
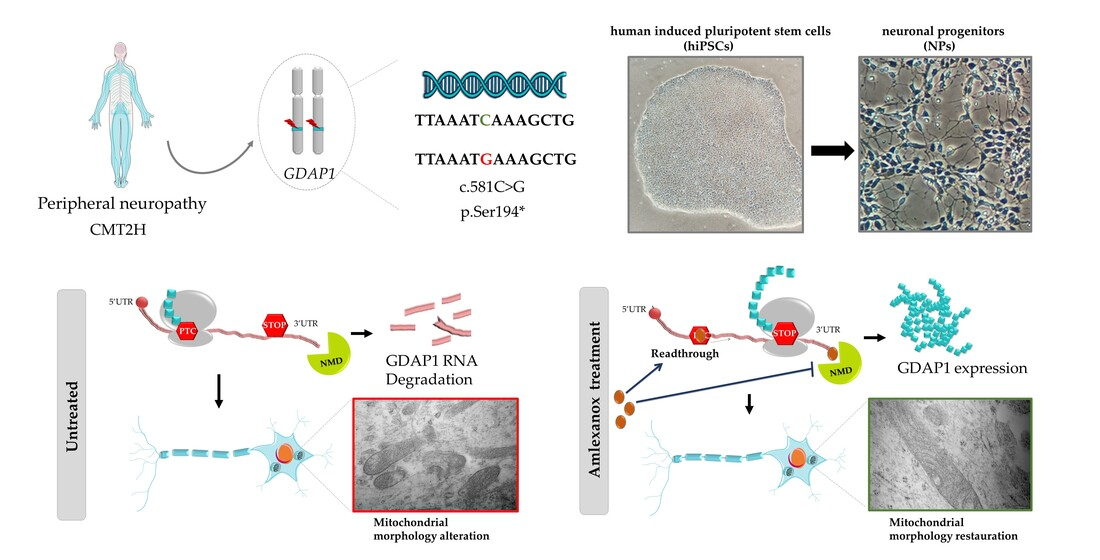
Amlexanox: Readthrough Induction and Nonsense-Mediated mRNA Decay Inhibition in a Charcot–Marie–Tooth Model of hiPSCs-Derived Neuronal Cells Harboring a Nonsense Mutation in GDAP1 Gene

 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Results
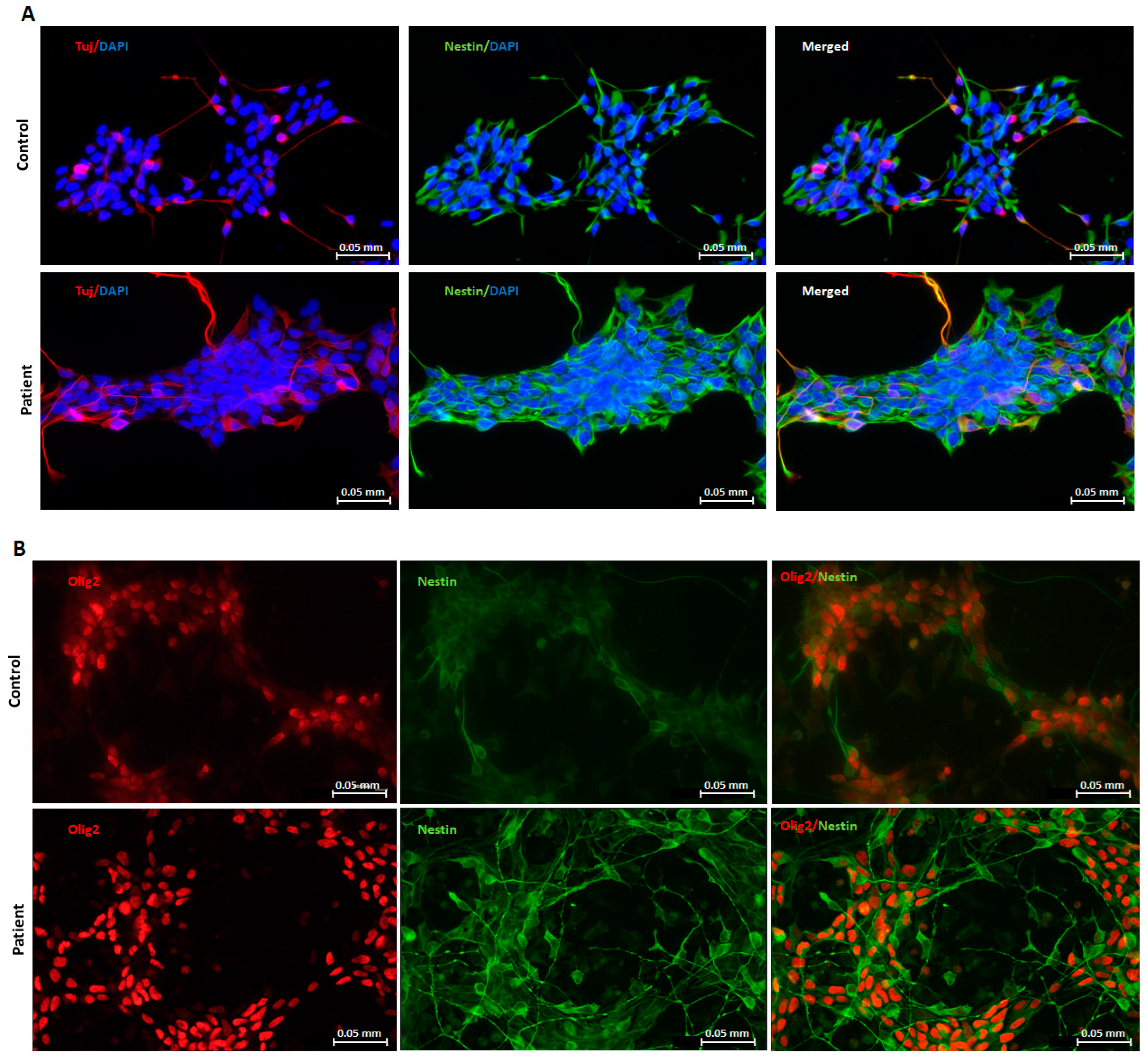
2.1. Generation and Characterization of Neuronal Progenitors (NPs) from hiPSCs
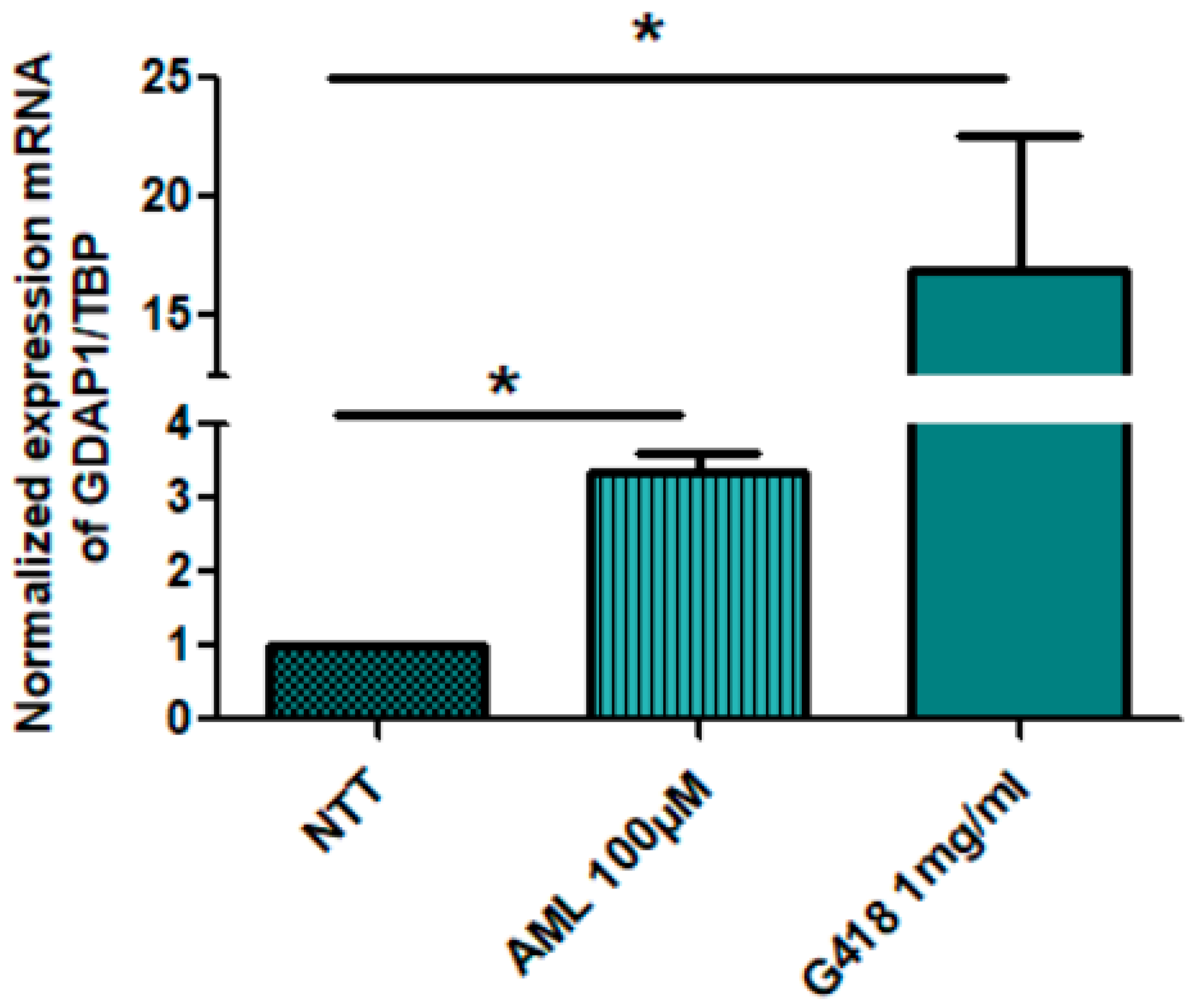
2.2. Restoration of GDAP1 mRNA Expression in NPs
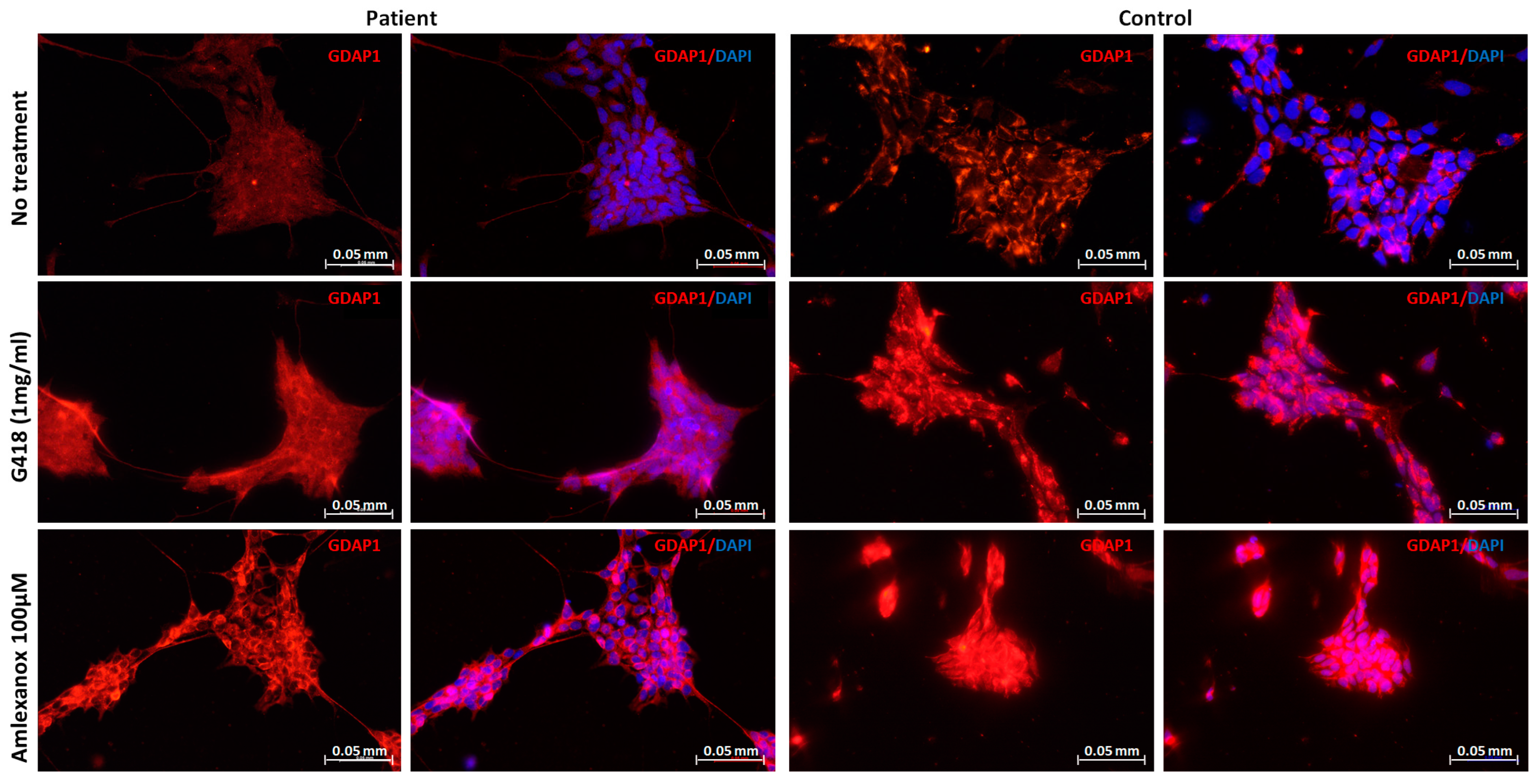
2.3. Restoration of GDAP1 Protein Expression in NPs
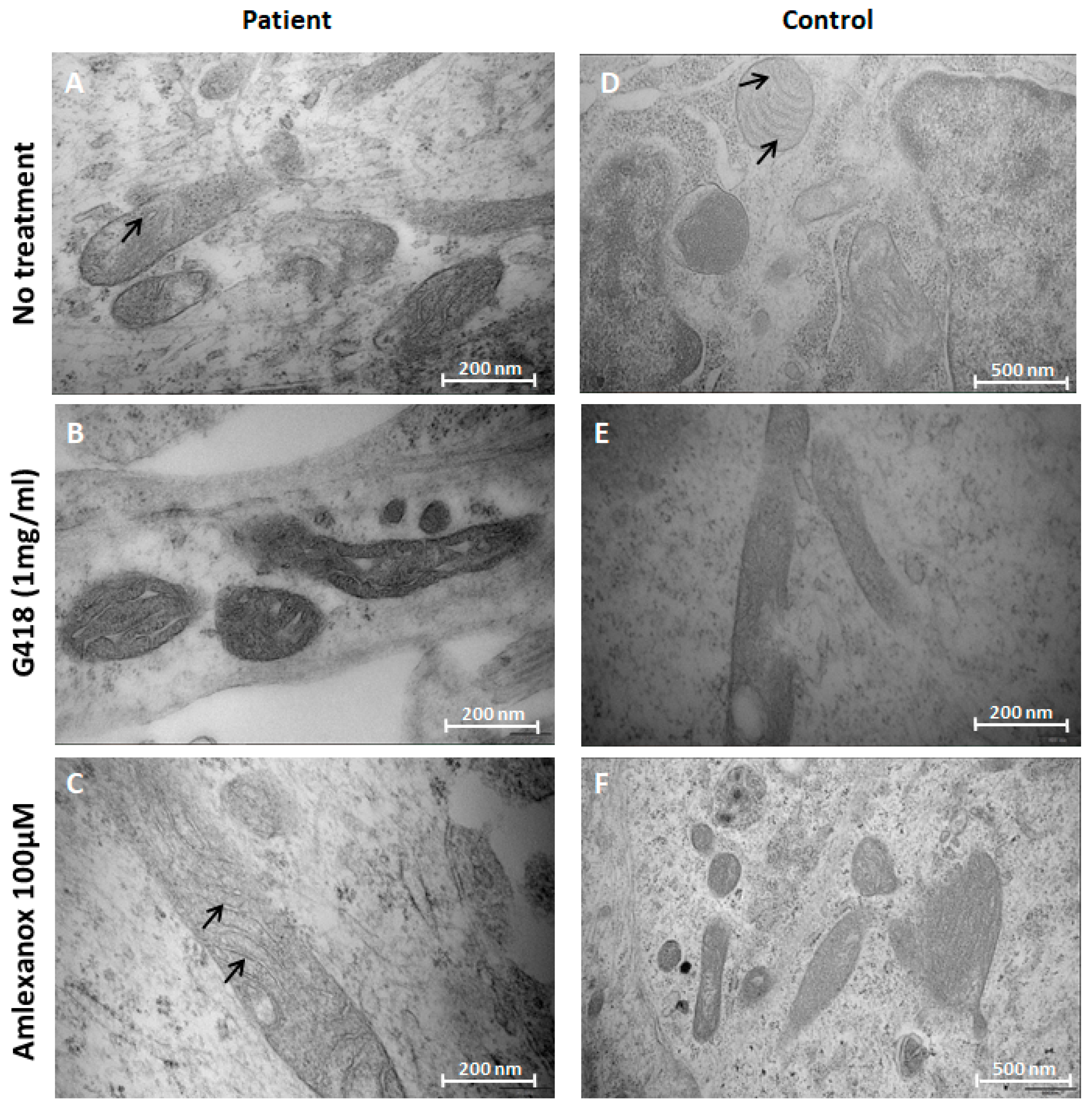
2.4. Amlexanox Effects on Mitochondrial Morphology in NPs
3. Discussion
4. Materials and Methods
4.1. Culture of Human iPS Cell Lines
4.2. Differentiation of hiPSCs into Neuronal Progenitors (NPs)
4.3. Amlexanox and G418 Preparation
4.4. Treatment of NPs
4.5. RNA Extraction and Analysis
4.6. Immunocytochemistry
4.7. Electron Microscopy
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A Meta-Analysis of Nonsense Mutations Causing Human Genetic Disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Guissart, C.; Mouzat, K.; Kantar, J.; Louveau, B.; Vilquin, P.; Polge, A.; Raoul, C.; Lumbroso, S. Premature Termination Codons in SOD1 Causing Amyotrophic Lateral Sclerosis Are Predicted to Escape the Nonsense-Mediated MRNA Decay. Sci. Rep. 2020, 10, 20738. [Google Scholar] [CrossRef] [PubMed]
- Martins-Dias, P.; Romão, L. Nonsense Suppression Therapies in Human Genetic Diseases. Cell. Mol. Life Sci. 2021, 78, 4677–4701. [Google Scholar] [CrossRef]
- Palma, M.; Lejeune, F. Deciphering the Molecular Mechanism of Stop Codon Readthrough. Biol. Rev. 2021, 96, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Déprez, B.; Lejeune, F. Rescue of Nonsense Mutations by Amlexanox in Human Cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef]
- Gómez-Grau, M.; Garrido, E.; Cozar, M.; Rodriguez-Sureda, V.; Domínguez, C.; Arenas, C.; Gatti, R.A.; Cormand, B.; Grinberg, D.; Vilageliu, L. Evaluation of Aminoglycoside and Non-Aminoglycoside Compounds for Stop-Codon Readthrough Therapy in Four Lysosomal Storage Diseases. PLoS ONE 2015, 10, e0135873. [Google Scholar] [CrossRef]
- Greenwood, G.J. Neomycin Ototoxicity; Report of a Case. AMA Arch. Otolaryngol. 1959, 69, 390–397. [Google Scholar] [CrossRef]
- Hock, R.; Anderson, R.J. Prevention of Drug-Induced Nephrotoxicity in the Intensive Care Unit. J. Crit. Care 1995, 10, 33–43. [Google Scholar] [CrossRef]
- McWilliam, S.J.; Antoine, D.J.; Smyth, R.L.; Pirmohamed, M. Aminoglycoside-Induced Nephrotoxicity in Children. Pediatr. Nephrol. Berl. Ger. 2017, 32, 2015–2025. [Google Scholar] [CrossRef]
- Lai, C.-H.; Chun, H.H.; Nahas, S.A.; Mitui, M.; Gamo, K.M.; Du, L.; Gatti, R.A. Correction of ATM Gene Function by Aminoglycoside-Induced Read-through of Premature Termination Codons. Proc. Natl. Acad. Sci. USA 2004, 101, 15676–15681. [Google Scholar] [CrossRef]
- Du, M.; Jones, J.R.; Lanier, J.; Keeling, K.M.; Lindsey, J.R.; Tousson, A.; Bebök, Z.; Whitsett, J.A.; Dey, C.R.; Colledge, W.H.; et al. Aminoglycoside Suppression of a Premature Stop Mutation in a Cftr-/- Mouse Carrying a Human CFTR-G542X Transgene. J. Mol. Med. Berl. Ger. 2002, 80, 595–604. [Google Scholar] [CrossRef]
- Dunant, P.; Walter, M.C.; Karpati, G.; Lochmüller, H. Gentamicin Fails to Increase Dystrophin Expression in Dystrophin-Deficient Muscle. Muscle Nerve 2003, 27, 624–627. [Google Scholar] [CrossRef]
- Meng, W.; Dong, Y.; Liu, J.; Wang, Z.; Zhong, X.; Chen, R.; Zhou, H.; Lin, M.; Jiang, L.; Gao, F.; et al. A Clinical Evaluation of Amlexanox Oral Adhesive Pellicles in the Treatment of Recurrent Aphthous Stomatitis and Comparison with Amlexanox Oral Tablets: A Randomized, Placebo Controlled, Blinded, Multicenter Clinical Trial. Trials 2009, 10, 30. [Google Scholar] [CrossRef]
- Frew, J.; Baradaran-Heravi, A.; Balgi, A.D.; Wu, X.; Yan, T.D.; Arns, S.; Shidmoossavee, F.S.; Tan, J.; Jaquith, J.B.; Jansen-West, K.R.; et al. Premature Termination Codon Readthrough Upregulates Progranulin Expression and Improves Lysosomal Function in Preclinical Models of GRN Deficiency. Mol. Neurodegener. 2020, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Martin, N.T.; Nakamura, K.; Azghadi, S.; Amiri, M.; Ben-David, U.; Perlman, S.; Gatti, R.A.; Hu, H.; Lowry, W.E. SMRT Compounds Abrogate Cellular Phenotypes of Ataxia Telangiectasia in Neural Derivatives of Patient-Specific HiPSCs. Nat. Commun. 2013, 4, 1824. [Google Scholar] [CrossRef]
- Sainio, M.T.; Rasila, T.; Molchanova, S.M.; Järvilehto, J.; Torregrosa-Muñumer, R.; Harjuhaahto, S.; Pennonen, J.; Huber, N.; Herukka, S.-K.; Haapasalo, A.; et al. Neurofilament Light Regulates Axon Caliber, Synaptic Activity, and Organelle Trafficking in Cultured Human Motor Neurons. Front. Cell Dev. Biol. 2021, 9, 820105. [Google Scholar] [CrossRef]
- Bird, T.D. Charcot-Marie-Tooth Hereditary Neuropathy Overview. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Yoshimura, A.; Yuan, J.-H.; Hashiguchi, A.; Ando, M.; Higuchi, Y.; Nakamura, T.; Okamoto, Y.; Nakagawa, M.; Takashima, H. Genetic Profile and Onset Features of 1005 Patients with Charcot-Marie-Tooth Disease in Japan. J. Neurol. Neurosurg. Psychiatry 2019, 90, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Marco, A.; Cuesta, A.; Pedrola, L.; Palau, F.; Marín, I. Evolutionary and Structural Analyses of GDAP1, Involved in Charcot-Marie-Tooth Disease, Characterize a Novel Class of Glutathione Transferase-Related Genes. Mol. Biol. Evol. 2004, 21, 176–187. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione Transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Niemann, A.; Huber, N.; Wagner, K.M.; Somandin, C.; Horn, M.; Lebrun-Julien, F.; Angst, B.; Pereira, J.A.; Halfter, H.; Welzl, H.; et al. The Gdap1 Knockout Mouse Mechanistically Links Redox Control to Charcot-Marie-Tooth Disease. Brain J. Neurol. 2014, 137 Pt 3, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the Protein Causing Charcot-Marie-Tooth Disease Type 4A, Is Expressed in Neurons and Is Associated with Mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Binięda, K.; Rzepnikowska, W.; Kolakowski, D.; Kaminska, J.; Szczepankiewicz, A.A.; Nieznańska, H.; Kochański, A.; Kabzińska, D. Mutations in GDAP1 Influence Structure and Function of the Trans-Golgi Network. Int. J. Mol. Sci. 2021, 22, 914. [Google Scholar] [CrossRef]
- Cantarero, L.; Juárez-Escoto, E.; Civera-Tregón, A.; Rodríguez-Sanz, M.; Roldán, M.; Benítez, R.; Hoenicka, J.; Palau, F. Mitochondria-Lysosome Membrane Contacts Are Defective in GDAP1-Related Charcot-Marie-Tooth Disease. Hum. Mol. Genet. 2021, 29, 3589–3605. [Google Scholar] [CrossRef]
- Huber, N.; Guimaraes, S.; Schrader, M.; Suter, U.; Niemann, A. Charcot-Marie-Tooth Disease-Associated Mutants of GDAP1 Dissociate Its Roles in Peroxisomal and Mitochondrial Fission. EMBO Rep. 2013, 14, 545–552. [Google Scholar] [CrossRef]
- Niemann, A.; Wagner, K.M.; Ruegg, M.; Suter, U. GDAP1 Mutations Differ in Their Effects on Mitochondrial Dynamics and Apoptosis Depending on the Mode of Inheritance. Neurobiol. Dis. 2009, 36, 509–520. [Google Scholar] [CrossRef]
- Miressi, F.; Benslimane, N.; Favreau, F.; Rassat, M.; Richard, L.; Bourthoumieu, S.; Laroche, C.; Magy, L.; Magdelaine, C.; Sturtz, F.; et al. GDAP1 Involvement in Mitochondrial Function and Oxidative Stress, Investigated in a Charcot-Marie-Tooth Model of HiPSCs-Derived Motor Neurons. Biomedicines 2021, 9, 945. [Google Scholar] [CrossRef] [PubMed]
- Pla-Martín, D.; Rueda, C.B.; Estela, A.; Sánchez-Piris, M.; González-Sánchez, P.; Traba, J.; de la Fuente, S.; Scorrano, L.; Renau-Piqueras, J.; Alvarez, J.; et al. Silencing of the Charcot-Marie-Tooth Disease-Associated Gene GDAP1 Induces Abnormal Mitochondrial Distribution and Affects Ca2+ Homeostasis by Reducing Store-Operated Ca2+ Entry. Neurobiol. Dis. 2013, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Cantarero, L.; García-Vargas, G.; Hoenicka, J.; Palau, F. Differential Effects of Mendelian GDAP1 Clinical Variants on Mitochondria-Lysosome Membrane Contacts Sites. Biol. Open 2023, 12, bio059707. [Google Scholar] [CrossRef]
- Labbé, K.; Murley, A.; Nunnari, J. Determinants and Functions of Mitochondrial Behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 357–391. [Google Scholar] [CrossRef]
- Li, S.; Xiong, G.-J.; Huang, N.; Sheng, Z.-H. The Crosstalk of Energy Sensing and Mitochondrial Anchoring Sustains Synaptic Efficacy by Maintaining Presynaptic Metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- Mannella, C.A. Structure and Dynamics of the Mitochondrial Inner Membrane Cristae. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2006, 1763, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; Selker, J.M.L.; Capaldi, R.A. The Cristal Membrane of Mitochondria Is the Principal Site of Oxidative Phosphorylation. FEBS Lett. 2003, 546, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Civera-Tregón, A.; Domínguez, L.; Martínez-Valero, P.; Serrano, C.; Vallmitjana, A.; Benítez, R.; Hoenicka, J.; Satrústegui, J.; Palau, F. Mitochondria and Calcium Defects Correlate with Axonal Dysfunction in GDAP1-Related Charcot-Marie-Tooth Mouse Model. Neurobiol. Dis. 2021, 152, 105300. [Google Scholar] [CrossRef]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-Induced Differentiation-Associated Protein-1 Is Mutant in Charcot-Marie-Tooth Disease Type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; García-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marín, I.; Vílchez, J.J.; Palau, F. The Gene Encoding Ganglioside-Induced Differentiation-Associated Protein 1 Is Mutated in Axonal Charcot-Marie-Tooth Type 4A Disease. Nat. Genet. 2002, 30, 22–25. [Google Scholar] [CrossRef]
- Sevilla, T.; Jaijo, T.; Nauffal, D.; Collado, D.; Chumillas, M.J.; Vilchez, J.J.; Muelas, N.; Bataller, L.; Domenech, R.; Espinós, C.; et al. Vocal Cord Paresis and Diaphragmatic Dysfunction Are Severe and Frequent Symptoms of GDAP1-Associated Neuropathy. Brain 2008, 131, 3051–3061. [Google Scholar] [CrossRef]
- Benhabiles, H.; Gonzalez-Hilarion, S.; Amand, S.; Bailly, C.; Prévotat, A.; Reix, P.; Hubert, D.; Adriaenssens, E.; Rebuffat, S.; Tulasne, D.; et al. Optimized Approach for the Identification of Highly Efficient Correctors of Nonsense Mutations in Human Diseases. PLoS ONE 2017, 12, e0187930. [Google Scholar] [CrossRef]
- Hörner, S.J.; Couturier, N.; Bruch, R.; Koch, P.; Hafner, M.; Rudolf, R. HiPSC-Derived Schwann Cells Influence Myogenic Differentiation in Neuromuscular Cocultures. Cells 2021, 10, 3292. [Google Scholar] [CrossRef]
- Bordonaro, M.; Lazarova, D. Amlexanox and UPF1 Modulate Wnt Signaling and Apoptosis in HCT-116 Colorectal Cancer Cells. J. Cancer 2019, 10, 287–292. [Google Scholar] [CrossRef]
- Cunha, D.L.; Eintracht, J.; Harding, P.; Zhou, J.H.; Moosajee, M. Restoration of Functional PAX6 in Aniridia Patient IPSC-Derived Ocular Tissue Models Using Repurposed Nonsense Suppression Drugs. bioRxiv 2022. [Google Scholar] [CrossRef]
- de Poel, E.; Spelier, S.; Suen, S.W.F.; Kruisselbrink, E.; Graeber, S.Y.; Mall, M.A.; Weersink, E.J.M.; van der Eerden, M.M.; Koppelman, G.H.; van der Ent, C.K.; et al. Functional Restoration of CFTR Nonsense Mutations in Intestinal Organoids. J. Cyst. Fibros. 2022, 21, 246–253. [Google Scholar] [CrossRef]
- Correa-Cerro, L.S. DHCR7 Nonsense Mutations and Characterisation of MRNA Nonsense Mediated Decay in Smith-Lemli-Opitz Syndrome. J. Med. Genet. 2005, 42, 350–357. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Jiang, Q.; Prisco, M.; Gruber, C.; Piñón Hofbauer, J.; Chen, M.; Has, C.; Bruckner-Tuderman, L.; McGrath, J.A.; Uitto, J.; et al. Amlexanox Enhances Premature Termination Codon Read-Through in COL7A1 and Expression of Full Length Type VII Collagen: Potential Therapy for Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2017, 137, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside Antibiotics Mediate Context-Dependent Suppression of Termination Codons in a Mammalian Translation System. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren Stimulates Ribosomal Selection of Near-Cognate TRNAs to Promote Nonsense Suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Direction | Exon |
|---|---|---|---|
| GDAP1 | GCTGCTTGATCATGACAATGT | F | 5–6 |
| CCTCTTCTGGGGTTTCTTCA | R | 5–6 | |
| TBP | ACAGGTGCTAAAGTCAGAGC | F | 7–8 |
| GAGGCAAGGGTACATGAGAG | R | 7–8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benslimane, N.; Miressi, F.; Loret, C.; Richard, L.; Nizou, A.; Pyromali, I.; Faye, P.-A.; Favreau, F.; Lejeune, F.; Lia, A.-S. Amlexanox: Readthrough Induction and Nonsense-Mediated mRNA Decay Inhibition in a Charcot–Marie–Tooth Model of hiPSCs-Derived Neuronal Cells Harboring a Nonsense Mutation in GDAP1 Gene. Pharmaceuticals 2023, 16, 1034. https://doi.org/10.3390/ph16071034
Benslimane N, Miressi F, Loret C, Richard L, Nizou A, Pyromali I, Faye P-A, Favreau F, Lejeune F, Lia A-S. Amlexanox: Readthrough Induction and Nonsense-Mediated mRNA Decay Inhibition in a Charcot–Marie–Tooth Model of hiPSCs-Derived Neuronal Cells Harboring a Nonsense Mutation in GDAP1 Gene. Pharmaceuticals. 2023; 16(7):1034. https://doi.org/10.3390/ph16071034
Chicago/Turabian StyleBenslimane, Nesrine, Federica Miressi, Camille Loret, Laurence Richard, Angélique Nizou, Ioanna Pyromali, Pierre-Antoine Faye, Frédéric Favreau, Fabrice Lejeune, and Anne-Sophie Lia. 2023. "Amlexanox: Readthrough Induction and Nonsense-Mediated mRNA Decay Inhibition in a Charcot–Marie–Tooth Model of hiPSCs-Derived Neuronal Cells Harboring a Nonsense Mutation in GDAP1 Gene" Pharmaceuticals 16, no. 7: 1034. https://doi.org/10.3390/ph16071034
APA StyleBenslimane, N., Miressi, F., Loret, C., Richard, L., Nizou, A., Pyromali, I., Faye, P.-A., Favreau, F., Lejeune, F., & Lia, A.-S. (2023). Amlexanox: Readthrough Induction and Nonsense-Mediated mRNA Decay Inhibition in a Charcot–Marie–Tooth Model of hiPSCs-Derived Neuronal Cells Harboring a Nonsense Mutation in GDAP1 Gene. Pharmaceuticals, 16(7), 1034. https://doi.org/10.3390/ph16071034