
Design and Synthesis of New Anthranyl Phenylhydrazides: Antileishmanial Activity and Structure–Activity Relationship


 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
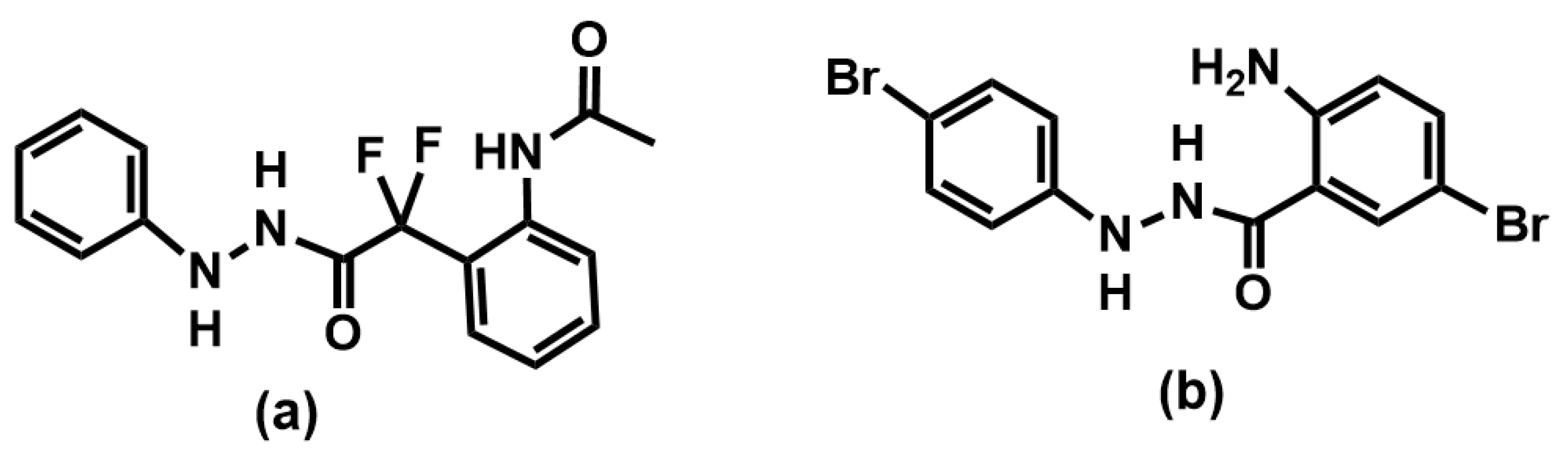
2.2. Planning of Anthranyl Hydrazides Series
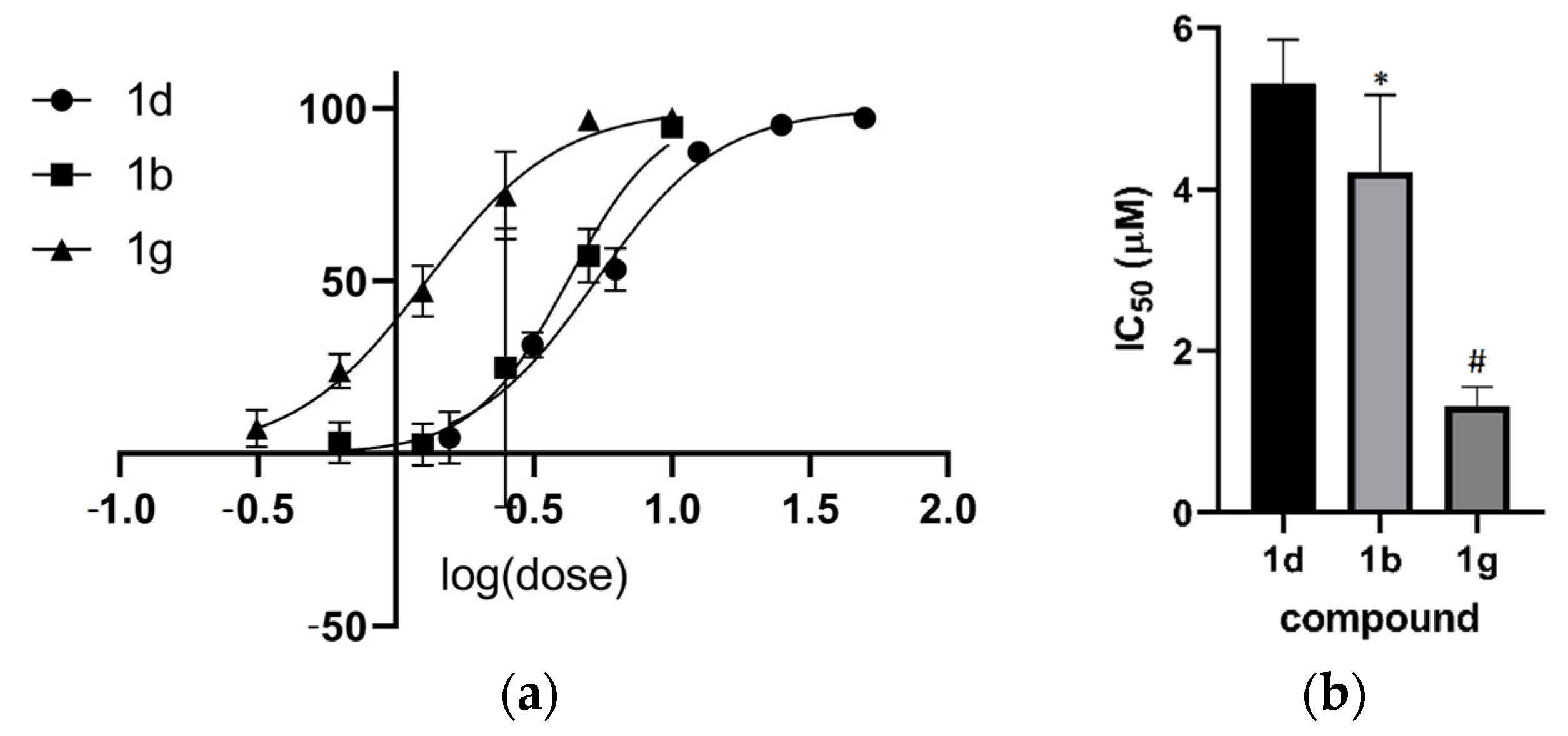
2.3. Activity in Promastigotes and Intracellular Amastigotes
2.4. Structure–Activity Relationship Analysis of the Anthranyl Hydrazides Series
2.5. Mechanism of Antileishmanial Activity
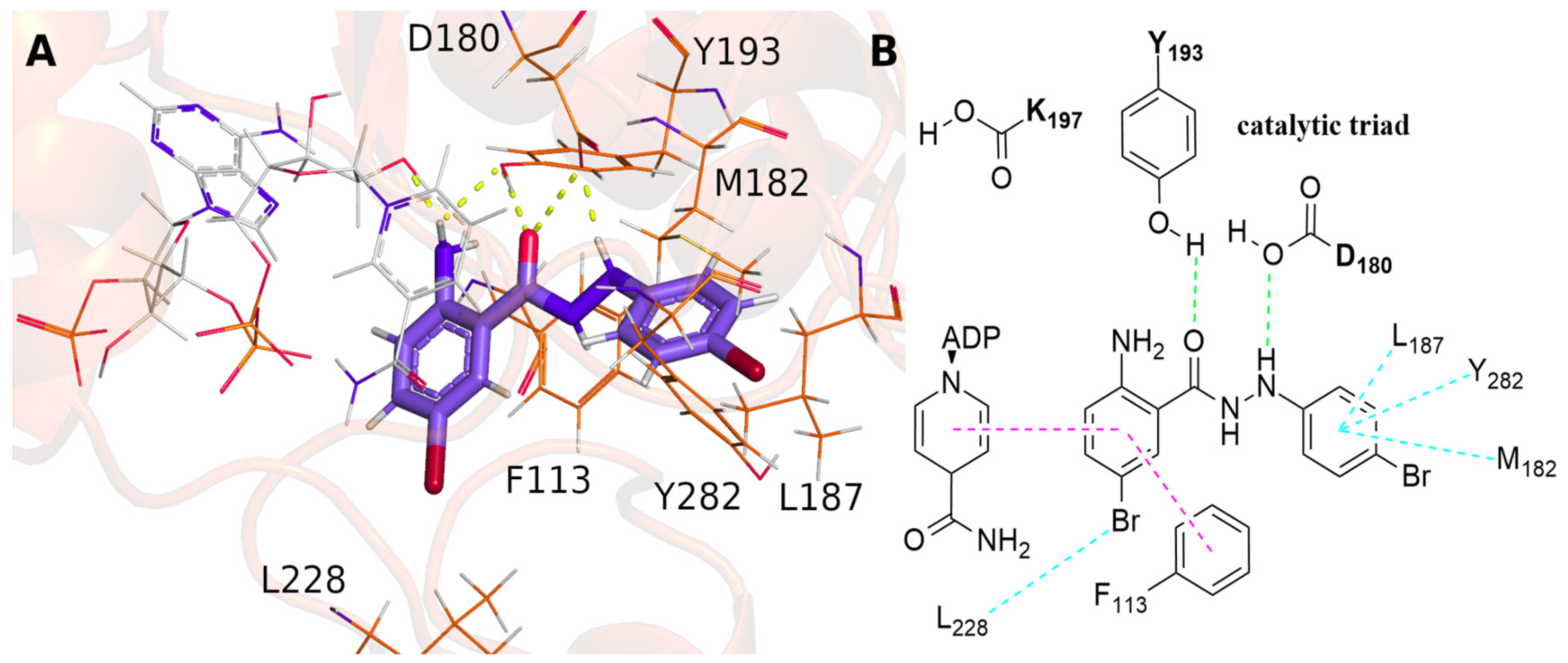
2.6. Target Fishing for Anthranyl Phenylhydrazides
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis
3.2.1. Synthesis of 2-amino-5-bromo-N′-phenylbenzohydrazide (1b)
3.2.2. Synthesis of 2-amino-N′-(2-bromophenyl)benzohydrazide (1c)
3.2.3. Synthesis of 2-amino-5-bromo-N′-(4-bromophenyl) benzohydrazide (1g)
3.2.4. Synthesis of 2-amino-N′-(m-tolyl)benzohydrazide (1i)
3.2.5. Synthesis of 2-amino-N′-(p-tolyl)benzohydrazide (1j)
3.2.6. Synthesis of 2-methyl-3-(phenylamino)quinazolin-4(3H)-one (2a)
3.2.7. 3-((4-bromophenyl)amino)-2-methylquinazolin-4(3H)-one (2b)
3.2.8. Synthesis of 6-bromo-3-((4-bromophenyl)amino)-2-methylquinazolin-4(3H)-one (2c)
3.3. Promastigotes Test
3.4. Arginase Assay
3.5. Target Fishing with Chemogenomics
3.6. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO|Leishmaniasis. WHO. 2019. Available online: https://www.who.int/leishmaniasis/en/ (accessed on 27 June 2019).
- Saha, P.; Chaudhury, A.; Maji, A.K. Sir U.N. Brahmachari and his battle against Kala-Azar. Trop. Parasitol. 2021, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Neglected Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Basselin, M.; Badet-Denisot, M.-A.; Lawrence, O.; Robert-Gero, M. Effects of Pentamidine on Polyamine Level and Biosynthesis in Wild-Type, Pentamidine-Treated, and Pentamidine-Resistant Leishmania. Exp. Parasitol. 1997, 85, 274–282. [Google Scholar] [CrossRef]
- Monge-Maillo, B.; Lopez-Velez, R. Miltefosine for Visceral and Cutaneous Leishmaniasis: Drug Characteristics and Evidence-Based Treatment Recommendations. Clin. Infect. Dis. 2015, 60, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Cuypers, B.; Bhattarai, N.R.; Uranw, S.; Berg, M.; Ostyn, B.; Dujardin, J.C.; Rijal, S.; Vanaerschot, M. Relapse after treatment with Miltefosine for visceral Leishmaniasis is associated with increased infectivity of the infecting Leishmania donovani strain. mBio 2013, 4, e00611-13. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Nogueira, F.; Avino, V.C.; Galvis-Ovallos, F.; Pereira-Chioccola, V.L.; Moreira, M.A.B.; Romariz, A.P.P.L.; Molla, L.M.; Menz, I. Use of miltefosine to treat canine visceral leishmaniasis caused by Leishmania infantum in Brazil. Parasites Vectors 2019, 12, 79. [Google Scholar] [CrossRef]
- Ginel, P.J.; Lucena, R.; López, R.; Molleda, J.M. Use of allopurinol for maintenance of remission in dogs with leishmaniasis. J. Small Anim. Pract. 1998, 39, 271–274. [Google Scholar] [CrossRef]
- Manna, L.; Vitale, F.; Reale, S.; Picillo, E.; Neglia, G.; Vescio, F.; Gravino, A.E. Study of efficacy of miltefosine and allopurinol in dogs with leishmaniosis. Vet. J. 2009, 182, 441–445. [Google Scholar] [CrossRef]
- Manna, L.; Corso, R.; Galiero, G.; Cerrone, A.; Muzj, P.; Gravino, A.E. Long-term follow-up of dogs with leishmaniosis treated with meglumine antimoniate plus allopurinol versus miltefosine plus allopurinol. Parasites Vectors 2015, 8, 289. [Google Scholar] [CrossRef]
- Trinconi, C.T.; Reimão, J.Q.; Bonano, V.I.; Espada, C.R.; Miguel, D.C.; Yokoyama-Yasunaka, J.K.U.; Uliana, S.R.B. Topical tamoxifen in the therapy of cutaneous leishmaniasis. Parasitology 2018, 145, 490–496. [Google Scholar] [CrossRef]
- Pineda, C.; Aguilera-Tejero, E.; Morales, M.C.; Belinchon-Lorenzo, S.; Gomez-Nieto, L.C.; Garcia, P.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Lopez, I. Treatment of canine leishmaniasis with marbofloxacin in dogs with renal disease. PLoS ONE 2017, 12, e0185981. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Dawood, K.F.; Fabrini, R.; Federici, G.; Gradoni, L.; Pedersen, J.Z.; Ricci, G. Trypanothione efficiently intercepts nitric oxide as a harmless iron complex in trypanosomatid parasites. FASEB J. 2010, 24, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.R.; Maquiaveli, C.D.C.; Magalhães, P.P. The leishmanicidal flavonols quercetin and quercitrin target Leishmania (Leishmania) amazonensis arginase. Exp. Parasitol. 2012, 130, 183–188. [Google Scholar] [CrossRef]
- Jiang, Y.; Roberts, S.C.; Jardim, A.; Carter, N.S.; Shih, S.; Ariyanayagam, M.; Fairlamb, A.H.; Ullman, B. Ornithine decarboxylase gene deletion mutants of Leishmania donovani. J. Biol. Chem. 1999, 274, 3781–3788. [Google Scholar] [CrossRef] [PubMed]
- de Lima, E.C.; Castelo-Branco, F.S.; Maquiaveli, C.C.; Farias, A.B.; Rennó, M.N.; Boechat, N.; Silva, E.R. Phenylhydrazides as inhibitors of Leishmania amazonensis arginase and antileishmanial activity. Bioorganic Med. Chem. 2019, 27, 3853–3859. [Google Scholar] [CrossRef]
- Paiva, J.P.B.; Cordeiro, M.S.; França, P.R.C.; Branco, L.O.P.; Santos, I.S.; Reis, N.F.; Pimentel, P.P.; Giorno, T.B.S.; Lima, E.C.; Fernandes, P.D. Phenylbenzohydrazides Obtained from Isatoic Anhydride Present Anti-Inflammatory Activity In Vivo and In Vitro. Biomolecules 2022, 12, 1901. [Google Scholar] [CrossRef]
- El Fadili, K.; Messier, N.; Leprohon, P.; Roy, G.; Guimond, C.; Trudel, N.; Saravia, N.G.; Papadopoulou, B.; Légaré, D.; Ouellette, M. Role of the ABC transporter MRPA (PGPA) in antimony resistance in Leishmania infantum axenic and intracellular amastigotes. Antimicrob. Agents Chemother. 2005, 49, 1988–1993. [Google Scholar] [CrossRef]
- Coelho, A.C.; Beverley, S.M.; Cotrim, P.C. Functional genetic identification of PRP1, an ABC transporter superfamily member conferring pentamidine resistance in Leishmania major. Mol. Biochem. Parasitol. 2003, 130, 83–90. [Google Scholar] [CrossRef]
- Maghsoudi, S.; Taghavi Shahraki, B.; Rameh, F.; Nazarabi, M.; Fatahi, Y.; Akhavan, O.; Rabiee, M.; Mostafavi, E.; Lima, E.C.; Saeb, M.R.; et al. A review on computer-aided chemogenomics and drug repositioning for rational COVID-19 drug discovery. Chem. Biol. Drug Des. 2022, 100, 699–721. [Google Scholar] [CrossRef]
- Jones, L.H.; Bunnage, M.E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 2017, 16, 285–296. [Google Scholar] [CrossRef]
- Nare, B.; Garraway, L.A.; Vickers, T.J.; Beverley, S.M. PTR1-dependent synthesis of tetrahydrobiopterin contributes to oxidant susceptibility in the trypanosomatid protozoan parasite Leishmania major. Curr. Genet. 2009, 55, 287. [Google Scholar] [CrossRef] [PubMed]
- Nare, B.; Hardy, L.W.; Beverley, S.M. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef] [PubMed]
- Opperdoes, F.R.; Coombs, G.H. Metabolism of Leishmania: Proven and predicted. Trends Parasitol. 2007, 23, 149–158. [Google Scholar] [CrossRef]
- Ouellette, M.; Drummelsmith, J.; El Fadili, A.; Kündig, C.; Richard, D.; Roy, G. Pterin transport and metabolism in Leishmania and related trypanosomatid parasites. Int. J. Parasitol. 2002, 32, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Das Neves, G.M.H.; Kagami, L.P.; Gonçalves, I.L.; Eifler-Lima, V.L. Targeting pteridine reductase 1 and dihydrofolate reductase: The old is a new trend for leishmaniasis drug discovery. Futur. Med. Chem. 2019, 11, 2107–2130. [Google Scholar] [CrossRef] [PubMed]
- de Souza Moreira, D.; Ferreira, R.F.; Murta, S.M.F. Molecular characterization and functional analysis of pteridine reductase in wild-type and antimony-resistant Leishmania lines. Exp. Parasitol. 2016, 160, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.H.A.; Froes, T.Q.; da Silva, S.G.; de Souza, E.I.M.; Vital-Fujii, D.G.; Trossini, G.H.G.; Pita, S.S.D.R.; Castilho, M.S. An integrated approach towards the discovery of novel non-nucleoside Leishmania major pteridine reductase 1 inhibitors. Eur. J. Med. Chem. 2017, 132, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar] [CrossRef]
- Nare, B.; Luba, J.; Hardy, L.W.; Beverley, S. New approaches to Leishmania chemotherapy: Pteridine reductase 1 (PTR1) as a target and modulator of antifolate sensitivity. Parasitology 1997, 114, 101–110. [Google Scholar] [CrossRef]
- Maquiaveli, C.C.; Lucon-Júnior, J.F.; Brogi, S.; Campiani, G.; Gemma, S.; Vieira, P.C.; Silva, E.R. Verbascoside Inhibits Promastigote Growth and Arginase Activity of Leishmania amazonensis. J. Nat. Prod. 2016, 79, 1459–1463. [Google Scholar] [CrossRef]
- da Silva, E.R.; da Silva, M.F.L.; Fischer, H.; Mortara, R.A.; Mayer, M.G.; Framesqui, K.; Silber, A.M.; Floeter-Winter, L.M. Biochemical and biophysical properties of a highly active recombinant arginase from Leishmania (Leishmania) amazonensis and subcellular localization of native enzyme. Mol. Biochem. Parasitol. 2008, 159, 104–111. [Google Scholar] [CrossRef]
- Fawcett, J.K.; Scott, J.E. A Rapid and Precise Method for the Determination of Urea. J. Clin. Pathol. 1960, 13, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Mervin, L.H.; Afzal, A.M.; Drakakis, G.; Lewis, R.; Engkvist, O.; Bender, A. Target prediction utilising negative bioactivity data covering large chemical space. J. Cheminform. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Mervin, L.H.; Bulusu, K.C.; Kalash, L.; Afzal, A.M.; Svensson, F.; Firth, M.A.; Barrett, I.; Engkvist, O.; Bender, A. Orthologue chemical space and its influence on target prediction. Bioinformatics 2018, 34, 72–79. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Simossis, V.A.; Heringa, J. PRALINE: A multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005, 33, W289–W294. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L. The PyMOL Molecular Graphics System. Available online: https://pymol.org/ (accessed on 20 April 2022).
- Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 20 April 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
| Compound 1 | Compound 2 | |||||
| R1 | R2 | R3 | R4 | Inhibition at 10 μM (%) | IC50(μM) (95% CI) | |
| 1a | H | H | H | H | 41.3 | >10 |
| 1b | Br | H | H | H | 67.7 | 4.2 (3.460–5.038) |
| 1c | H | Br | H | H | 30.0 | >10 |
| 1d | H | H | H | Br | 55.2 | 5.3 (4.963–5.670) |
| 1e | H | H | H | Cl | 59.2 | <10 |
| 1f | H | H | H | F | 12.8 | >10 |
| 1g | Br | H | H | Br | 97.9 | 1.3 (1.177–1.411) |
| 1h | H | CH3 | H | H | 4.4 | >10 |
| 1i | H | H | CH3 | H | nd | ~20 |
| 1j | H | H | H | CH3 | nd | ~20 |
| 2a | H | H | H | H | inactive | - |
| 2b | H | H | H | Br | inactive | - |
| 2c | Br | H | H | Br | inactive | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
do Carmo Maquiaveli, C.; da Silva, E.R.; Hild de Jesus, B.; Oliveira Monteiro, C.E.; Rodrigues Navarro, T.; Pereira Branco, L.O.; Souza dos Santos, I.; Figueiredo Reis, N.; Portugal, A.B.; Mendes Wanderley, J.L.; et al. Design and Synthesis of New Anthranyl Phenylhydrazides: Antileishmanial Activity and Structure–Activity Relationship. Pharmaceuticals 2023, 16, 1120. https://doi.org/10.3390/ph16081120
do Carmo Maquiaveli C, da Silva ER, Hild de Jesus B, Oliveira Monteiro CE, Rodrigues Navarro T, Pereira Branco LO, Souza dos Santos I, Figueiredo Reis N, Portugal AB, Mendes Wanderley JL, et al. Design and Synthesis of New Anthranyl Phenylhydrazides: Antileishmanial Activity and Structure–Activity Relationship. Pharmaceuticals. 2023; 16(8):1120. https://doi.org/10.3390/ph16081120
Chicago/Turabian Styledo Carmo Maquiaveli, Claudia, Edson Roberto da Silva, Barbara Hild de Jesus, Caio Eduardo Oliveira Monteiro, Tiago Rodrigues Navarro, Luiz Octavio Pereira Branco, Isabela Souza dos Santos, Nanashara Figueiredo Reis, Arieli Bernardo Portugal, João Luiz Mendes Wanderley, and et al. 2023. "Design and Synthesis of New Anthranyl Phenylhydrazides: Antileishmanial Activity and Structure–Activity Relationship" Pharmaceuticals 16, no. 8: 1120. https://doi.org/10.3390/ph16081120
APA Styledo Carmo Maquiaveli, C., da Silva, E. R., Hild de Jesus, B., Oliveira Monteiro, C. E., Rodrigues Navarro, T., Pereira Branco, L. O., Souza dos Santos, I., Figueiredo Reis, N., Portugal, A. B., Mendes Wanderley, J. L., Borges Farias, A., Correia Romeiro, N., & de Lima, E. C. (2023). Design and Synthesis of New Anthranyl Phenylhydrazides: Antileishmanial Activity and Structure–Activity Relationship. Pharmaceuticals, 16(8), 1120. https://doi.org/10.3390/ph16081120