju, A.Y.; Ukor, N.A.; Lucero-Prisno, D.E., 3rd. COVID-19 and Antimicrobial Resistance: A Review. Infect. Dis. 2021, 14, 11786337211033870. [Google Scholar] [CrossRef]
- Ghosh, S.; Bornman, C.; Zafer, M.M. Antimicrobial Resistance Threats in the emerging COVID-19 pandemic: Where do we stand? J. Infect. Public Health 2021, 14, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Founou, R.C.; Blocker, A.J.; Noubom, M.; Tsayem, C.; Choukem, S.P.; Van Dongen, M.; Founou, L.L. The COVID-19 pandemic: A threat to antimicrobial resistance containment. Future Sci. OA 2021, 7, FSO736. [Google Scholar] [CrossRef] [PubMed]
- Rawson, T.M.; Moore, L.S.P.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and Fungal Coinfection in Individuals with Coronavirus: A Rapid Review to Support COVID-19 Antimicrobial Prescribing. Clin. Infect. Dis. 2020, 71, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Clancy, C.J.; Nguyen, M.H. Coronavirus Disease 2019, Superinfections, and Antimicrobial Development: What Can We Expect? Clin. Infect. Dis. 2020, 71, 2736–2743. [Google Scholar] [CrossRef] [PubMed]
- WHO. Media Centre. News Release. WHO Publishes List of Bacteria for which New Anti-Biotics are Urgently Needed. Available online: http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/ (accessed on 25 June 2022).
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, S.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Nandhini, P.; Kumar, P.; Mickymaray, S.; Alothaim, A.S.; Somasundaram, J.; Rajan, M. Recent Developments in Methicillin-Resistant Staphylococcus aureus (MRSA) Treatment: A Review. Antibiotics 2022, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R. The shikimate pathway a metabolic tree with many branches. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 307–384. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.A.; Duque, M.A.; De Freitas, T.F.; Galina, L.; Timmers, L.F.S.M.; Bizarro, C.V.; Machado, P.; Basso, L.A.; Ducati, R.G. Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs. Molecules 2020, 25, 1259. [Google Scholar] [CrossRef] [PubMed]
- Ducati, R.G.; Basso, L.A.; Santos, D.S. Mycobacterial shikimate pathway enzymes as targets for drug design. Curr. Drug Targets 2007, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Roszak, A.W.; Robinson, D.A.; Krell, T.; Hunter, I.S.; Fredrickson, M.; Abell, C.; Coggins, J.R.; Lapthorn, A.J. The structure and mechanism of the type II dehydroquinase from Streptomyces coelicolor. Structure 2002, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Y.M.; Sun, Q.L.; Jiang, C.Y.; Liu, S.J. Unraveling the kinetic diversity of microbial 3-dehydroquinate dehydratases of shikimate pathway. AMB Express 2015, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.E.; Lockyer, M.; Hawkins, A.R.; Stammers, D.K. Crystal structures of Staphylococcus aureus type I dehydroquinase from enzyme turnover experiments. Proteins 2004, 56, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Light, S.H.; Minasov, G.; Shuvalova, L.; Duban, M.E.; Caffrey, M.; Anderson, W.F.; Lavie, A. Insights into the mechanism of type I dehydroquinate dehydratases from structures of reaction intermediates. J. Biol. Chem. 2015, 290, 19008. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Perles, L.A.; Nagem, R.A.; Shrive, A.K.; Hawkins, A.; Sawyer, L.; Polikarpov, I. Comparison of different crystal forms of 3-dehydroquinase from Salmonella typhi and its implication for the enzyme activity. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.R.; Rose, I.A. The absolute stereochemical course of citric acid biosynthesis. Proc. Natl. Acad. Sci. USA 1963, 50, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Shneier, A.; Kleanthous, C.; Deka, R.; Coggins, J.R.; Abell, C. Observation of an imine intermediate on dehydroquinase by electrospray mass spectrometry. J. Am. Chem. Soc. 1991, 113, 9416–9418. [Google Scholar] [CrossRef]
- González-Bello, C.; Tizón, L.; Lence, E.; Otero, J.M.; van Raaij, M.J.; Martinez-Guitian, M.; Beceiro, A.; Thompson, P.; Hawkins, A.R. Chemical Modification of a Dehydratase Enzyme Involved in Bacterial Virulence by an Ammonium Derivative: Evidence of its Active Site Covalent Adduct. J. Am. Chem. Soc. 2015, 137, 9333–9343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wright, J.A.; Osman, A.A.; Nair, S.P. An aroD Ochre Mutation Results in a Staphylococcus aureus Small Colony Variant That Can Undergo Phenotypic Switching via Two Alternative Mechanisms. Front. Microbiol. 2017, 8, 1001. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Mann, B.J.; Qin, A.; Santiago, A.E.; Grassel, C.; Lipsky, M.; Vogel, S.N.; Barry, E.M. Characterization of Schu S4 aro mutants as live attenuated tularemia vaccine candidates. Virulence 2020, 11, 283–294. [Google Scholar] [CrossRef]
- González-Bello, C.; Harris, J.M.; Manthey, M.K.; Coggins, J.R.; Abell, C. Irreversible inhibition of type I dehydroquinase by substrates for type II dehydroquinase. Bioorg. Med. Chem. Lett. 2000, 10, 407–409. [Google Scholar] [CrossRef]
- Ratia, K.; Light, S.H.; Antanasijevic, A.; Anderson, W.F.; Caffrey, M.; Lavie, A. Discovery of selective inhibitors of the Clostridium difficile dehydroquinate dehydratase. PLoS ONE 2014, 9, e89356. [Google Scholar] [CrossRef] [PubMed]
- Cheung, V.W.; Xue, B.; Hernandez-Valladares, M.; Go, M.K.; Tung, A.; Aguda, A.H.; Robinson, R.C.; Yew, W.S. Identification of polyketide inhibitors targeting 3-dehydroquinate dehydratase in the shikimate pathway of Enterococcus faecalis. PLoS ONE 2014, 9, e103598. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bender, A.; GTresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Cerón-Carrasco, J.P. When Virtual Screening Yields Inactive Drugs: Dealing with False Theoretical Friends. Chem. Med. Chem. 2022, 17, e202200278. [Google Scholar] [CrossRef] [PubMed]
- Breznik, M.; Ge, Y.; Bluck, J.P.; Briem, H.; Hahn, D.F.; Christ, C.D.; Mortier, J.; Mobley, D.L.; Meier, K. Prioritizing Small Sets of Molecules for Synthesis through in-silico Tools: A Comparison of Common Ranking Methods. ChemMedChem 2023, 18, e202200425. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Webster Andrews, C.; Capelli, A.-N.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, R.F.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB. MedChemComm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef]
- Wang, S.; Sun, X.; Cui, W.; Yuan, S. MM/PB(GB)SA benchmarks on soluble proteins and membrane proteins. Front. Pharmacol. 2022, 13, 1018351. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, L.; Santos-Martins, D.; Sasmal, S.; Eberhardt, J.; Bianco, G.; Ambrosio, F.A.; Solis-Vasquez, L.; Koch, A.; Forli, S.; Mobley, D.L. Comparison of affinity ranking using AutoDock-GPU and MM-GBSA scores for BACE-1 inhibitors in the D3R Grand Challenge 4. J. Comput. Aided Mol. Des. 2019, 33, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. Free Energy Calculations: A Breakthrough for Modeling Organic Chemistry in Solution. Acc. Chem. Res. 1989, 22, 184–189. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Genheden, S.; Nilsson, I.; Ryde, U. Binding affinities of factor Xa inhibitors estimated by thermodynamic integration and MM/GBSA. J. Chem. Inf. Model 2011, 51, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15 Ligand Discovery for Everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Christ, C.D.; Christen, M.; Eichenberger, A.P.; van Gunsteren, W.F. Architecture, Implementation and Parallelisation of the GROMOS Software for Biomolecular Simulation. Comp. Phys. Commun. 2012, 183, 890–903. [Google Scholar] [CrossRef]
- Sharp, K.A.; Honig, B. Calculating Total Electrostatic Energies with the Nonlinear Pois-son-Boltzmann Equation. J. Phys. Chem. 1990, 94, 7684–7692. [Google Scholar] [CrossRef]
- Paissoni, C.; Spiliotopoulos, D.; Musco, G.; Spitaleri, A. GMXPBSA 2.1: A GROMACS Tool to Perform MM/PBSA and Computational Alanine Scanning. Comput. Phys. Commun. 2015, 186, 105–107. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Seul, South Corea: Bioinformatics and Molecular Design Research Center. Pre-ADMET Program. Available online: http://preadmet.bmdrc.org (accessed on 6 June 2022).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Docking Score (kcal/mol) | Structure |
|---|---|---|
| 3-dehydroquinate | −6.1 ± 0.0 |  |
| Compound 1 | −7.5 ± 0.0 |  |
| Compound 2 | −7.5 ± 0.0 |  |
| Compound 3 | −7.5 ± 0.0 |  |
| Compound 4 | −7.8 ± 0.0 |  |
| Ligand | Total (kcal/mol) | Polar Contribution | Apolar Contribution |
|---|---|---|---|
| 3-dehydroquinate | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Compound 1 | 86.88 ± 8.96 | 219.17 ± 7.68 | −132.29 ± 4.61 |
| Compound 2 | 68.67 ± 9.39 | 218.77 ± 8.51 | −150.10 ± 4.04 |
| Compound 3 | 29.08 ± 9.33 | 237.14 ± 8.41 | −208.06 ± 4.09 |
| Compound 4 | 4.80 ± 8.90 | 172.73 ± 7.85 | −168.03 ± 4.15 |
| 3-dehydroquinate | Compound 1 | Compound 2 | Compound 3 | Compound 4 | |
|---|---|---|---|---|---|
| THR9 * | −1.35 ± 2.96 (88.67) | −3.97 ± 3.75 (65.33) | −3.93 ± 3.77 (89.33) | −2.28 ± 1.71 (73.67) | |
| GLU35 * | −89.04 ± 48.34 (88.67) | 64.91 ± 47.03 (67.00) | −52.03 ± 29.74 (88.33) | −43.42 ± 27.92 (85.67) | |
| ARG37 * | −45.26 ± 21.24 (88.67) | −12.50 ± 9.63 (67.00) | −10.91 ± 8.45 (89.33) | −22.15 ± 21.41 (72.67) | |
| THR68 * | 0.08 ± 1.48 (52.67) | −4.34 ± 4.27 (63.00) | |||
| ARG70 * | −19.21 ± 16.73 (71.67) | −16.05 ± 9.22 (89.33) | −7.47 ± 8.01 (74.33) | −19.87 ± 6.70 (89.33) | −37.71 ± 15.75 (86.00) |
| GLN74 | −8.36 ± 8.11 (86.67) | −4.28 ± 4.86 (59.00) | −3.99 ± 5.35 (59.33) | ||
| GLY75 | −4.38 ± 3.40 (83.67) | −8.05 ± 5.10 (89.33) | −8.45 ± 4.22 (85.67) | ||
| GLY76 | −3.42 ± 2.00 (86.33) | −2.98 ± 2.13 (67.33) | |||
| ASP102 * | −6.03 ± 8.87 (57.00) | 5.97 ± 4.50 (57.67) | |||
| SER131 * | 1.93 ± 3.36 (78.33) | ||||
| HIS133 * | −8.65 ± 6.69 (97.33) | −3.83 ± 3.84 (61.67) | −31.24 ± 16.88 (86.00) | ||
| PHE135 | −5.86 ± 6.24 (70.00) | −1.90 ± 2.03 (62.33) | |||
| LYS160 * | −65.90 ± 21.63 (88.67) | −2.07 ± 3.98 (58.67) | −17.53 ± 14.40 (68.67) | 1.60 ± 5.04 (62.67) | −35.25 ± 25.62 (86.00) |
| ALA162 * | −1.81 ± 1.02 (67.00) | −5.34 ± 4.63 (80.00) | −2.42 ± 1.81 (58.33) | −4.63 ± 1.99 (86.00) | |
| MET164 | −3.36 ± 2.73 (63.00) | ||||
| ILE192 * | −4.65 ± 1.96 (88.00) | −4.46 ± 1.73 (83.00) | −9.06 ± 2.36 (86.00) | ||
| SER193 | 2.73 ± 2.05 (59.33) | ||||
| MET194 * | −16.97 ± 10.48 (87.33) | −9.89 ± 4.22 (98.67) | −12.99 ± 13.97 (91.33) | −19.99 ± 7.40 (89.33) | −33.32 ± 7.31 (86.00) |
| ARG202 * | −35.99 ± 29.86 (76.00) | ||||
| TYR214 * | −8.48 ± 5.76 (88.33) | −6.06 ± 5.32 (91.67) | −14.17 ± 7.95 (89.33) | −10.07 ± 7.73 (86.00) | |
| GLN221 * | −19.07 ± 13.41 (73.67) | −7.14 ± 8.21 (55.33) | |||
| ALA222 * | −6.12 ± 4.98 (60.67) | −3.28 ± 2.88 (61.33) | −3.12 ± 2.48 (56.67) | −12.81 ± 6.59 (86.67) | −9.28 ± 2.89 (86.00) |
| PRO223 | 3.71 ± 3.00 (55.67) | −4.76 ± 2.70 (83.33) | −2.74 ± 2.81 (83.33) | ||
| GLN225 * | −40.92 ± 27.09 (72.67) | −8.89 ± 11.19 (72.67) | −10.35 ± 7.63 (89.33) | −22.02 ± 9.30 (86.00) | |
| Total interaction energy | −345.01 ± 12.26 | −90.67 ± 22.37 | −181.24 ± 21.01 | −202.39 ± 23.87 | −284.03 ± 19.98 |
| Parameter | Compound 1 | Compound 2 | Compound 3 | Compound 4 |
|---|---|---|---|---|
| Pharmacokinetics | ||||
| GI absortion | Low | High | High | Low |
| Log Kp (Skin permeation) | −7.36 cm/s | −8.33 cm/s | −8.27 cm/s | −8.90 cm/s |
| Druglikeness | ||||
| Ghose | No; 2 violations: WLOGP < −0.4, #atoms < 20 | No; 3 violations: WLOGP < −0.4, MR < 40, #atoms < 20 | No; 1 violation: WLOGP < −0.4 | No; 1 violation: WLOGP < −0.4 |
| Veber | Yes | Yes | Yes | Yes |
| Egan | Yes | Yes | Yes | Yes |
| Muegge | No; 2 violations: MW < 200, #C < 5 | No; 2 violations: MW < 200, #C < 5 | Yes | Yes |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 |
| Medicinal chemistry | ||||
| PAINS | 0 alert | 0 alert | 0 alert | 0 alert |
| Brenk | 3 alerts: imine_1, oxime_1, oxygen-nitrogen_single_bond | 0 alert | 0 alert | 1 alert: beta_keto_anhydride |
| Leadlikeness | No; 1 violation: MW < 250 | No; 1 violation: MW < 250 | No; 1 violation: MW < 250 | No; 1 violation: MW < 250 |
| Synthetic accesibility | 3.60 | 2.42 | 2.78 | 2.61 |
| BBB | 0.31 | 0.08 | 0.05 | 0.05 |
| In vitro Caco2 cell permeability | 3.94 | 0.73 | 1.28 | 6.54 |
| In vitro CYP 2C19 inhibition | Inhibitor | Inhibitor | Non | Inhibitor |
| In vitro CYP 2C9 inhibition | Non | Non | Non | Non |
| In vitro CYP 2D6 inhibition | Non | Non | Non | Non |
| In vitro CYP 2D6 substrate | Non | Non | Non | Non |
| In vitro CYP 3A4 inhibition | Non | Non | Non | Inhibitor |
| In vitro CYP 3A4 substrate | Weakly | Non | Substrate | Non |
| HIA | 40.23 | 70.25 | 70.86 | 27.24 |
| MDCK | 1.42 | 0.72 | 0.60 | 0.59 |
| Pgp inhibition | Non | Non | Non | Non |
| Plasma Protein Binding | 2.68 | 14.37 | 60.17 | 5.91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millán-Pacheco, C.; Rios-Soto, L.; Corral-Rodríguez, N.; Sierra-Campos, E.; Valdez-Solana, M.; Téllez-Valencia, A.; Avitia-Domínguez, C. Discovery of Potential Noncovalent Inhibitors of Dehydroquinate Dehydratase from Methicillin-Resistant Staphylococcus aureus through Computational-Driven Drug Design. Pharmaceuticals 2023, 16, 1148. https://doi.org/10.3390/ph16081148
Millán-Pacheco C, Rios-Soto L, Corral-Rodríguez N, Sierra-Campos E, Valdez-Solana M, Téllez-Valencia A, Avitia-Domínguez C. Discovery of Potential Noncovalent Inhibitors of Dehydroquinate Dehydratase from Methicillin-Resistant Staphylococcus aureus through Computational-Driven Drug Design. Pharmaceuticals. 2023; 16(8):1148. https://doi.org/10.3390/ph16081148
Chicago/Turabian StyleMillán-Pacheco, César, Lluvia Rios-Soto, Noé Corral-Rodríguez, Erick Sierra-Campos, Mónica Valdez-Solana, Alfredo Téllez-Valencia, and Claudia Avitia-Domínguez. 2023. "Discovery of Potential Noncovalent Inhibitors of Dehydroquinate Dehydratase from Methicillin-Resistant Staphylococcus aureus through Computational-Driven Drug Design" Pharmaceuticals 16, no. 8: 1148. https://doi.org/10.3390/ph16081148
APA StyleMillán-Pacheco, C., Rios-Soto, L., Corral-Rodríguez, N., Sierra-Campos, E., Valdez-Solana, M., Téllez-Valencia, A., & Avitia-Domínguez, C. (2023). Discovery of Potential Noncovalent Inhibitors of Dehydroquinate Dehydratase from Methicillin-Resistant Staphylococcus aureus through Computational-Driven Drug Design. Pharmaceuticals, 16(8), 1148. https://doi.org/10.3390/ph16081148