

Curcumin Stereoisomer, Cis-Trans Curcumin, as a Novel Ligand to A1 and A3 Adenosine Receptors

 ,
,  ,
, 
Abstract
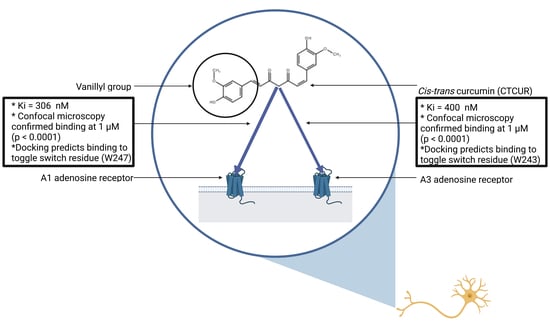
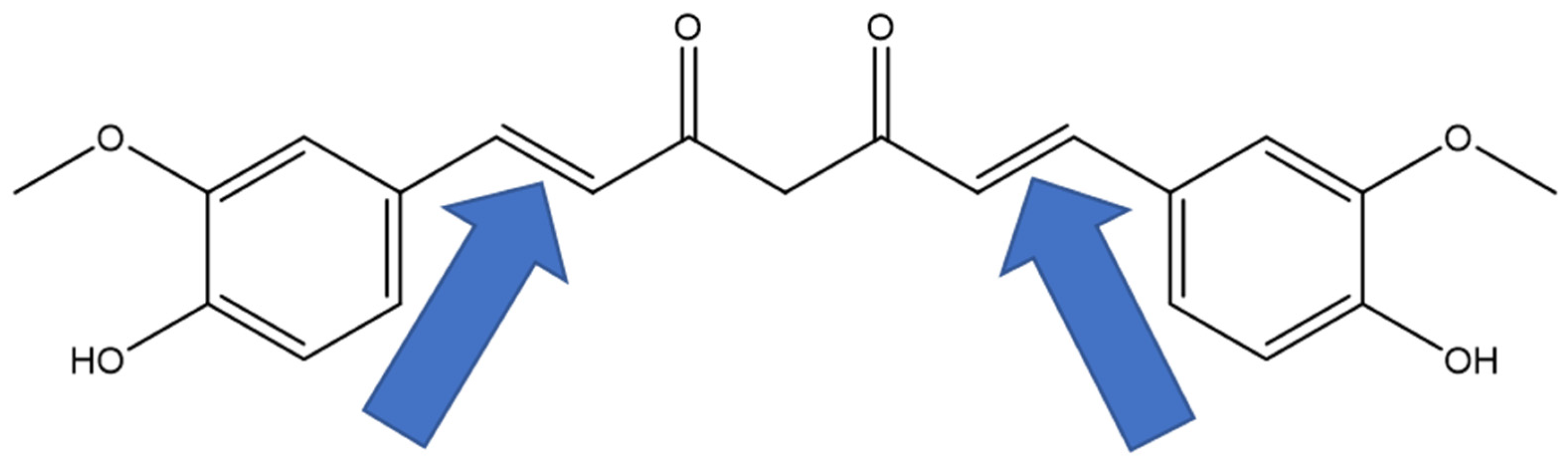
1. Introduction
2. Results
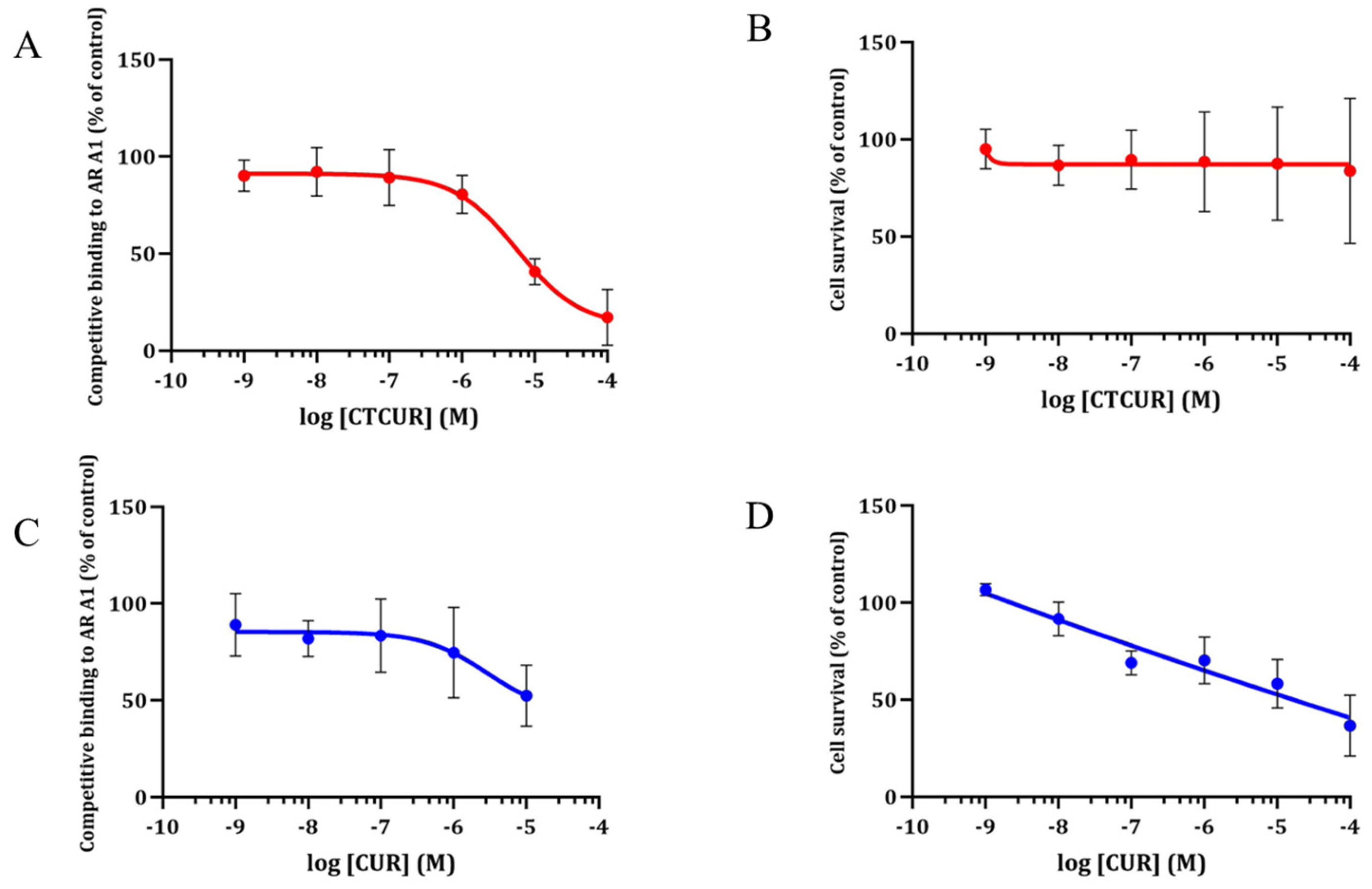
2.1. Receptor Binding and Cell Survival with CTCUR or CUR at A1AR
2.2. Receptor Binding and Cell Survival with CTCUR or CUR at A3AR
2.3. Binding Assay with CTCUR at Adenosine Receptors using Fluorescence Microscopy
2.4. Docking Analysis with CTCUR at Adenosine Receptors
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.2. Cell Culture
4.3. Competitive Binding Assays
4.4. Docking Studies
4.4.1. Preparation of the Protein Structures
4.4.2. Preparation of the Ligand Structures
4.4.3. Glide Docking Procedures
4.5. Confocal Fluorescence Microscopy
4.6. Cytotoxicity Assays
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Imm, J.; Zhang, G.; Chan, L.-Y.; Nitteranon, V.; Parkin, K.L. [6]-Dehydroshogaol, a minor component in ginger rhizome, exhibits quinone reductase inducing and anti-inflammatory activities that rival those of curcumin. Food Res. Int. 2010, 43, 2208–2213. [Google Scholar] [CrossRef]
- Mai, C.W.; Kang, Y.-B.; Nadarajah, V.D.V.; Hamzah, A.S.; Pichika, M.R. Drug-like dietary vanilloids induce anticancer activity through proliferation inhibition and regulation of bcl-related apoptotic proteins. Phytother. Res. 2018, 32, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Sim, Y.-B.; Choi, S.-M.; Seo, Y.-J.; Kwon, M.-S.; Lee, J.-K.; Suh, H.-W. Antinociceptive profiles and mechanisms of orally administered vanillin in the mice. Arch. Pharm. Res. 2009, 32, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Hayman, M.; Kam, P.C.A. Capsaicin: A review of its pharmacology and clinical applications. Curr. Anesth. Crit. Care 2008, 19, 338–343. [Google Scholar] [CrossRef]
- Mata-Bermudez, A.; Izquierdo, T.; Monteros-Zuñiga, E.D.L.; Coen, A.; Godínez-Chaparro, B. Antiallodynic effect induced by [6]-gingerol in neuropathic rats is mediated by activation of the serotoninergic system and the nitric oxide-cyclic guanosine monophosphate-adenosine triphosphate-sensitive K(+) channel pathway. Phytother. Res. 2018, 32, 2520–2530. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, C.; Yang, G.; Yang, Y. Biological properties of 6-gingerol: A brief review. Nat. Prod. Commun. 2014, 9, 1027–1030. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, D.; Yu, X.; Xie, X.; Wang, L.; Lian, L.; Fei, N.; Chen, J.; Zhu, N.; Wang, G.; et al. The antinociceptive effects of ferulic acid on neuropathic pain: Involvement of descending monoaminergic system and opioid receptors. Oncotarget 2016, 7, 20455–20468. [Google Scholar] [CrossRef]
- Chi, Y.-M.; Nakamura, M.; Yoshizawa, T.; Zhao, X.-Y.; Yan, W.-M.; Hashimoto, F.; Kinjo, J.; Nohara, T.; Sakurada, S. Strong antinociceptive effect of incarvillateine, a novel monoterpene alkaloid from Incarvillea sinensis. J. Nat. Prod. 1999, 62, 1293–1294. [Google Scholar]
- Wang, M.-L.; Yu, G.; Yi, S.-P.; Zhang, F.-Y.; Wang, Z.-T.; Huang, B.; Su, R.-B.; Jia, Y.-X.; Gong, Z.-H. Antinociceptive effects of incarvillateine, a monoterpene alkaloid from Incarvillea sinensis, and possible involvement of the adenosine system. Sci. Rep. 2015, 5, 16107. [Google Scholar] [CrossRef]
- Saxena, A.K.; Jain, P.N.; Bhatnagar, S. The Prevalence of Chronic Pain among Adults in India. Indian J. Palliat. Care 2018, 24, 472–477. [Google Scholar]
- Chen, L.; Yang, G.; Grosser, T. Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat. 2013, 104–105, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Kerstein, P.C.; del Camino, D.; Moran, M.M.; Stucky, C.L. Pharmacological blockade of TRPA1 inhibits mechanical firing in nociceptors. Mol. Pain 2009, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Labuz, D.; Mousa, S.A.; Schäfer, M.; Stein, C.; Machelska, H. Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res. 2007, 1160, 30–38. [Google Scholar] [CrossRef]
- Chen, Z.; Janes, K.; Chen, C.; Doyle, T.; Bryant, L.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012, 26, 1855–1865. [Google Scholar] [CrossRef]
- Godfrey, L.; Yan, L.; Clarke, G.D.; Ledent, C.; Kitchen, I.; Hourani, S.M. Modulation of paracetamol antinociception by caffeine and by selective adenosine A2 receptor antagonists in mice. Eur. J. Pharmacol. 2006, 531, 80–86. [Google Scholar] [CrossRef]
- Little, J.W.; Ford, A.; Symons-Liguori, A.M.; Chen, Z.; Janes, K.; Doyle, T.; Xie, J.; Luongo, L.; Tosh, D.K.; Maione, S.; et al. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138 Pt 1, 28–35. [Google Scholar] [CrossRef]
- Savegnago, L.; Jesse, C.R.; Nogueira, C.W. Caffeine and a selective adenosine A(2B) receptor antagonist but not imidazoline receptor antagonists modulate antinociception induced by diphenyl diselenide in mice. Neurosci. Lett. 2008, 436, 120–123. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets–what are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Choi, D.S.; Cunha, R.A. Striatopallidal adenosine A(2A) receptor modulation of goal-directed behavior: Homeostatic control with cognitive flexibility. Neuropharmacology 2023, 226, 109421. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Targa, M.; Romagnoli, R.; Merighi, S.; Gessi, S.; Baraldi, P.G.; Borea, P.A.; Varani, K. TRR469, a potent A(1) adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 2014, 81, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.M.; Largent-Milnes, T.M.; Chen, Z.; Staikopoulos, V.; Esposito, E.; Dalgarno, R. Chronic Morphine-Induced Changes in Signaling at the A3 Adenosine Receptor Contribute to Morphine-Induced Hyperalgesia, Tolerance, and Withdrawal. J. Pharmacol. Exp. Ther. 2020, 374, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Katz, N.K.; Ryals, J.M.; Wright, D.E. Central or peripheral delivery of an adenosine A1 receptor agonist improves mechanical allodynia in a mouse model of painful diabetic neuropathy. Neuroscience 2015, 285, 312–323. [Google Scholar] [CrossRef]
- Nascimento, F.P.; Macedo-Junior, S.J.; Pamplona, F.A.; Luiz-Cerutti, M.; Córdova, M.M.; Constantino, L.; Tasca, C.I.; Dutra, R.C.; Calixto, J.B.; Reid, A.; et al. Adenosine A1 receptor-dependent antinociception induced by inosine in mice: Pharmacological, genetic and biochemical aspects. Mol. Neurobiol. 2015, 51, 1368–1378. [Google Scholar] [CrossRef]
- Petrelli, R.; Scortichini, M.; Belardo, C.; Boccella, S.; Luongo, L.; Capone, F. Structure-Based Design, Synthesis, and In Vivo Antinociceptive Effects of Selective A(1) Adenosine Receptor Agonists. J. Med. Chem. 2018, 61, 305–318. [Google Scholar] [CrossRef]
- Ford, A.; Castonguay, A.; Cottet, M.; Little, J.W.; Chen, Z.; Symons-Liguori, A.M.; Doyle, T.; Egan, T.M.; Vanderah, T.W.; De Koninck, Y.; et al. Engagement of the GABA to KCC2 signaling pathway contributes to the analgesic effects of A3AR agonists in neuropathic pain. J. Neurosci. 2015, 35, 6057–6067. [Google Scholar] [CrossRef]
- Janes, K.; Symons-Liguori, A.; Jacobson, K.A.; Salvemini, D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br. J. Pharmacol. 2016, 173, 1253–1267. [Google Scholar] [CrossRef]
- Stemmer, S.M.; Benjaminov, O.; Medalia, G.; Ciuraru, N.B.; Silverman, M.H.; Bar-Yehuda, S.; Fishman, S.; Harpaz, Z.; Farbstein, M.; Cohen, S.; et al. CF102 for the treatment of hepatocellular carcinoma: A phase I/II, open-label, dose-escalation study. Oncologist 2013, 18, 25–26. [Google Scholar] [CrossRef]
- David, M.; Akerman, L.; Ziv, M.; Kadurina, M.; Gospodinov, D.; Pavlotsky, F.; Yankova, R.; Kouzeva, V.; Ramon, M.; Silverman, M.; et al. Treatment of plaque-type psoriasis with oral CF101: Data from an exploratory randomized phase 2 clinical trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Priebe, A.; Hunke, M.; Tonello, R.; Sonawane, Y.; Berta, T.; Natarajan, A.; Bhuvanesh, N.; Pattabiraman, M.; Chandra, S. Ferulic acid dimer as a non-opioid therapeutic for acute pain. J. Pain Res. 2018, 11, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Hajialilo, M.; Dolati, S.; Eghbal-Fard, S.; Heydarlou, H.; Ghaebi, M.; Ghassembaglou, A.; Aghebati-Maleki, L.; Kafil, H.S.; Kamrani, A.; et al. The effects of nanocurcumin on Treg cell responses and treatment of ankylosing spondylitis patients: A randomized, double-blind, placebo-controlled clinical trial. J. Cell Biochem. 2020, 121, 103–110. [Google Scholar] [CrossRef]
- Agarwal, K.A.; Tripathi, C.D.; Agarwal, B.B.; Saluja, S. Efficacy of turmeric (curcumin) in pain and postoperative fatigue after laparoscopic cholecystectomy: A double-blind, randomized placebo-controlled study. Surg. Endosc. 2011, 25, 3805–3810. [Google Scholar] [CrossRef]
- Zhu, X.; Li, Q.; Chang, R.; Yang, D.; Song, Z.; Guo, Q.; Huang, C. Curcumin alleviates neuropathic pain by inhibiting p300/CBP histone acetyltransferase activity-regulated expression of BDNF and cox-2 in a rat model. PLoS ONE 2014, 9, e91303. [Google Scholar] [CrossRef] [PubMed]
- Yeon, K.; Kim, S.; Kim, Y.; Lee, M.; Ahn, D.; Kim, H.; Kim, J.; Jung, S.; Oh, S. Curcumin produces an antihyperalgesic effect via antagonism of TRPV1. J. Dent. Res. 2010, 89, 170–174. [Google Scholar] [CrossRef]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the Adenosine A1 Receptor Reveals the Basis for Subtype Selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef]
- Hamilton, L.J.; Walker, M.; Pattabiraman, M.; Zhong, H.A.; Luedtke, B.; Chandra, S. Novel curcumin analog (cis-trans curcumin) as ligand to adenosine receptors A2A and A2B: Potential for therapeutics. Pharmacol. Res. 2021, 165, 105410. [Google Scholar] [CrossRef]
- Muller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 200, 151–199. [Google Scholar]
- Cappellacci, L.; Franchetti, P.; Vita, P.; Petrelli, R.; Lavecchia, A.; Costa, B. 5′-Carbamoyl derivatives of 2′-C-methyl-purine nucleosides as selective A1 adenosine receptor agonists: Affinity, efficacy, and selectivity for A1 receptor from different species. Bioorg. Med. Chem. 2008, 16, 336–353. [Google Scholar] [CrossRef]
- Peterman, C.; Sanoski, C.A. Tecadenoson: A novel, selective A1 adenosine receptor agonist. Cardiol. Rev. 2005, 13, 315–321. [Google Scholar] [CrossRef]
- Fishman, P. Drugs Targeting the A3 Adenosine Receptor: Human Clinical Study Data. Molecules 2022, 27, 3680. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.; Furness, S.G.; Venugopal, H.; Baltos, J.-A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A(1) receptor-G(i) complex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Nygaard, R.; Valentin-Hansen, L.; Bach, A.; Engelstoft, M.S.; Petersen, P.S.; Frimurer, T.M.; Schwartz, T.W. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 2010, 285, 3973–3985. [Google Scholar] [CrossRef] [PubMed]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Irenius, E.; Kull, B.; Schulte, G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem. Pharmacol. 2001, 61, 443–448. [Google Scholar] [CrossRef]
- Zylka, M.J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 2011, 17, 188–196. [Google Scholar] [CrossRef]
- Lucarini, E.; Coppi, E.; Micheli, L.; Parisio, C.; Vona, A.; Cherchi, F.; Pugliese, A.M.; Pedata, F.; Failli, P.; Palomino, S.; et al. Acute visceral pain relief mediated by A3AR agonists in rats: Involvement of N-type voltage-gated calcium channels. Pain 2020, 161, 2179–2190. [Google Scholar] [CrossRef]
- Starowicz, K.; Maione, S.; Cristino, L.; Palazzo, E.; Marabese, I.; Rossi, F.; de Novellis, V.; Di Marzo, V. Tonic endovanilloid facilitation of glutamate release in brainstem descending antinociceptive pathways. J. Neurosci. 2007, 27, 13739–13749. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Haddad, M.; Alsalem, M.; Aldossary, S.A.; Kalbouneh, H.; Jaffal, S.M.; Alshawabkeh, Q.; Al Hayek, S.; Abdelhai, O.; Barakat, N.A.; El-Salem, K. The role of adenosine receptor ligands on inflammatory pain: Possible modulation of TRPV1 receptor function. Inflammopharmacology 2023, 31, 337–347. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Prystowsky, E.N.; Niazi, I.; Curtis, A.B.; Wilber, D.J.; Bahnson, T.; Ellenbogen, K.; Dhala, A.; Bloomfield, D.M.; Gold, M.; Kadish, A.; et al. Termination of paroxysmal supraventricular tachycardia by tecadenoson (CVT-510), a novel A1-adenosine receptor agonist. J. Am. Coll. Cardiol. 2003, 42, 1098–1102. [Google Scholar] [CrossRef]
- Tendera, M.; Gaszewska-Żurek, E.; Parma, Z.; Ponikowski, P.; Jankowska, E.; Kawecka-Jaszcz, K.; Czarnecka, D.; Krzemińska-Pakuła, M.; Bednarkiewicz, Z.; Sosnowski, M.; et al. The new oral adenosine A1 receptor agonist capadenoson in male patients with stable angina. Clin. Res. Cardiol. 2012, 101, 585–591. [Google Scholar] [CrossRef]
- Myers, J.S.; Sall, K.N.; DuBiner, H.; Slomowitz, N.; McVicar, W.; Rich, C.C.; Baumgartner, R.A. A Dose-Escalation Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Efficacy of 2 and 4 Weeks of Twice-Daily Ocular Trabodenoson in Adults with Ocular Hypertension or Primary Open-Angle Glaucoma. J. Ocul. Pharmacol. Ther. 2016, 32, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, M.; Kim, J.Y.; Brown, K.M.; Haase, V.H.; D’Agati, V.D.; Lee, H.T. Proximal tubule sphingosine kinase-1 has a critical role in A1 adenosine receptor-mediated renal protection from ischemia. Kidney Int. 2012, 82, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Tabrizi, M.A.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-J.; Harvey, B.K.; Shen, H.; Chou, J.; Victor, A.; Wang, Y. Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J. Neurosci. Res. 2006, 84, 1848–1855. [Google Scholar] [CrossRef]
- Arruda, M.A.; Stoddart, L.A.; Gherbi, K.; Briddon, S.J.; Kellam, B.; Hill, S.J. A Non-imaging High Throughput Approach to Chemical Library Screening at the Unmodified Adenosine-A3 Receptor in Living Cells. Front. Pharmacol. 2017, 8, 908. [Google Scholar] [CrossRef]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Nabb, D.L.; Song, S.; Kluthe, K.E.; Daubert, T.A.; Luedtke, B.E.; Nuxoll, A.S. Polymicrobial Interactions Induce Multidrug Tolerance in Staphylococcus aureus Through Energy Depletion. Front. Microbiol. 2019, 10, 2803. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound * | A1 | A3 | A2A | A2B |
|---|---|---|---|---|
| CTCUR | 306 | 400 | 5107 a | 6722 a |
| CUR | - | >10,000,000 | >10,000,000 a | 5,700,000 a |
| Adenosine | 100 b | 290 b | 310 b | 15,000 b |
| NECA | 14 c | 25 c | 20 c | 140 c |
| Tecadenoson | 2 d | 227 d | 6390 d | 25,800 d |
| IB-MECA | 51 c | 1.8 c | 2900 c | 11,000 c |
| Caffeine | 10,700 c | 13,300 c | 23,400 c | 33,800 c |
| Theophylline | 6770 c | 22,300 c | 1710 c | 9070 c |
| Method | A1 | A3 | A2A | A2B |
|---|---|---|---|---|
| CBA (Ki in nM) | 306 | 400 | 5107 | 6722 |
| Confocal Microscopy | Confirms binding (p < 0.0001) | Confirms binding (p < 0.0001) | No difference (N/A) | Confirms binding (p < 0.05) |
| Docking | ||||
| (ΔG in kcal/mol) | −10.22 | −10.16 | −9.6 | −7.3 |
| Compounds | Docking (A1) | Docking (A3) | ΔGexp a (A1) | ΔGexp a (A3) | ΔΔG (A1) | ΔΔG (A3) |
|---|---|---|---|---|---|---|
| CTCUR | −10.22 | −10.16 | −8.89 | −8.73 | −1.33 | −1.43 |
| Tecadenoson | −7.00 | −9.29 | −11.87 | −9.06 | 4.87 | −0.23 |
| Adenosin | −7.60 | −8.08 | −9.55 | −8.92 | 1.95 | 0.84 |
| Theophylline | −7.42 | −7.68 | −7.04 | −6.35 | −0.38 | −0.63 |
| Caffeine | −5.77 | −6.73 | −5.93 | −6.65 | 0.16 | −0.08 |
| Vanillin | −6.39 | −7.30 | ||||
| Vapsaicin | −7.19 | −8.53 | ||||
| 6-gingerol | −8.68 | −9.33 | ||||
| Ferulic acid | −6.64 | −7.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamilton, L.J.; Pattabiraman, M.; Zhong, H.A.; Walker, M.; Vaughn, H.; Chandra, S. Curcumin Stereoisomer, Cis-Trans Curcumin, as a Novel Ligand to A1 and A3 Adenosine Receptors. Pharmaceuticals 2023, 16, 917. https://doi.org/10.3390/ph16070917
Hamilton LJ, Pattabiraman M, Zhong HA, Walker M, Vaughn H, Chandra S. Curcumin Stereoisomer, Cis-Trans Curcumin, as a Novel Ligand to A1 and A3 Adenosine Receptors. Pharmaceuticals. 2023; 16(7):917. https://doi.org/10.3390/ph16070917
Chicago/Turabian StyleHamilton, Luke J., Mahesh Pattabiraman, Haizhen A. Zhong, Michaela Walker, Hilary Vaughn, and Surabhi Chandra. 2023. "Curcumin Stereoisomer, Cis-Trans Curcumin, as a Novel Ligand to A1 and A3 Adenosine Receptors" Pharmaceuticals 16, no. 7: 917. https://doi.org/10.3390/ph16070917
APA StyleHamilton, L. J., Pattabiraman, M., Zhong, H. A., Walker, M., Vaughn, H., & Chandra, S. (2023). Curcumin Stereoisomer, Cis-Trans Curcumin, as a Novel Ligand to A1 and A3 Adenosine Receptors. Pharmaceuticals, 16(7), 917. https://doi.org/10.3390/ph16070917