
Recent Advances in Covalent Drug Discovery


Abstract
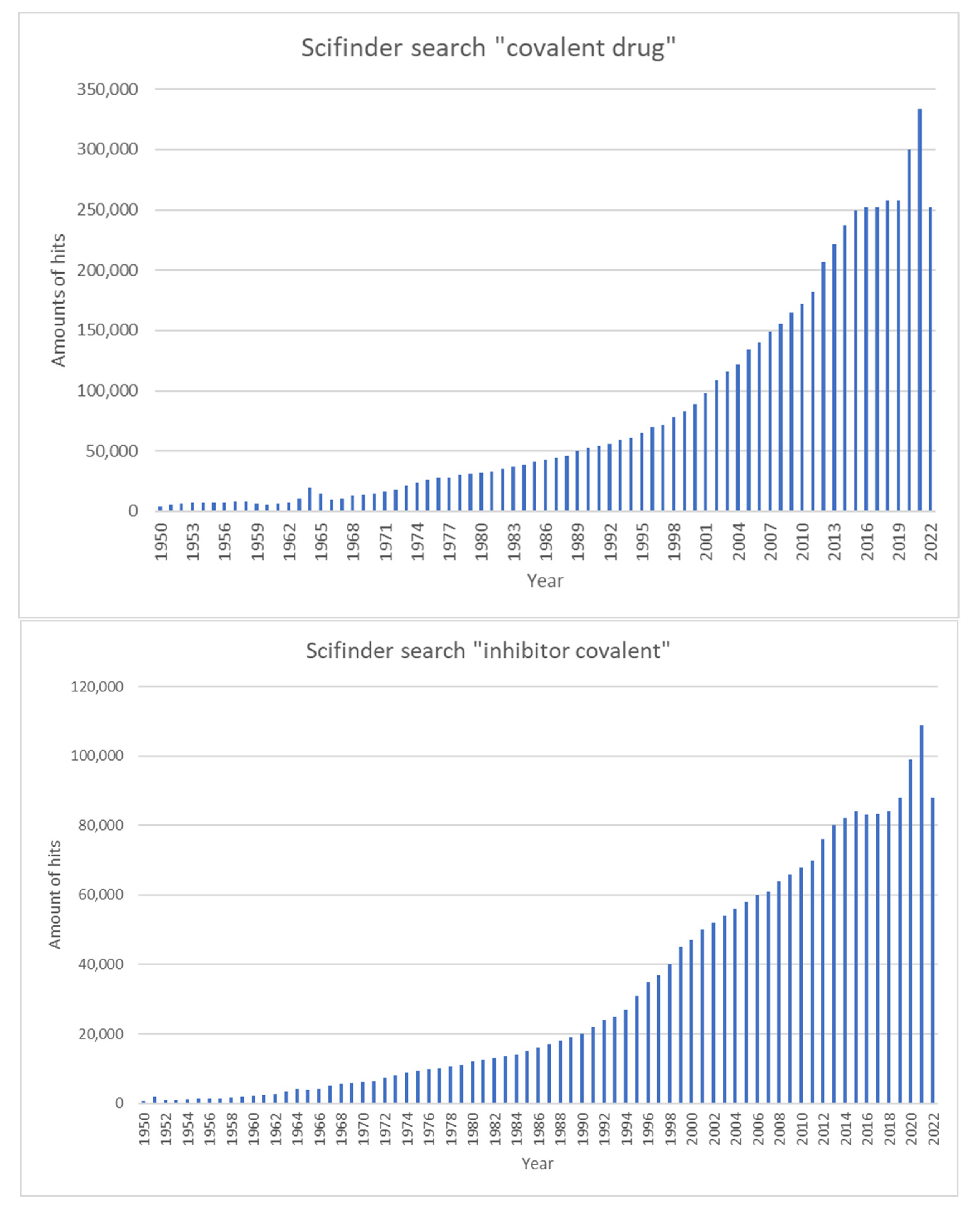
1. Introduction
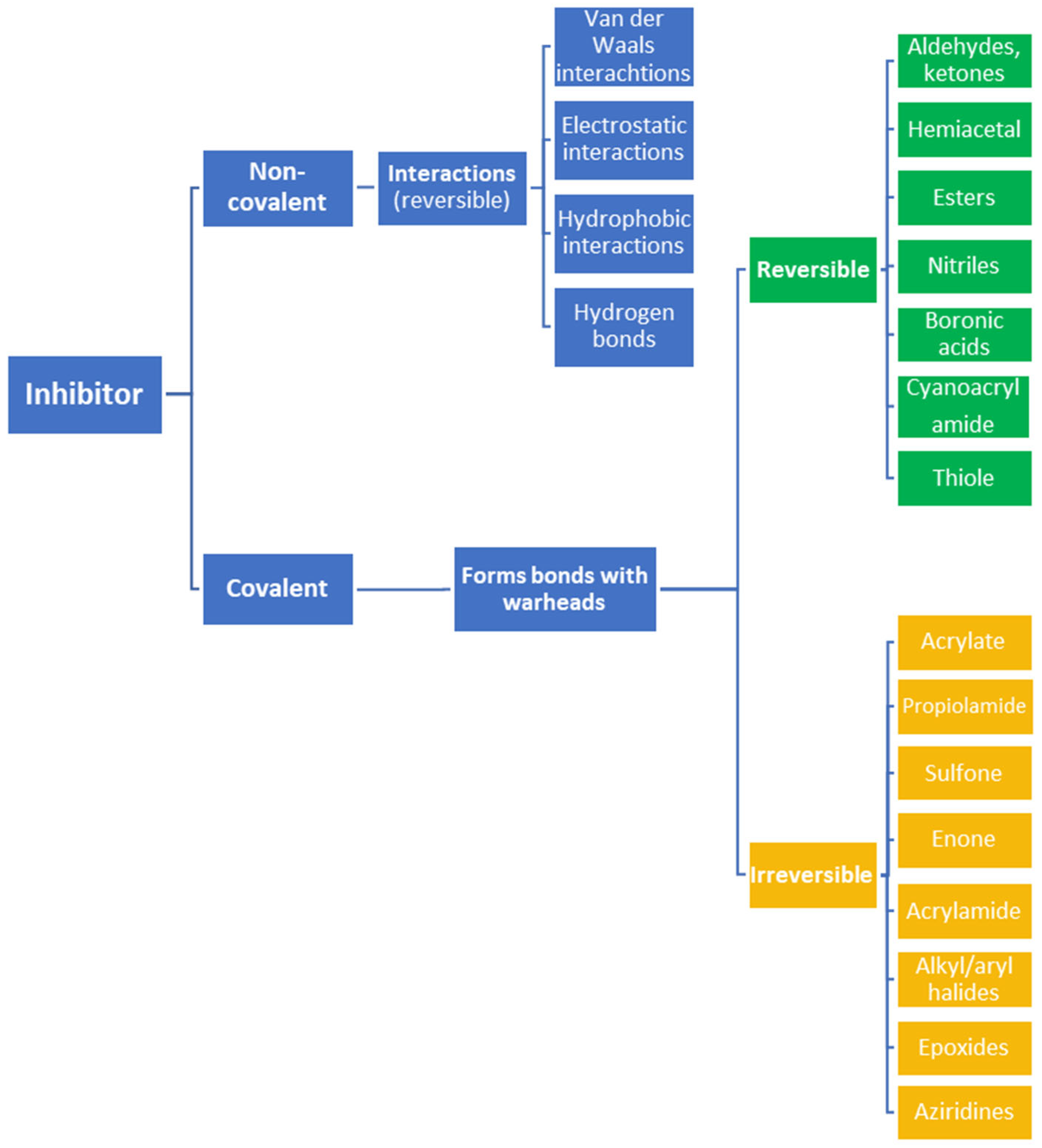
2. Advantages and Disadvantages of Covalent Inhibitors
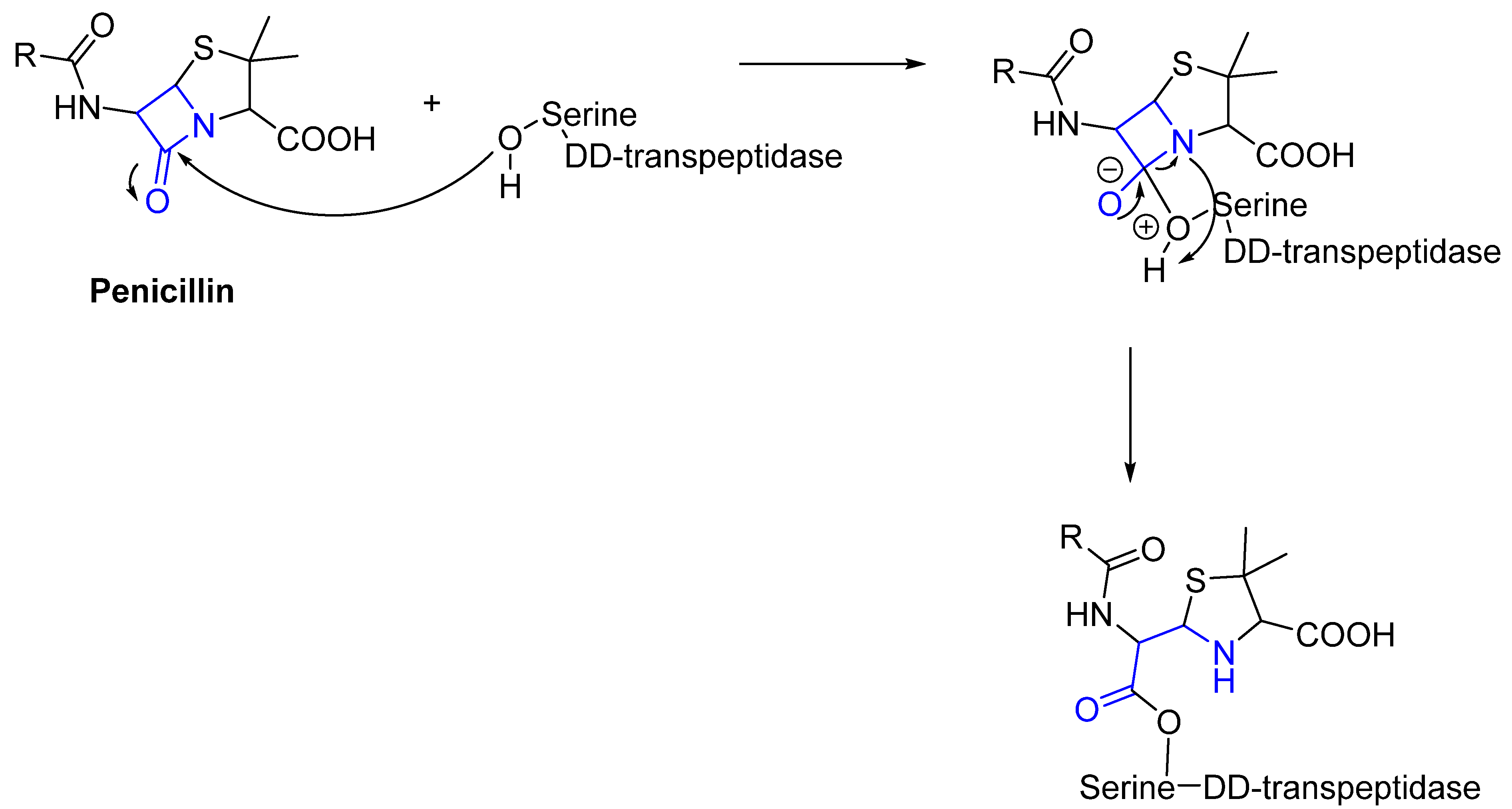
3. Mechanisms of Action and Chemical Designs
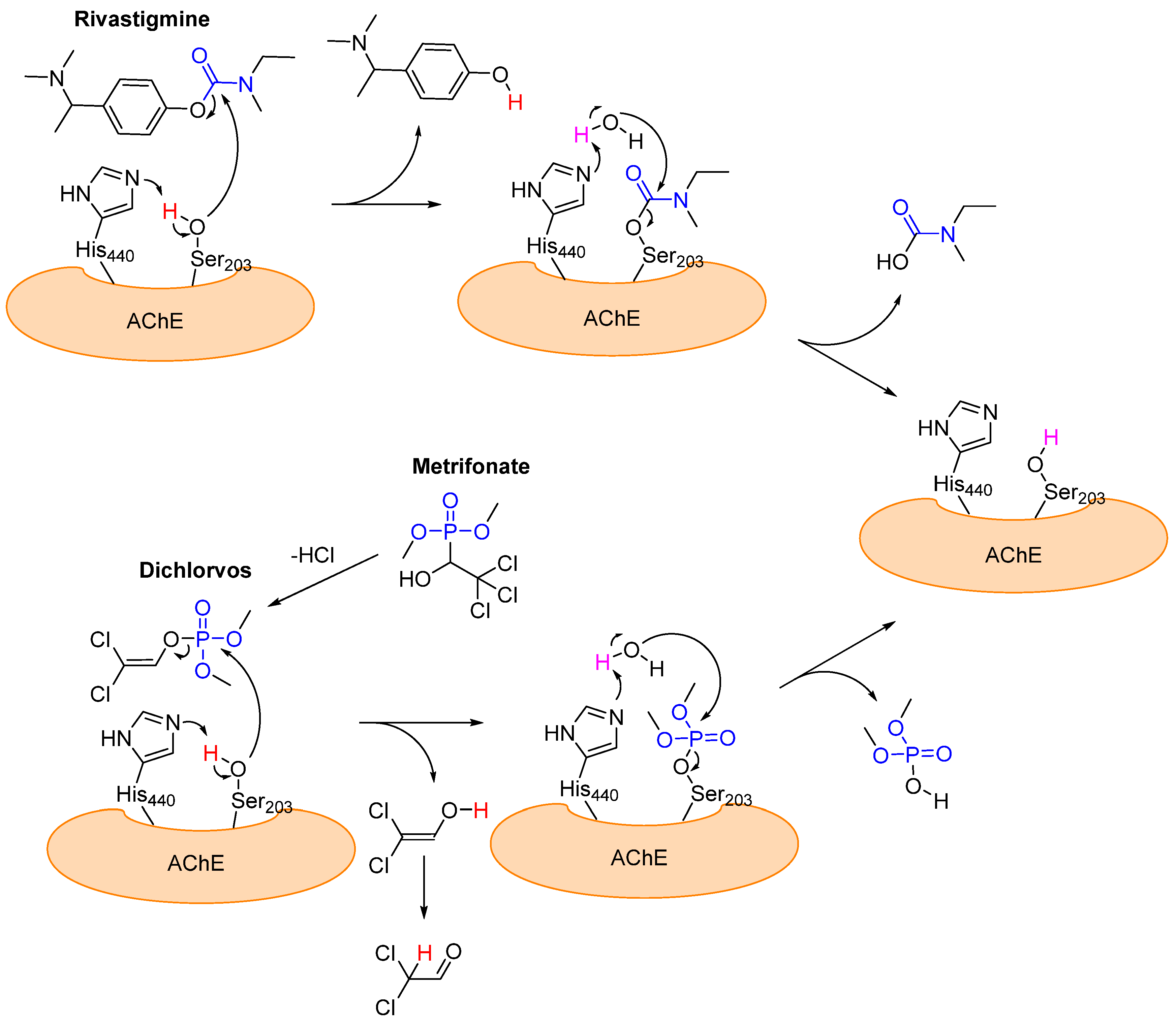
3.1. Alzheimer’s Disease: Acetylcholinesterase
3.2. X-Linked Agammaglobulinemia [67], B-Cell Leukemia [68], and B-Cell Lymphoma [69]: Bruton’s Tyrosine Kinase (BTK) [70,71]
3.3. Pancreatic Cancer [77], Colorectal Cancer [78], and Lung Cancer [79,80]: KRAS G12C Mutation [81]
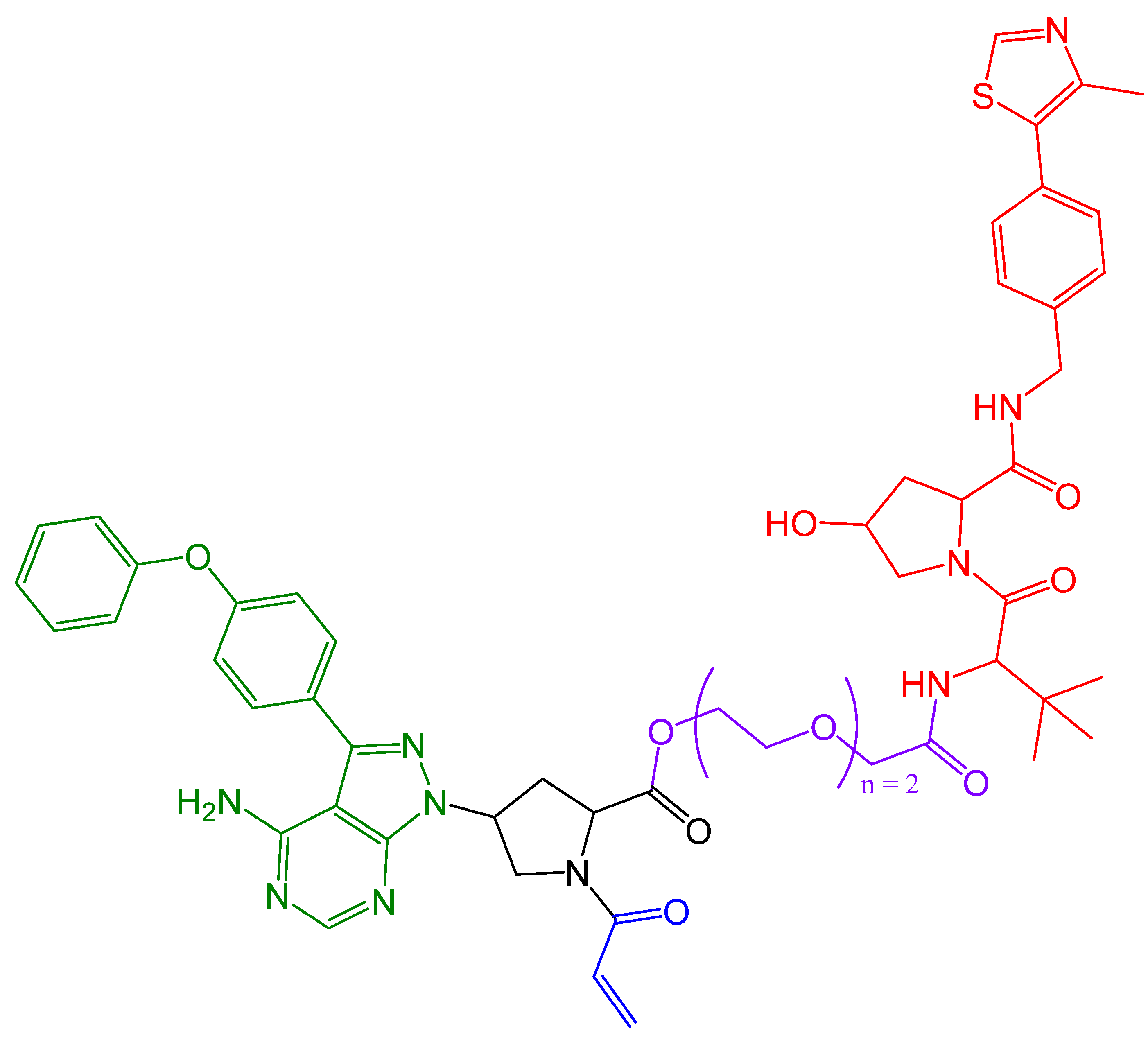
4. Covalent PROTACs
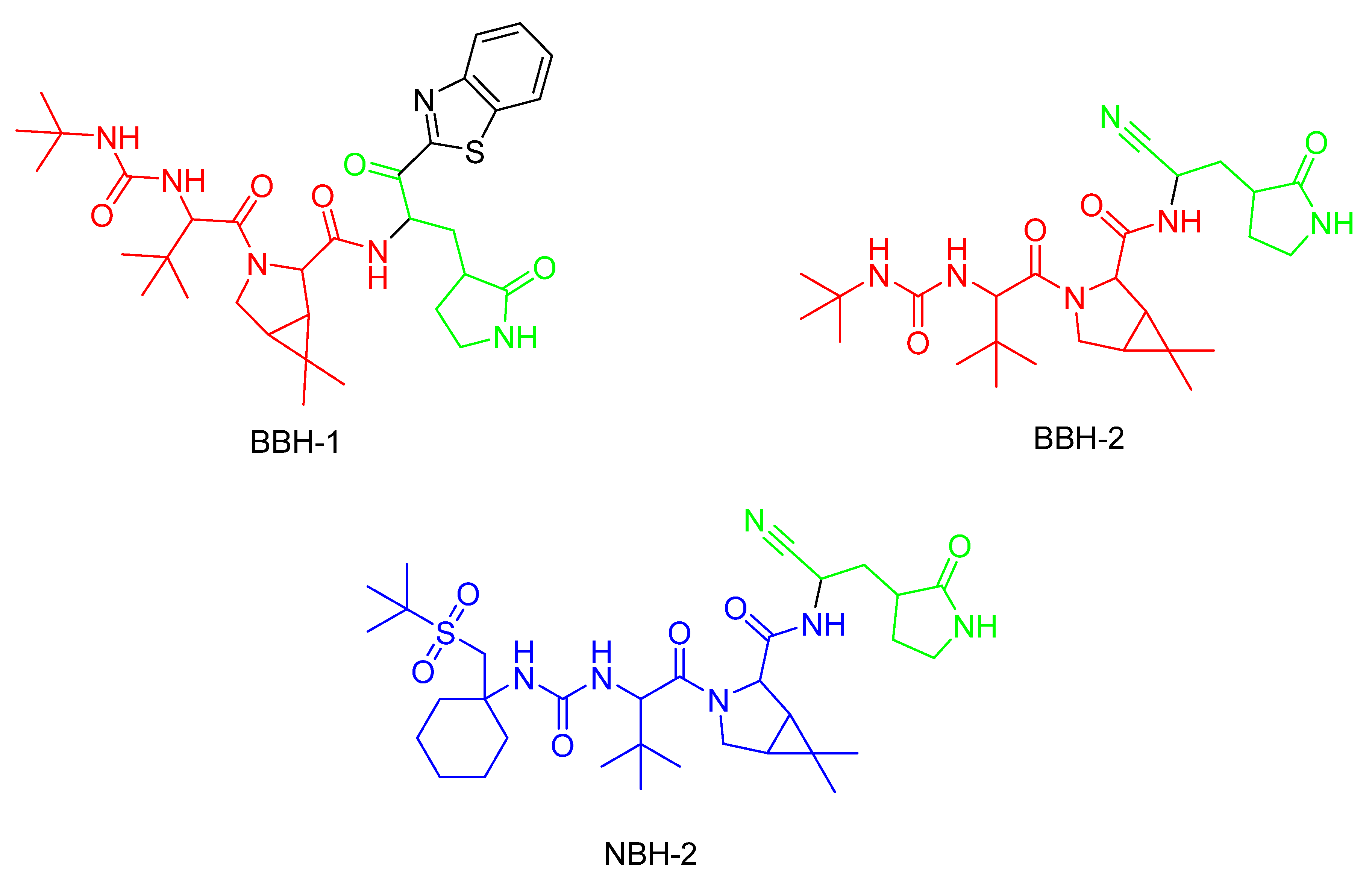
5. Covalent Inhibitors That Target the Main SARS-CoV-2 Protease Mpro
6. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, J.R.; Williams, G.; Pazdur, R. End points and United States Food and Drug Administration approval of oncology drugs. J. Clin. Oncol. 2003, 21, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Dumit, J. Drugs for Life: How Pharmaceutical Companies Define Our Health; Duke University Press: Durham, NC, USA, 2012. [Google Scholar]
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics/Pharmacodynamics; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Aljoundi, A.; Bjij, I.; El Rashedy, A.; Soliman, M.E. Covalent versus non-covalent enzyme inhibition: Which route should we take? A justification of the good and bad from molecular modelling perspective. Protein J. 2020, 39, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Jollow, D.; Mitchell, J.; Potter, W.; Davis, D.; Gillette, J.; Brodie, B. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202. [Google Scholar] [PubMed]
- Baillie, T.A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef] [PubMed]
- Jack, D.B. One hundred years of aspirin. Lancet 1997, 350, 437–439. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Baillie, T.A. The contributions of Sidney D. Nelson to drug metabolism research. Drug Metab. Rev. 2015, 47, 4–11. [Google Scholar] [CrossRef]
- Lei, J.; Zhou, Y.; Xie, D.; Zhang, Y. Mechanistic Insights into a Classic Wonder Drug Aspirin. J. Am. Chem. Soc. 2015, 137, 70–73. [Google Scholar] [CrossRef]
- Fleming, A. Penicillin. Br. Med. J. 1941, 2, 386. [Google Scholar] [CrossRef]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E. Theory and applications of covalent docking in drug discovery: Merits and pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef]
- Vane, J.; Botting, R. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Zhang, F.; Diao, H.; Wu, R. Covalent Inhibition Mechanism of Antidiabetic Drugs—Vildagliptin vs Saxagliptin. ACS Catal. 2019, 9, 2292–2302. [Google Scholar] [CrossRef]
- Kahne, D.; Still, W.C. Hydrolysis of a peptide bond in neutral water. J. Am. Chem. Soc. 1988, 110, 7529–7534. [Google Scholar] [CrossRef]
- Smith, R.M.; Hansen, D.E. The pH-rate profile for the hydrolysis of a peptide bond. J. Am. Chem. Soc. 1998, 120, 8910–8913. [Google Scholar] [CrossRef]
- Sun, Y.; Frenkel-Pinter, M.; Liotta, C.L.; Grover, M.A. The pH dependent mechanisms of non-enzymatic peptide bond cleavage reactions. Phys. Chem. Chem. Phys. 2020, 22, 107–113. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, C.; Zhang, Z.; Zheng, R.; Zheng, Y. Amidase as a versatile tool in amide-bond cleavage: From molecular features to biotechnological applications. Biotechnol. Adv. 2020, 43, 107574. [Google Scholar] [CrossRef]
- Bengtsson, G.; Olivecrona, T.J. Lipoprotein lipase: Mechanism of product inhibition. Eur. J. Biochem. 1980, 106, 557–562. [Google Scholar] [CrossRef]
- Herzog, K.; Müller, R.-J.; Deckwer, W.-D. Mechanism and kinetics of the enzymatic hydrolysis of polyester nanoparticles by lipases. Polym. Degrad. Stab. 2006, 91, 2486–2498. [Google Scholar] [CrossRef]
- Morar, M.; Pengelly, K.; Koteva, K.; Wright, G.D. Mechanism and diversity of the erythromycin esterase family of enzymes. Biochemistry 2012, 51, 1740–1751. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Yang, B. Chemical transformations of quaternary ammonium salts via C–N bond cleavage. Org. Biomol. Chem. 2020, 18, 1057–1072. [Google Scholar] [CrossRef]
- Smith, A.J.; Zhang, X.; Leach, A.G.; Houk, K. Beyond picomolar affinities: Quantitative aspects of noncovalent and covalent binding of drugs to proteins. J. Med. Chem. 2009, 52, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, A.A.; Muthusamy, R.; Soliman, M.E. New drug design with covalent modifiers. Expert Opin. Drug Discov. 2016, 11, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent inhibition in drug discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Nakamura, Y.; Oka, M.; Soda, H.; Shiozawa, K.; Yoshikawa, M.; Itoh, A.; Ikegami, Y.; Tsurutani, J.; Nakatomi, K.; Kitazaki, T. Gefitinib (“Iressa”, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2–mediated drug resistance. Cancer Res. 2005, 65, 1541–1546. [Google Scholar] [CrossRef]
- Koehler, J.; Schuler, M. Treatment, Afatinib, erlotinib and gefitinib in the first-line therapy of EGFR mutation-positive lung adenocarcinoma: A review. Oncol. Res. 2013, 36, 510–518. [Google Scholar]
- Dungo, R.T.; Keating, G.M. Afatinib: First global approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef]
- Singh, J. The Ascension of Targeted Covalent Inhibitors. J. Med. Chem. 2022, 65, 5886–5901. [Google Scholar] [CrossRef]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef]
- Shaikh, M.; Shinde, Y.; Pawara, R.; Noolvi, M.; Surana, S.; Ahmad, I.; Patel, H. Emerging Approaches to Overcome Acquired Drug Resistance Obstacles to Osimertinib in Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 1008–1046. [Google Scholar] [CrossRef]
- Cohen, S.D.; Pumford, N.R.; Khairallah, E.A.; Boekelheide, K.; Pohl, L.R.; Amouzadeh, H.; Hinson, J.A. Selective protein covalent binding and target organ toxicity. Toxicol. Appl. Pharmacol. 1997, 143, 1–12. [Google Scholar] [CrossRef]
- Coukell, A.J.; Markham, A. Clopidogrel. Drugs 1997, 54, 745–750. [Google Scholar] [CrossRef]
- Jarvis, B.; Simpson, K. Clopidogrel. Drugs 2000, 60, 347–377. [Google Scholar] [CrossRef]
- Bryant, J.; Post, J.M.; Alexander, S.; Wang, Y.-X.; Kent, L.; Schirm, S.; Tseng, J.-L.; Subramanyam, B.; Buckman, B.; Islam, I. Novel P2Y12 adenosine diphosphate receptor antagonists for inhibition of platelet aggregation (I): In vitro effects on platelets. Thromb. Res. 2008, 122, 523–532. [Google Scholar] [CrossRef]
- Bluet, G.; Blankenstein, J.; Brohan, E.; Prévost, C.; Chevé, M.; Schofield, J.; Roy, S. Synthesis of the stabilized active metabolite of clopidogrel. Tetrahedron 2014, 70, 3893–3900. [Google Scholar] [CrossRef]
- Shringarpure, R.; Davies, K.J.A. Protein turnover by the proteasome in aging and disease. Free Radic. Biol. Med. 2002, 32, 1084–1089. [Google Scholar] [CrossRef]
- Davies, A.; Nixon, A.; Muhammed, R.; Tsintzas, K.; Kirkham, S.; Stephens, F.B.; Moran, G.W. Reduced skeletal muscle protein balance in paediatric Crohn’s disease. Clin. Nutr. 2020, 39, 1250–1257. [Google Scholar] [CrossRef]
- Perry, C.M. Telaprevir. Drugs 2012, 72, 619–641. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef]
- Ouertani, A.; Neifar, M.; Ouertani, R.; Masmoudi, A.S.; Mosbah, A.; Cherif, A. Effectiveness of enzyme inhibitors in biomedicine and pharmacotherapy. Adv. Tissue Eng. Regen. Med. Open Access 2019, 5, 85–90. [Google Scholar] [CrossRef]
- Hajizadeh, M.; Moosavi-Movahedi, Z.; Sheibani, N.; Moosavi-Movahedi, A.A. An outlook on suicide enzyme inhibition and drug design. J. Iran. Chem. Soc. 2022, 19, 1575–1592. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Shindo, N.; Ojida, A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Bioorg. Med. Chem. 2021, 47, 116386. [Google Scholar] [CrossRef] [PubMed]
- Hartenfeller, M.; Schneider, G. De novo drug design. Chemoinform. Comput. Chem. Biol. 2010, 672, 299–323. [Google Scholar]
- Mandal, S.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef]
- Xu, D.; Xu, Z.; Han, L.; Liu, C.; Zhou, Z.; Qiu, Z.; Lin, X.; Tang, G.; Shen, H.; Aebi, J. Identification of new ATG4B inhibitors based on a novel high-throughput screening platform. SLAS Discov. 2017, 22, 338–347. [Google Scholar] [CrossRef]
- McAulay, K.; Bilsland, A.; Bon, M. Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery. Pharmaceuticals 2022, 15, 1366. [Google Scholar] [CrossRef]
- Pouliot, M.; Jeanmart, S. Pan Assay Interference Compounds (PAINS) and other promiscuous compounds in antifungal research: Miniperspective. J. Med. Chem. 2016, 59, 497–503. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Sotriffer, C. Docking of Covalent Ligands: Challenges and Approaches. Mol. Inform. 2018, 37, 1800062. [Google Scholar] [CrossRef]
- Kairys, V.; Baranauskiene, L.; Kazlauskiene, M.; Matulis, D.; Kazlauskas, E. Binding affinity in drug design: Experimental and computational techniques. Expert Opin. Drug Discov. 2019, 14, 755–768. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef]
- Berger, A.B.; Vitorino, P.M.; Bogyo, M. Activity-based protein profiling. Am. J. Pharm. 2004, 4, 371–381. [Google Scholar] [CrossRef]
- Lonsdale, R.; Ward, R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [Google Scholar] [CrossRef]
- Talesa, V.N. Acetylcholinesterase in Alzheimer’s disease. Mech. Ageing Dev. 2001, 122, 1961–1969. [Google Scholar] [CrossRef]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2012, 2012, 728983. [Google Scholar] [CrossRef]
- Eagger, S.A.; Levy, R.; Sahakian, B.J. Tacrine in Alzheimer’s disease. Lancet 1991, 337, 989–992. [Google Scholar] [CrossRef]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 6, CD001190. [Google Scholar] [CrossRef]
- Spencer, C.M.; Noble, S. Rivastigmine. Drugs Aging 1998, 13, 391–411. [Google Scholar] [CrossRef]
- Müller, T. Treatment, Rivastigmine in the treatment of patients with Alzheimer’s disease. Neuropsychiatr. Dis. 2007, 3, 211. [Google Scholar] [CrossRef] [PubMed]
- Holmstedt, B.; Nordgren, I.; Sandoz, M.; Sundwall, A. Metrifonate. Arch. Toxicol. 1978, 41, 3–29. [Google Scholar] [CrossRef] [PubMed]
- López-Arrieta, J.; Schneider, L. Metrifonate for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, 2, CD003155. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Metrifonate: A new agent for the treatment of Alzheimer’; s disease. Am. J. Health-Syst. Pharm. 1999, 56, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Smith, C. X-linked agammaglobulinemia. A clinical and molecular analysis. Medicine 1996, 75, 287–299. [Google Scholar] [CrossRef]
- Vogler, L.B.; Crist, W.M.; Bockman, D.E.; Pearl, E.R.; Lawton, A.R.; Cooper, M.D. Pre-B-cell leukemia: A new phenotype of childhood lymphoblastic leukemia. N. Engl. J. Med. 1978, 298, 872–878. [Google Scholar] [CrossRef]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Mohamed, A.J.; Yu, L.; Bäckesjö, C.M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglöf, A.; Vihinen, M.; Nore, B.F. Bruton’s tyrosine kinase (Btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef]
- Maas, A.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cell development. Dev. Immunol. 2001, 8, 171–181. [Google Scholar] [CrossRef]
- Thibaud, S.; Tremblay, D.; Bhalla, S.; Zimmerman, B.; Sigel, K.; Gabrilove, J. Protective role of Bruton tyrosine kinase inhibitors in patients with chronic lymphocytic leukaemia and COVID-19. Br. J. Haematol. 2020, 190, e73. [Google Scholar] [CrossRef]
- Cameron, F.; Sanford, M. Ibrutinib: First global approval. Drugs 2014, 74, 263–271. [Google Scholar] [CrossRef]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef]
- Markham, A.; Dhillon, S. Acalabrutinib: First global approval. Drugs 2018, 78, 139–145. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- Jaffee, E.M.; Hruban, R.H.; Canto, M.; Kern, S.E. Focus on pancreas cancer. Cancer Cell 2002, 2, 25–28. [Google Scholar] [CrossRef]
- Center, M.M.; Jemal, A.; Smith, R.A.; Ward, E. Worldwide variations in colorectal cancer. CA Cancer J. Clin. 2009, 59, 366–378. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Gazdar, A.F. Lung cancer preneoplasia. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 331–348. [Google Scholar] [CrossRef]
- Minna, J.D.; Roth, J.A.; Gazdar, A.F. Focus on lung cancer. Cancer Cell 2002, 1, 49–52. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- McCormick, F. KRAS as a therapeutic target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef]
- Park, J.T.; Johnson, N.; Liu, S.; Levesque, M.; Wang, Y.J.; Ho, H.; Huso, D.; Maitra, A.; Parsons, M.J.; Prescott, J.D. Differential in vivo tumorigenicity of diverse KRAS mutations in vertebrate pancreas: A comprehensive survey. Oncogene 2015, 34, 2801–2806. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Adjei, A.A. The ras/raf/mapk pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Gurbani, D.; Ficarro, S.B.; Carrasco, M.A.; Lim, S.M.; Choi, H.G.; Xie, T.; Marto, J.A.; Chen, Z.; Gray, N.S. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 2014, 111, 8895–8900. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Shokat, K.M. Targeting KRAS G12C with Covalent Inhibitors. Annu. Rev. Cancer Biol. 2022, 6, 49–64. [Google Scholar] [CrossRef]
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef]
- Dhillon, S. Adagrasib: First Approval. Drugs 2023, 83, 275–285. [Google Scholar] [CrossRef]
- Ou, S.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients with Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef]
- Purkey, H. Discovery of GDC-6036, a clinical stage treatment for KRAS G12C-positive cancers. Cancer Res. 2022, 82, ND11. [Google Scholar] [CrossRef]
- Wang, J.; Martin-Romano, P.; Cassier, P.; Johnson, M.; Haura, E.; Lenox, L.; Guo, Y.; Bandyopadhyay, N.; Russell, M.; Shearin, E.; et al. Phase I Study of JNJ-74699157 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. Oncologist 2022, 27, e536–e553. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Ye, J. Commentary: PROTACs make undruggable targets druggable: Challenge and opportunity. Acta Pharm. Sin. B 2021, 11, 3335. [Google Scholar] [CrossRef]
- Schreiber, S.L. The rise of molecular glues. Cell 2021, 184, 3–9. [Google Scholar] [CrossRef]
- Geiger, T.M.; Schäfer, S.C.; Dreizler, J.K.; Walz, M.; Hausch, F. Clues to molecular glues. Curr. Res. Chem. Biol. 2022, 2, 100018. [Google Scholar] [CrossRef]
- Toriki, E.S.; Papatzimas, J.W.; Nishikawa, K.; Dovala, D.; McGregor, L.M.; Hesse, M.J.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Nomura, D.K. Rational Chemical Design of Molecular Glue Degraders. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yardley, D.A. MONALEESA clinical program: A review of ribociclib use in different clinical settings. Future Oncol. 2019, 15, 2673–2686. [Google Scholar] [CrossRef]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs RD 2019, 19, 73–92. [Google Scholar] [CrossRef]
- Wedam, S.; Fashoyin-Aje, L.; Bloomquist, E.; Tang, S.; Sridhara, R.; Goldberg, K.B.; Theoret, M.R.; Amiri-Kordestani, L.; Pazdur, R.; Beaver, J.A. FDA Approval Summary: Palbociclib for Male Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 1208–1212. [Google Scholar] [CrossRef]
- Słabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.-D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Volkov, O.A.; Czerwinski, R.M.; Tan, H.; Huerta, C.; Morton, E.R.; Rizzi, J.P.; Wehn, P.M.; Xu, R.; Nijhawan, D. Structural basis and kinetic pathway of RBM39 recruitment to DCAF15 by a sulfonamide molecular glue E7820. Structure 2019, 27, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Kiely-Collins, H.; Winter, G.E.; Bernardes, G.J.L. The role of reversible and irreversible covalent chemistry in targeted protein degradation. Cell Chem. Biol. 2021, 28, 952–968. [Google Scholar] [CrossRef]
- Gabizon, R.; London, N. The rise of covalent proteolysis targeting chimeras. Curr. Opin. Chem. Biol. 2021, 62, 24–33. [Google Scholar] [CrossRef]
- Grimster, N.P. Covalent PROTACs: The best of both worlds? RSC Med. Chem. 2021, 12, 1452–1458. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, X.; He, X.; Zhou, Y.; Jiang, X.; Chen-Kiang, S.; Jaffrey, S.R.; Xu, G. A novel effect of thalidomide and its analogs: Suppression of cereblon ubiquitination enhances ubiquitin ligase function. FASEB J. 2015, 29, 4829–4839. [Google Scholar] [CrossRef]
- Cardote, T.A.; Gadd, M.S.; Ciulli, A. Crystal structure of the Cul2-Rbx1-EloBC-VHL ubiquitin ligase complex. Structure 2017, 25, 901–911. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Klein, V.G.; Townsend, C.E.; Testa, A.; Zengerle, M.; Maniaci, C.; Hughes, S.J.; Chan, K.-H.; Ciulli, A.; Lokey, R.S. Understanding and improving the membrane permeability of VH032-based PROTACs. ACS Med. Chem. Lett. 2020, 11, 1732–1738. [Google Scholar] [CrossRef]
- Tao, Y.; Remillard, D.; Vinogradova, E.V.; Yokoyama, M.; Banchenko, S.; Schwefel, D.; Melillo, B.; Schreiber, S.L.; Zhang, X.; Cravatt, B.F. Targeted Protein Degradation by Electrophilic PROTACs that Stereoselectively and Site-Specifically Engage DCAF1. J. Am. Chem. Soc. 2022, 144, 18688–18699. [Google Scholar] [CrossRef]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L. Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem. Biol. 2020, 27, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Chen, J.; Liu, L.; Zhou, D.; Zuo, Y.; Fu, T.; Pan, Z. Protein degradation through covalent inhibitor-based PROTACs. Chem. Commun. 2020, 56, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Welborn, M.; Zhu, T.; Yang, N.J.; Santos, M.S.; Van Voorhis, T.; Pentelute, B.L. π-Clamp-mediated cysteine conjugation. Nat. Chem. 2016, 8, 120–128. [Google Scholar] [CrossRef]
- Gama-Brambila, R.A.; Chen, J.; Dabiri, Y.; Tascher, G.; Němec, V.; Münch, C.; Song, G.; Knapp, S.; Cheng, X. A chemical toolbox for labeling and degrading engineered cas proteins. JACS Au 2021, 1, 777–785. [Google Scholar] [CrossRef]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef]
- Amanat, F.; Krammer, F. SARS-CoV-2 vaccines: Status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nat. Commun. 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Wang, L.; Yu, Z.; Wang, S.; Guo, Z.; Sun, Q.; Lai, L. Discovery of novel SARS-CoV-2 3CL protease covalent inhibitors using deep learning-based screen. Eur. J. Med. Chem. 2022, 244, 114803. [Google Scholar] [CrossRef]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Höbartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Flury, P.; Krüger, N.; Su, H.; Schäkel, L.; Barbosa Da Silva, E.; Eppler, O.; Kronenberger, T.; Nie, T.; Luedtke, S. Small-Molecule Thioesters as SARS-CoV-2 Main Protease Inhibitors: Enzyme Inhibition, Structure–Activity Relationships, Antiviral Activity, and X-ray Structure Determination. J. Med. Chem. 2022, 65, 9376–9395. [Google Scholar] [CrossRef]
- Kneller, D.W.; Li, H.; Phillips, G.; Weiss, K.L.; Zhang, Q.; Arnould, M.A.; Jonsson, C.B.; Surendranathan, S.; Parvathareddy, J.; Blakeley, M.P. Covalent narlaprevir-and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022, 13, 2268. [Google Scholar] [CrossRef]
- Lamb, Y.N. Remdesivir: First approval. Drugs 2020, 80, 1355–1363. [Google Scholar] [CrossRef]
- Lamb, Y.N. Nirmatrelvir plus Ritonavir: First approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef]
- Joyce, R.P.; Hu, V.W.; Wang, J. The history, mechanism, and perspectives of nirmatrelvir (PF-07321332): An orally bioavailable main protease inhibitor used in combination with ritonavir to reduce COVID-19-related hospitalizations. Med. Chem. Res. 2022, 31, 1637–1646. [Google Scholar] [CrossRef]
- Halford, B. The Path to Paxlovid. ACS Cent. Sci. 2022, 8, 405–407. [Google Scholar] [CrossRef]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. [Google Scholar] [CrossRef]
- Wong, C.K.H.; Au, I.C.H.; Lau, K.T.K.; Lau, E.H.Y.; Cowling, B.J.; Leung, G.M. Real-world effectiveness of molnupiravir and nirmatrelvir plus ritonavir against mortality, hospitalisation, and in-hospital outcomes among community-dwelling, ambulatory patients with confirmed SARS-CoV-2 infection during the omicron wave in Hong Kong: An observational study. Lancet 2022, 400, 1213–1222. [Google Scholar]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.-C.; et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. bioRxiv 2022. [Google Scholar] [CrossRef]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 Main Protease for Treatment of COVID-19: Covalent Inhibitors Structure–Activity Relationship Insights and Evolution Perspectives. J. Med. Chem. 2022, 65, 12500–12534. [Google Scholar] [CrossRef]
- Kumar, P.; Ratia, K.M.; Richner, J.M.; Thatcher, G.R.J.; Kadam, R.; Smieszek, S.P.; Przychodzen, B.P.; Koprivica, V.; Birznieks, G.; Polymeropoulos, M.H.; et al. Dual Inhibition of Cathepsin L and 3CL-Pro by GC-376 Constrains SARS Cov2 Infection Including Omicron Variant. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liu, H.; Iketani, S.; Zask, A.; Khanizeman, N.; Bednarova, E.; Forouhar, F.; Fowler, B.; Hong, S.J.; Mohri, H.; Nair, M.S.; et al. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022, 13, 1891. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.A.; Talwani, R.; Nehra, V.; Temesgen, Z. Boceprevir. Drugs Today 2011, 47, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Arasappan, A.; Bennett, F.; Bogen, S.L.; Venkatraman, S.; Blackman, M.; Chen, K.X.; Hendrata, S.; Huang, Y.; Huelgas, R.M.; Nair, L. Discovery of narlaprevir (SCH 900518): A potent, second generation HCV NS3 serine protease inhibitor. ACS Med. Chem. Lett. 2010, 1, 64–69. [Google Scholar] [CrossRef]
- Pan, Z.; Scheerens, H.; Li, S.J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.; Grothaus, P.G. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem Chem. Enabling Drug Discov. 2007, 2, 58–61. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Name of Drug | Warhead | Function |
|---|---|---|---|
| 2010 | Ceftaroline (β-lactam) |  | β-lactam antibiotic |
| 2011 | Telaprevir (α-ketoamide) |  | HCV protease inhibitor |
| 2011 | Boceprevir (α-ketoamide) |  | HCV protease inhibitor |
| 2011 | Abiraterone (-) | - | Prostate cancer treatment |
| 2012 | Carfilzomib (epoxide) |  | Proteasome inhibitor (cancer) |
| 2013 | Afatinib (α,β-unsaturated carbonyl) |  | EGFR tyrosine kinase inhibitor |
| 2013 | Dimethyl fumarate (α,β-unsaturated carbonyl) |  | Immunomodulatory drug |
| 2013 | Neostigmine (carbonyl group) |  | Acetylcholinesterase inhibitor |
| 2013 | Ibrutinib (α,β-unsaturated carbonyl) |  | EGFR tyrosine kinase inhibitor |
| 2014 | Ceftolozane (β-lactam) |  | β-lactam antibiotic |
| 2015 | Osimertinib (α,β-unsaturated carbonyl) |  | EGFR tyrosine kinase inhibitor |
| 2015 | Olmutinib (α,β-unsaturated carbonyl) |  | EGFR tyrosine kinase inhibitor |
| 2016 | Narlaprevir (α-ketoamide) |  | HCV protease inhibitor |
| 2017 | Acalabrutinib (α,β-unsaturated propargylamide) |  | Bruton’s tyrosine kinase inhibitor |
| 2017 | Neratinib (α,β-unsaturated carbonyl) |  | EGFR tyrosine kinase inhibitor |
| 2017 | Vaborbactam (boronic acid) |  | Non-β-lactam β-lactamase inhibitor |
| 2018 | Dacomitinib (α,β-unsaturated carbonyl) |  | EGFR tyrosine kinase inhibitor |
| 2019 | Selinexor (α,β-unsaturated carbonyl) |  | Nuclear export inhibitor |
| 2019 | Zanubrutinib (α,β-unsaturated carbonyl) |  | Bruton’s tyrosine kinase inhibitor |
| 2019 | Cerfiderocol (β-lactam) |  | β-lactam antibiotic |
| 2019 | Voxelotor (aldehyde) |  | Hemoglobin oxygen-affinity modulator |
| 2021 | Sotorasib (α,β-unsaturated carbonyl) |  | KRAS G12C inhibitor |
| 2021 | Nirmatrevir (nitrile) |  | SARS-CoV-2 main protease inhibitor |
| Type of Inhibitor | Advantages | Disadvantages |
|---|---|---|
| Non-Covalent |
|
|
| Covalent |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaefer, D.; Cheng, X. Recent Advances in Covalent Drug Discovery. Pharmaceuticals 2023, 16, 663. https://doi.org/10.3390/ph16050663
Schaefer D, Cheng X. Recent Advances in Covalent Drug Discovery. Pharmaceuticals. 2023; 16(5):663. https://doi.org/10.3390/ph16050663
Chicago/Turabian StyleSchaefer, Daniel, and Xinlai Cheng. 2023. "Recent Advances in Covalent Drug Discovery" Pharmaceuticals 16, no. 5: 663. https://doi.org/10.3390/ph16050663
APA StyleSchaefer, D., & Cheng, X. (2023). Recent Advances in Covalent Drug Discovery. Pharmaceuticals, 16(5), 663. https://doi.org/10.3390/ph16050663