

PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer


Abstract
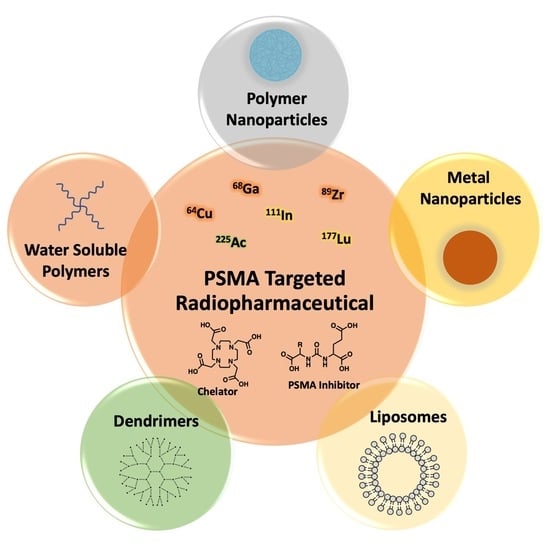
1. Introduction
2. Radiometals and Chelators for Imaging and Therapy
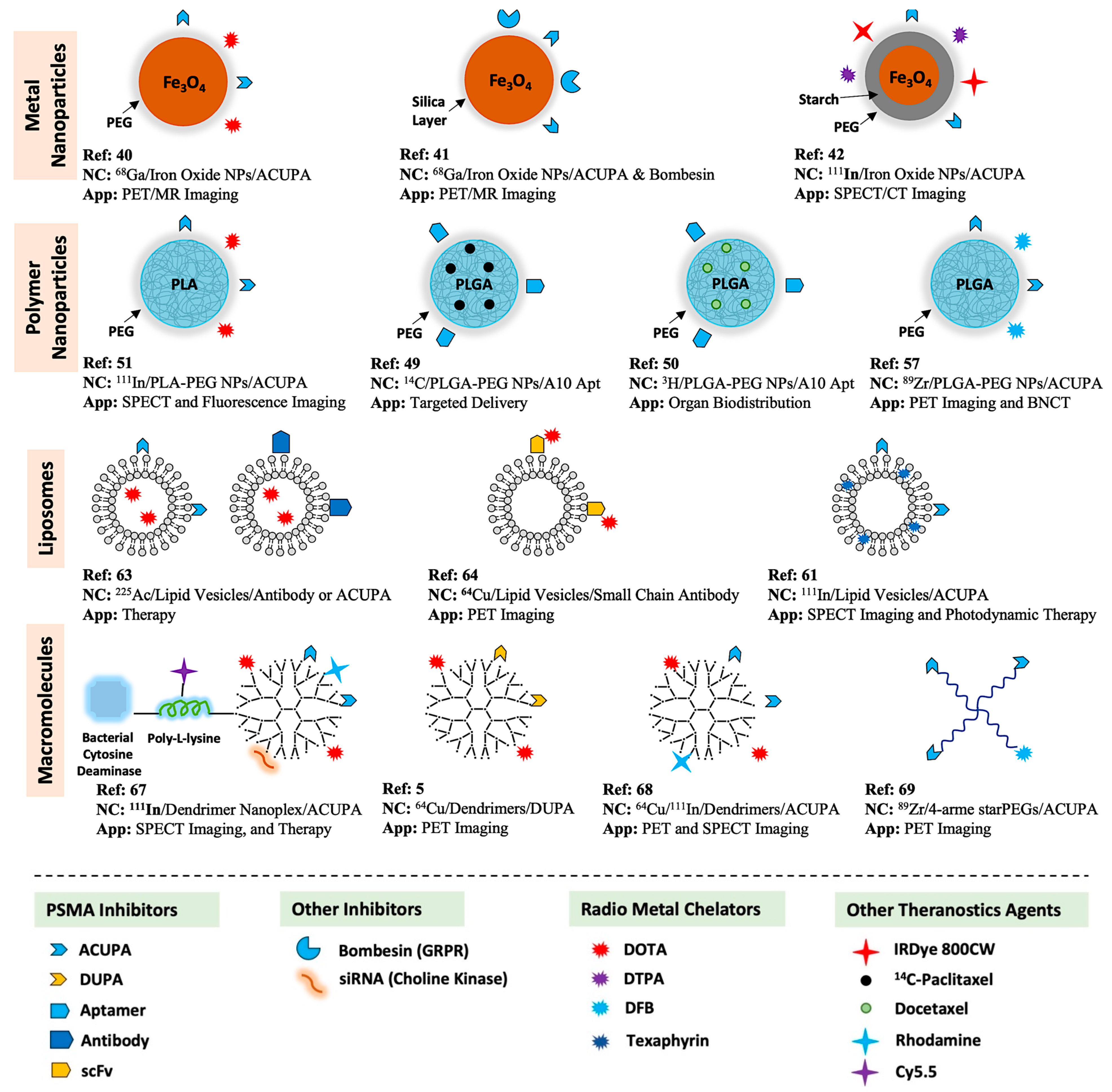
3. PSMA and Its Targeting Ligands
4. EPR Effect and Need of Targeted Nanomedicine
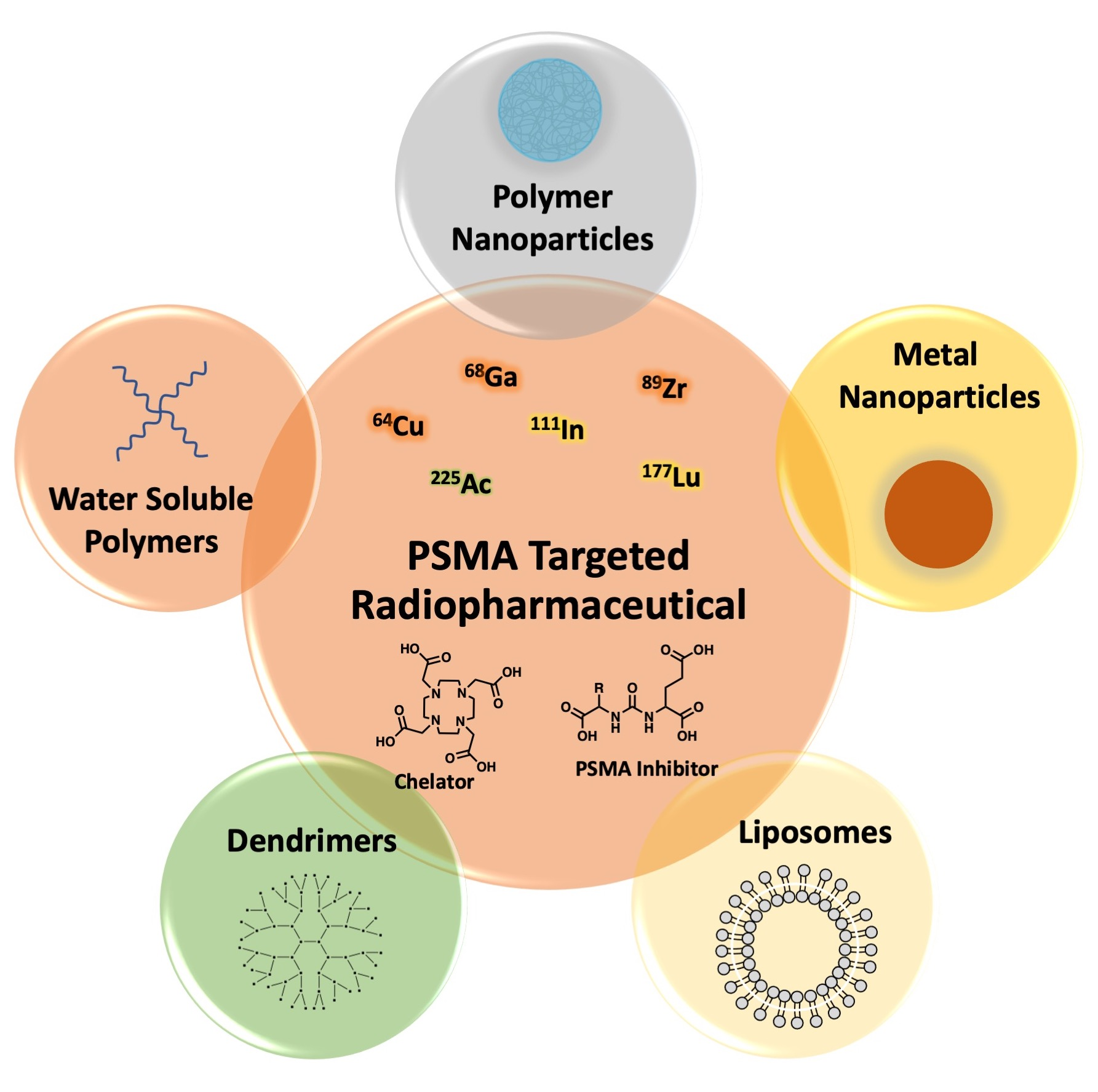
5. PSMA-Targeted Nanocarriers
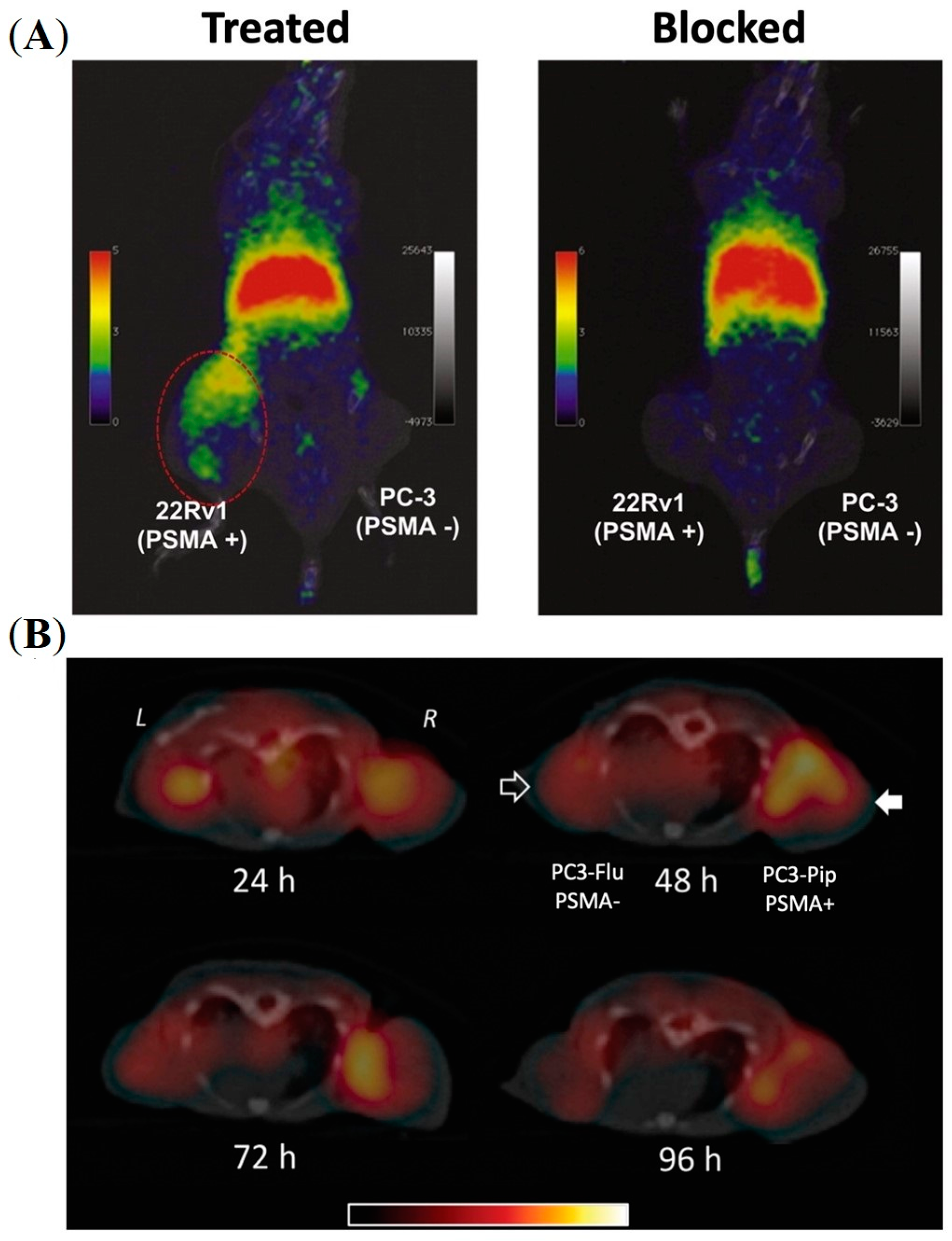
5.1. PSMA-Targeted Metal NPs

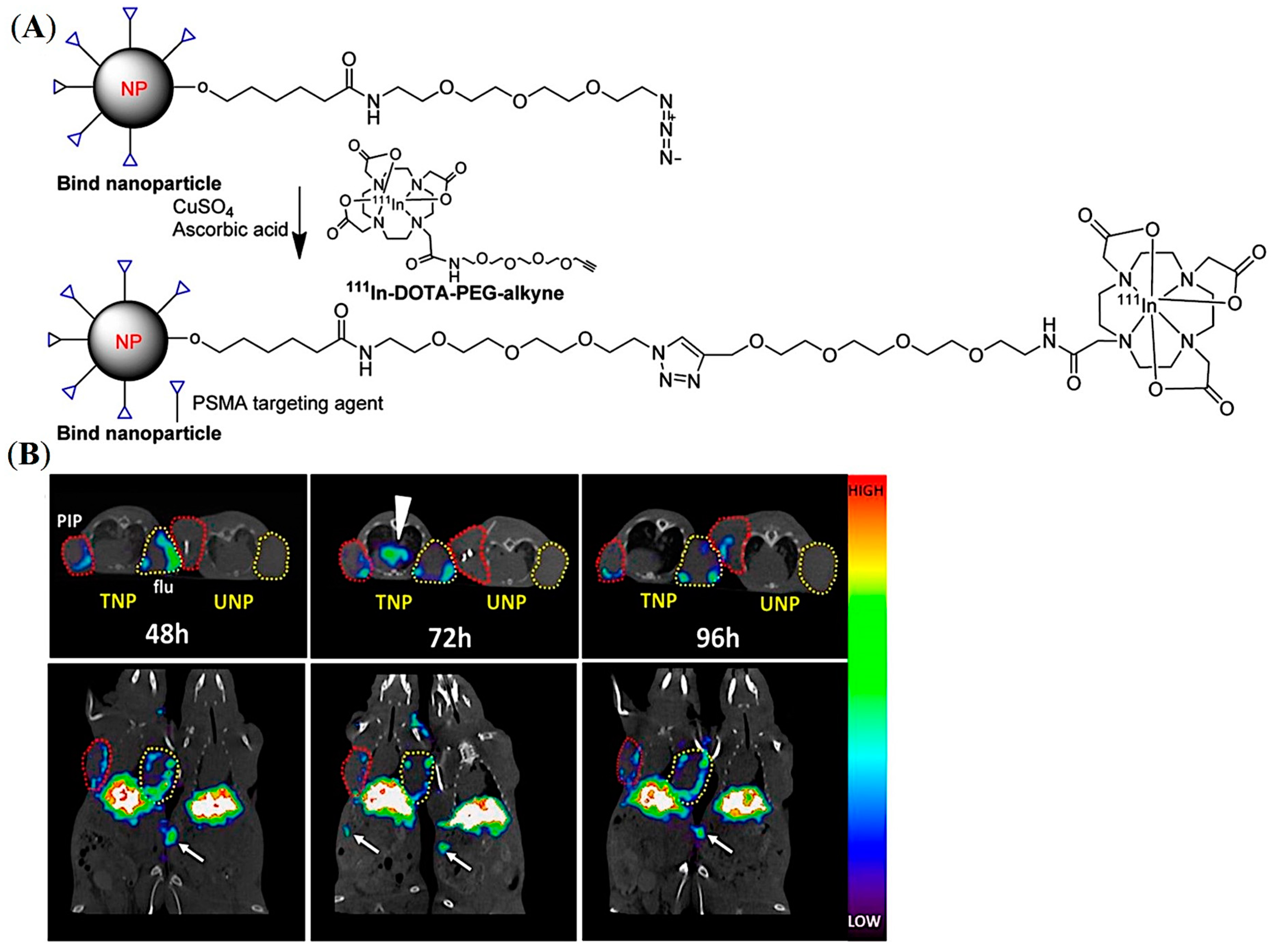
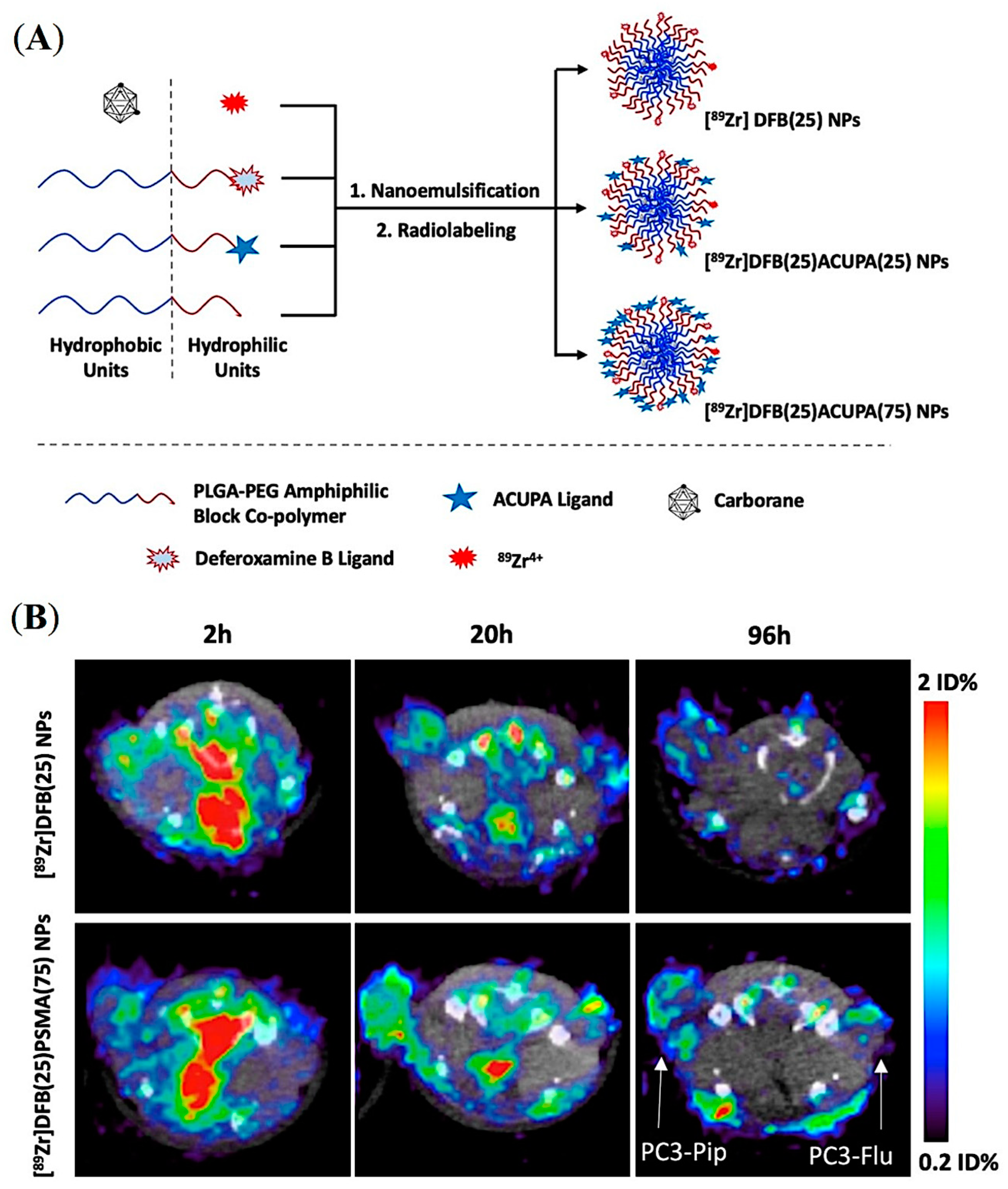
5.2. PSMA-Targeted Amphiphilic Block Copolymers
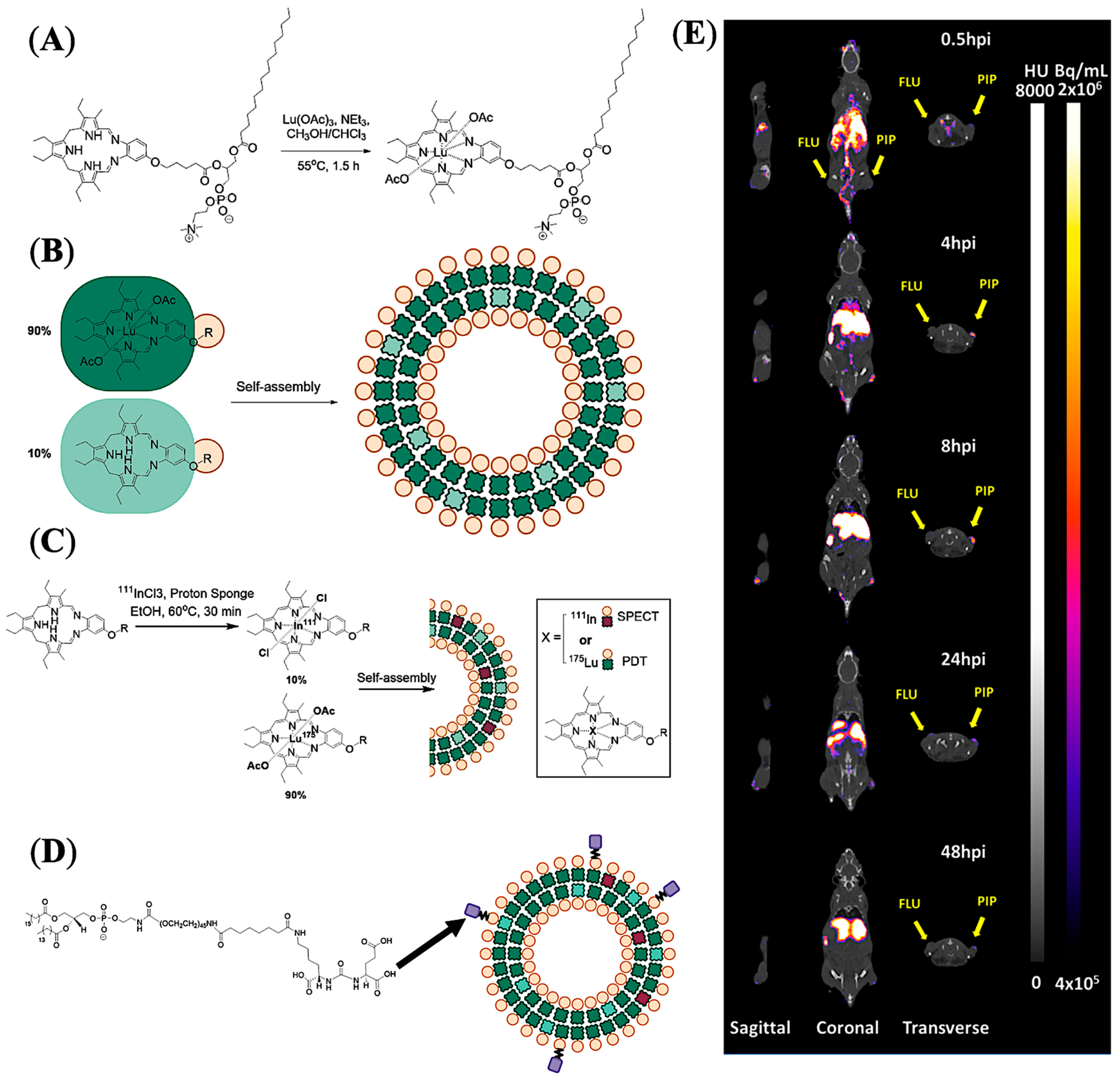
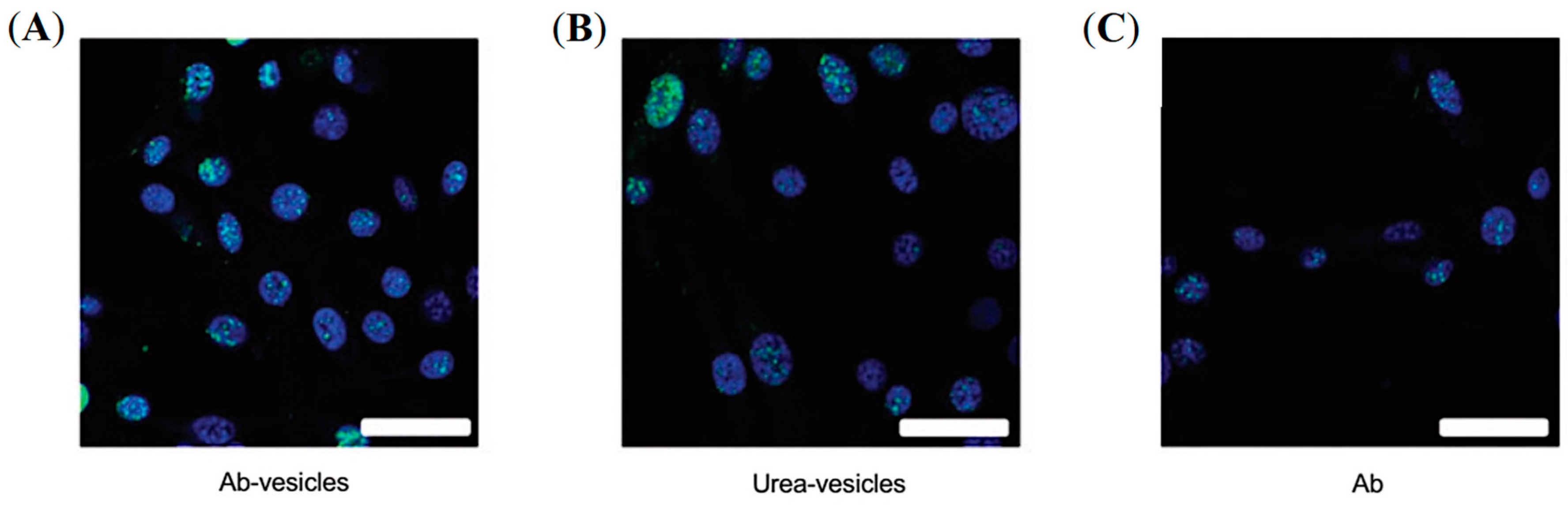
5.3. PSMA-Targeted Liposomes
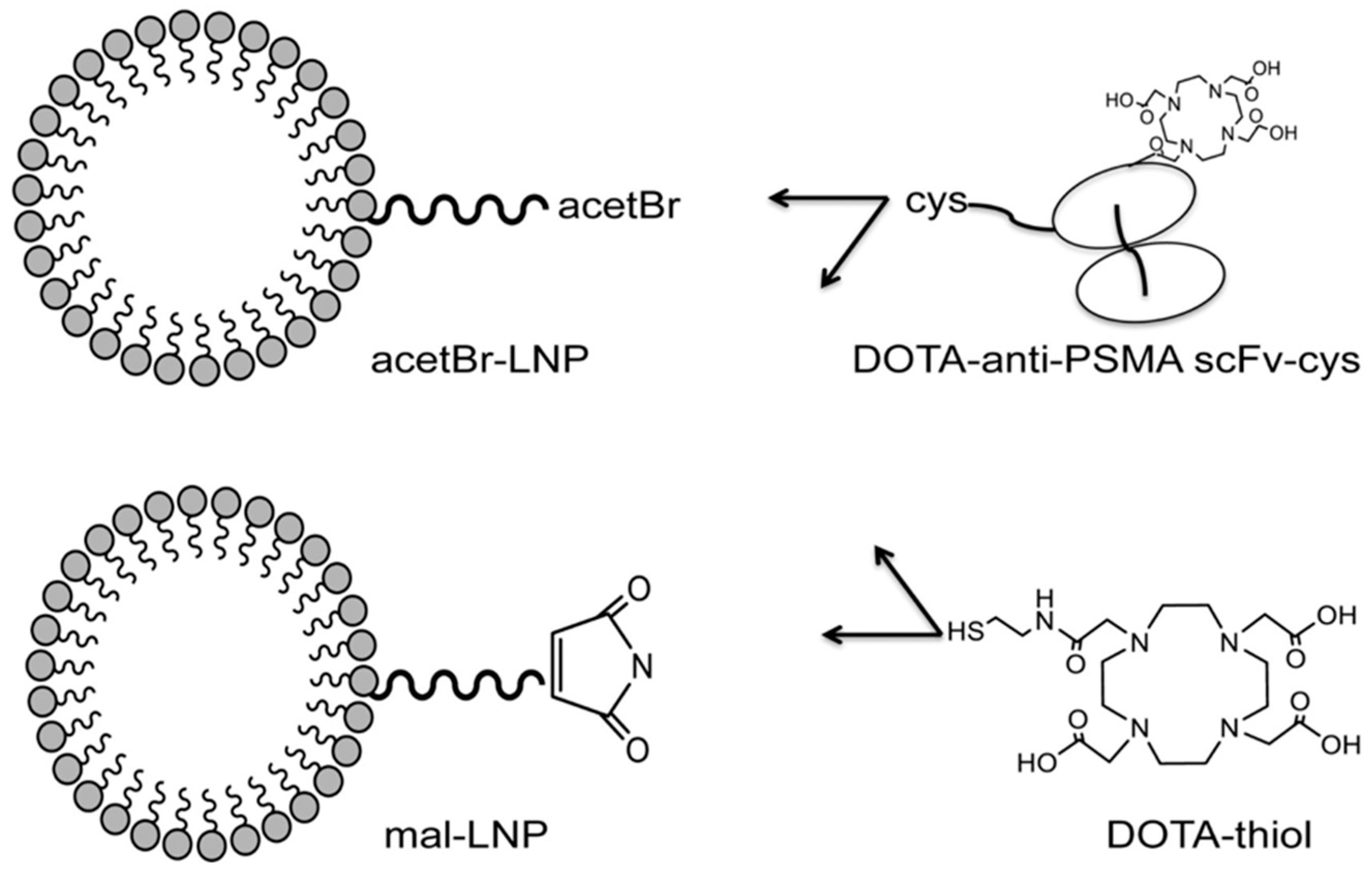
5.4. PSMA-Targeted Nanoplex
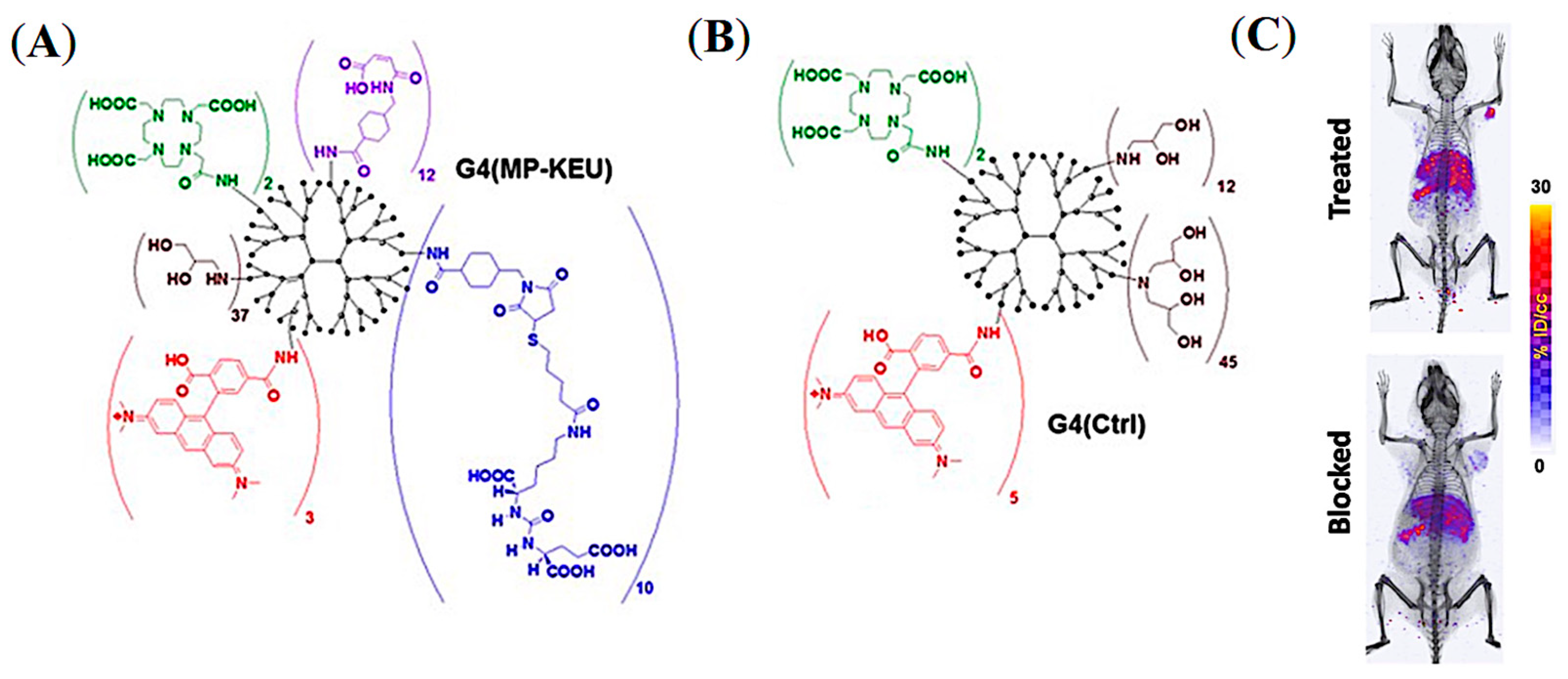
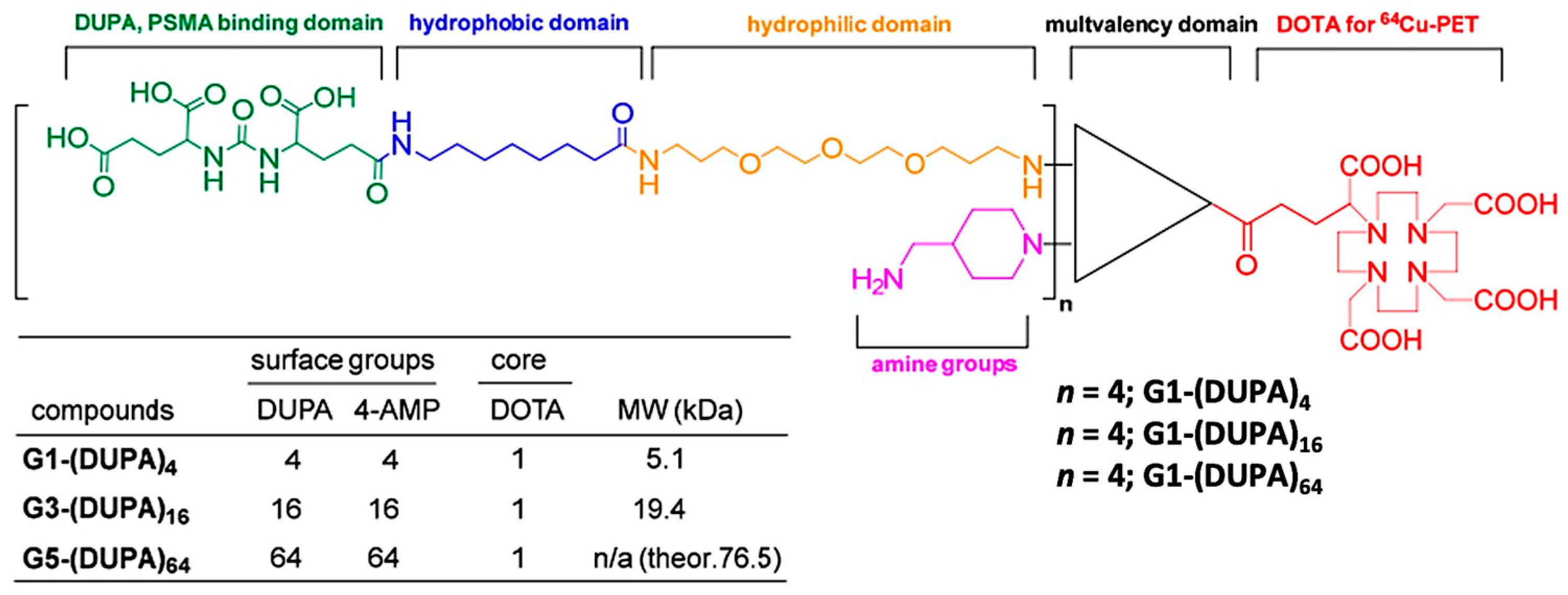
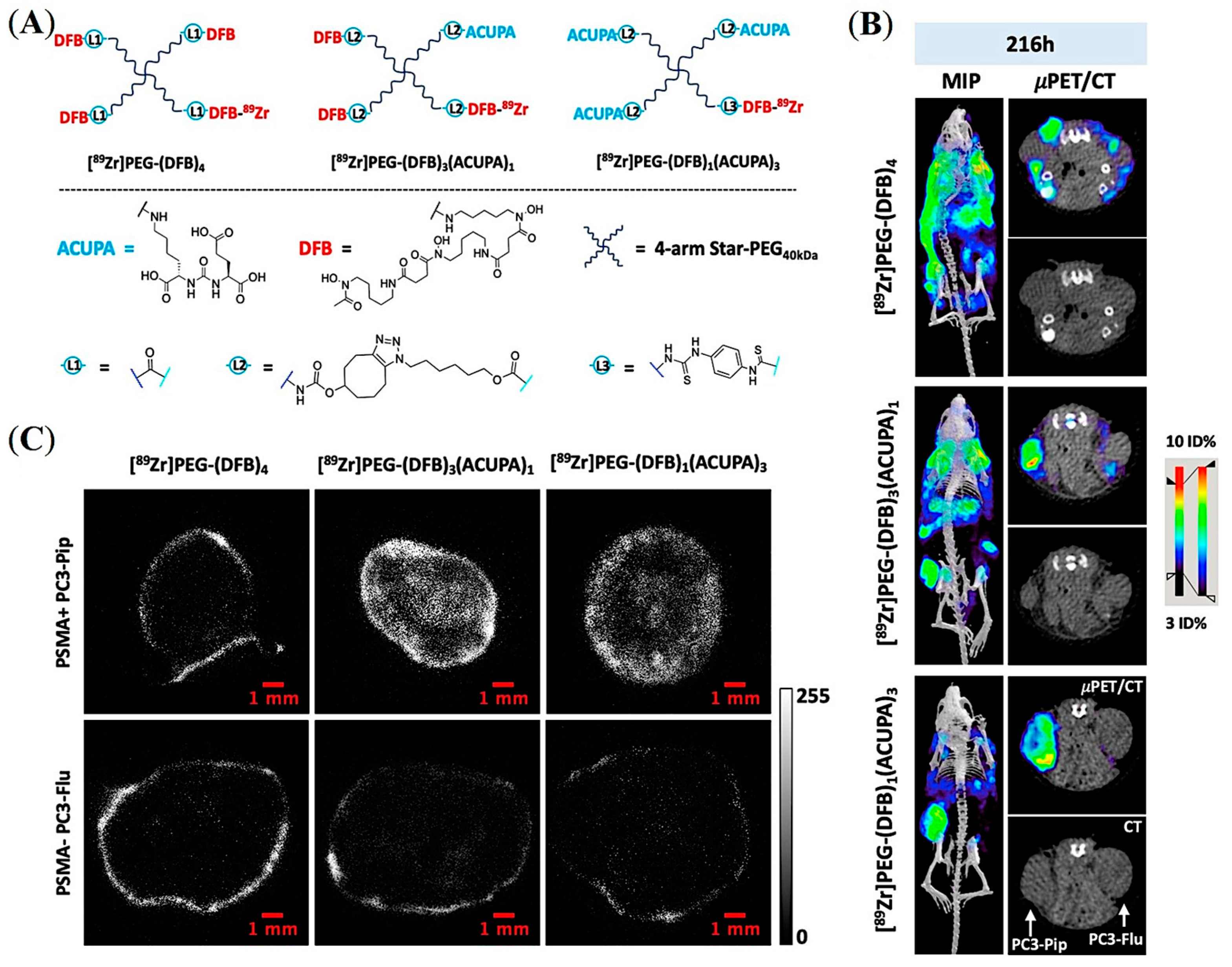
5.5. PSMA-Targeted Multivalent Dendrimers and starPEG Nanocarriers
6. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACUPA | ((S)-2-(3-((S)-5-amino-1- carboxypentyl) ureido) pentanedioic acid |
| BNCT | boron neutron capture therapy |
| BSB | binding site barrier |
| Chk | choline kinase |
| DFB | deferoxamine B |
| DUPA | 2-[3-(1,3-dicarboxypropyl)-ureido]pentanedioic acid |
| EPR | enhanced permeability and retention |
| FDA | Food and Drug Administration |
| GCPII | glutamate carboxypeptidase II |
| GRPR | gastrin-releasing peptide receptors |
| NPs | nanoparticles |
| PCa | prostate cancer |
| PEI | polyethyleneimine |
| PEG | polyethylene glycol |
| PET | positron emission tomography |
| PLA | polylactic acid |
| PLGA | poly(D,L-lactide-co-glycolide) |
| PSMA | prostate specific membrane antigen |
| SPECT | single-photon emission computerized tomography |
| TFA | trifluoroacetic acid |
References
- Mitchell, M.; Billingsley, M.; Haley, R.; Wechsler, M.; Peppas, N.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, B.; Hojjat-Farsangi, M.; Mohammadi, H.; Anvari, E.; Ghalamfarsa, G.; Yousefi, M.; Jadidi-Niaragh, F. Nanoparticles and targeted drug delivery in cancer therapy. Immunol. Lett. 2017, 190, 64–83. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Senter, P.; Steinhagen, H. Drug Delivery in Oncology: From Basic Research to Cancer Therapy, Vols 1–3; Wiley: Hoboken, NJ, USA, 2012; pp. 1–1689. [Google Scholar]
- Chauhan, V.; Stylianopoulos, T.; Martin, J.; Popovic, Z.; Chen, O.; Kamoun, W.; Bawendi, M.; Fukumura, D.; Jain, R. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef]
- Lim, J.; Guan, B.; Nham, K.; Hao, G.; Sun, X.; Simanek, E. Tumor Uptake of Triazine Dendrimers Decorated with Four, Sixteen, and Sixty-Four PSMA-Targeted Ligands: Passive versus Active Tumor Targeting. Biomolecules 2019, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Goos, J.; Cho, A.; Carter, L.; Dilling, T.; Davydova, M.; Mandleywala, K.; Puttick, S.; Gupta, A.; Price, W.; Quinn, J.; et al. Delivery of polymeric nanostars for molecular imaging and endoradiotherapy through the enhanced permeability and retention (EPR) effect. Theranostics 2020, 10, 567–584. [Google Scholar] [CrossRef]
- Heneweer, C.; Holland, J.; Divilov, V.; Carlin, S.; Lewis, J. Magnitude of Enhanced Permeability and Retention Effect in Tumors with Different Phenotypes: Zr-89-Albumin as a Model System. J. Nucl. Med. 2011, 52, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, Z.; Liu, X.; Huang, Y.; Hou, J.; Zhang, W.; Ding, D. Advances in Prostate-Specific Membrane Antigen (PSMA)-Targeted Phototheranostics of Prostate Cancer. Small Struct. 2022, 3, 2200036. [Google Scholar] [CrossRef]
- Zhou, J.; Neale, J.; Pomper, M.; Kozikowski, A. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat. Rev. Drug Discov. 2005, 4, 1015–1026. [Google Scholar] [CrossRef]
- Carter, R.; Feldman, A.; Coyle, J. Prostate-specific membrane antigen is a hydrolase with substrate and pharmacologic characteristics of a neuropeptidase. Proc. Natl. Acad. Sci. USA 1996, 93, 749–753. [Google Scholar] [CrossRef]
- Davis, M.; Bennett, M.; Thomas, L.; Bjorkman, P. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [Google Scholar] [CrossRef]
- Filippi, L.; Bagni, O.; Nervi, C. Aptamer-based technology for radionuclide targeted imaging and therapy: A promising weapon against cancer. Expert Rev. Med. Devices 2020, 17, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Sah, B.; Burger, I.; Schibli, R.; Friebe, M.; Dinkelborg, L.; Graham, K.; Borkowski, S.; Bacher-Stier, C.; Valencia, R.; Srinivasan, A.; et al. Dosimetry and First Clinical Evaluation of the New F-18-Radiolabeled Bombesin Analogue BAY 864367 in Patients with Prostate Cancer. J. Nucl. Med. 2015, 56, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.; Zettlitz, K.; Tavare, R.; Kobayashi, N.; Reiter, R.; Wu, A. Dual-Modality ImmunoPET/Fluorescence Imaging of Prostate Cancer with an Anti-PSCA Cys-Minibody. Theranostics 2018, 8, 5903–5914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Chopra, S.; Trepka, K.; Wang, Y.; Sakhamuri, S.; Hooshdaran, N.; Kim, H.; Zhou, J.; Lim, S.; Leung, K.; et al. CUB Domain-Containing Protein 1 (CDCP1) Is a Target for Radioligand Therapy in Castration-Resistant Prostate Cancer, including PSMA Null Disease. Clin. Cancer Res. 2022, 28, 3066–3075. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Hua, J.; Su, Y.; Beckford-Vera, D.; Zhao, W.; Jayaraman, M.; Huynh, T.; Zhao, N.; Wang, Y.; et al. Molecular Imaging of Prostate Cancer Targeting CD46 Using ImmunoPET. Clin. Cancer Res. 2021, 27, 1305–1315. [Google Scholar] [CrossRef]
- Timmermand, O.; Elgqvist, J.; Beattie, K.; Orbom, A.; Larsson, E.; Eriksson, S.; Thorek, D.; Beattie, B.; Tran, T.; Ulmert, D.; et al. Preclinical efficacy of hK2 targeted [Lu-177] hu11B6 for prostate cancer theranostics. Theranostics 2019, 9, 2129–2142. [Google Scholar] [CrossRef]
- Korsen, J.; Kalidindi, T.; Khitrov, S.; Samuels, Z.; Chakraborty, G.; Gutierrez, J.; Poirier, J.; Rudin, C.; Chen, Y.; Morris, M.; et al. Molecular Imaging of Neuroendocrine Prostate Cancer by Targeting Delta-Like Ligand 3. J. Nucl. Med. 2022, 63, 1401–1407. [Google Scholar] [CrossRef]
- Korsen, J.; Gutierrez, J.; Tully, K.; Carter, L.; Samuels, Z.; Khitrov, S.; Poirier, J.; Rudin, C.; Chen, Y.; Morris, M.; et al. Delta-like ligand 3-targeted radioimmunotherapy for neuroendocrine prostate cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2203820119. [Google Scholar] [CrossRef]
- Chou, J.; Egusa, E.A.; Wang, S.; Badura, M.L.; Lee, F.; Bidkar, A.P.; Zhu, J.; Shenoy, T.; Trepka, K.; Robinson, T.M.; et al. Immunotherapeutic Targeting and PET Imaging of DLL3 in Small-Cell Neuroendocrine Prostate Cancer. Cancer Res. 2023, 83, 301–315. [Google Scholar] [CrossRef]
- van Rij, C.; Frielink, C.; Goldenberg, D.; Sharkey, R.; Franssen, G.; Lutje, S.; McBride, W.; Oyen, W.; Boerman, O. Pretargeted ImmunoPET of Prostate Cancer with an Anti-TROP-2 x Anti-HSG Bispecific Antibody in Mice with PC3 Xenografts. Mol. Imaging Biol. 2015, 17, 94–101. [Google Scholar] [CrossRef]
- Filippi, L.; Evangelista, L.; Sathekge, M.; Schillaci, O. ImmunoPET for prostate cancer in the PSMA era: Do we need other targets? Clin. Transl. Imaging 2022, 10, 587–596. [Google Scholar] [CrossRef]
- Virgolini, I.; Decristoforo, C.; Haug, A.; Fanti, S.; Uprimny, C. Current status of theranostics in prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 471–495. [Google Scholar] [CrossRef]
- Pastorino, S.; Riondato, M.; Uccelli, L.; Giovacchini, G.; Giovannini, E.; Duce, V.; Ciarmiello, A. Toward the Discovery and Development of PSMA Targeted Inhibitors for Nuclear Medicine Applications. Curr. Radiopharm. 2020, 13, 63–79. [Google Scholar] [CrossRef]
- Fendler, W.; Calais, J.; Eiber, M.; Flavell, R.; Mishoe, A.; Feng, F.; Nguyen, H.; Reiter, R.; Rettig, M.; Okamoto, S.; et al. Assessment of Ga-68-PSMA-11 PET Accuracy in Localizing Recurrent Prostate Cancer: A Prospective Single-Arm Clinical Trial. Jama Oncol. 2019, 5, 856–863. [Google Scholar] [CrossRef]
- Morris, M.; Rowe, S.; Gorin, M.; Saperstein, L.; Pouliot, F.; Josephson, D.; Wong, J.; Pantel, A.; Cho, S.; Gage, K.; et al. Diagnostic Performance of F-18-DCFPyL-PET/CT in Men with Biochemically Recurrent Prostate Cancer: Results from the CONDOR Phase III, Multicenter Study. Clin. Cancer Res. 2021, 27, 3674–3682. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.; Chi, K.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.; Nordquist, L.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.; Cardinale, J.; Aulsebrook, M.; Gasser, G.; Mindt, T. An Overview of PET Radiochemistry, Part 2: Radiometals. J. Nucl. Med. 2018, 59, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Price, E.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Crisan, G.; Moldovean-Cioroianu, N.; Timaru, D.; Andries, G.; Cainap, C.; Chis, V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. FDA Approves Pluvicto/Locametz for Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2022, 63, 13N. [Google Scholar]
- Filippi, L.; Chiaravalloti, A.; Schillaci, O.; Bagni, O. The potential of PSMA-targeted alpha therapy in the management of prostate cancer. Expert Rev. Anticancer Ther. 2020, 20, 823–829. [Google Scholar] [CrossRef]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer-Chemotherapy—Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Vakoc, B.; Lanning, R.; Tyrrell, J.; Padera, T.; Bartlett, L.; Stylianopoulos, T.; Munn, L.; Tearney, G.; Fukumura, D.; Jain, R.; et al. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat. Med. 2009, 15, 1219–1223. [Google Scholar] [CrossRef]
- Miao, L.; Newby, J.; Lin, C.; Zhang, L.; Xu, F.; Kim, W.; Forest, M.; Lai, S.; Milowsky, M.; Wobker, S.; et al. The Binding Site Barrier Elicited by Tumor Associated Fibroblasts Interferes Disposition of Nanoparticles in Stroma-Vessel Type Tumors. Acs Nano 2016, 10, 9243–9258. [Google Scholar] [CrossRef] [PubMed]
- Regmi, S.; Sathianathen, N.; Stout, T.; Konety, B. MRI/PET Imaging in elevated PSA and localized prostate cancer: A narrative review. Transl. Androl. Urol. 2021, 10, 3117–3129. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.; Lawrentschuk, N.; Francis, R.; Tang, C.; Vela, I.; Thomas, P.; Rutherford, N.; Martin, J.; Frydenberg, M.; Shakher, R.; et al. Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): A prospective, randomised, multicentre study. Lancet 2020, 395, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Yang, B.; Kim, Y.; Hong, M.; Lee, Y.; Lee, D.; Chung, J.; Jeong, J. Development of a complementary PET/MR dual-modal imaging probe for targeting prostate-specific membrane antigen (PSMA). Nanomed.-Nanotechnol. Biol. Med. 2016, 12, 871–879. [Google Scholar] [CrossRef]
- Liolios, C.; Koutsikou, T.; Salvanou, E.; Kapiris, F.; Machairas, E.; Stampolaki, M.; Kolocouris, A.; Efthimiadou, E.; Bouziotis, P. Synthesis and in vitro proof-of-concept studies on bispecific iron oxide magnetic nanoparticles targeting PSMA and GRP receptors for PET/MR imaging of prostate cancer. Int. J. Pharm. 2022, 624, 122008. [Google Scholar] [CrossRef]
- Azad, B.; Banerjee, S.; Pullambhatla, M.; Lacerda, S.; Foss, C.; Wang, Y.; Ivkov, R.; Pomper, M. Evaluation of a PSMA-targeted BNF nanoparticle construct. Nanoscale 2015, 7, 4432–4442. [Google Scholar] [CrossRef] [PubMed]
- Ancira-Cortez, A.; Ferro-Flores, G.; Jimenez-Mancilla, N.; Morales-Avila, E.; Trujillo-Benitez, D.; Ocampo-Garcia, B.; Santos-Cuevas, C.; Escudero-Castellanos, A.; Luna-Gutierrez, M. Synthesis, chemical and biochemical characterization of Lu2O3-iPSMA nanoparticles activated by neutron irradiation. Mater. Sci. Eng. C-Mater. Biol. Appl. 2020, 117, 111335. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Jimenez, T.; Cruz-Nova, P.; Ancira-Cortez, A.; Gibbens-Bandala, B.; Lara-Almazan, N.; Ocampo-Garcia, B.; Santos-Cuevas, C.; Morales-Avila, E.; Ferro-Flores, G. Toxicity Assessment of [Lu-177] Lu-iFAP/iPSMA Nanoparticles Prepared under GMP-Compliant Radiopharmaceutical Processes. Nanomaterials 2022, 12, 4181. [Google Scholar] [CrossRef] [PubMed]
- Czerwinska, M.; Fracasso, G.; Pruszynski, M.; Bilewicz, A.; Kruszewski, M.; Majkowska-Pilip, A.; Lankoff, A. Design and Evaluation of 223Ra-Labeled and Anti-PSMA Targeted NaA Nanozeolites for Prostate Cancer Therapy-Part I. Materials 2020, 13, 3875. [Google Scholar] [CrossRef]
- Lankoff, A.; Czerwinska, M.; Walczak, R.; Karczmarczyk, U.; Tomczyk, K.; Brzoska, K.; Fracasso, G.; Garnuszek, P.; Mikolajczak, R.; Kruszewski, M. Design and Evaluation of Ra-223-Labeled and Anti-PSMA Targeted NaA Nanozeolites for Prostate Cancer Therapy-Part II. Toxicity, Pharmacokinetics and Biodistribution. Int. J. Mol. Sci. 2021, 22, 5702. [Google Scholar] [CrossRef]
- Hrkach, J.; Von Hoff, D.; Ali, M.; Andrianova, E.; Auer, J.; Campbell, T.; De Witt, D.; Figa, M.; Figueiredo, M.; Horhota, A.; et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci. Transl. Med. 2012, 4, 128–139. [Google Scholar] [CrossRef]
- Afsharzadeh, M.; Hashemi, M.; Babaei, M.; Abnous, K.; Ramezani, M. PEG-PLA nanoparticles decorated with small-molecule PSMA ligand for targeted delivery of galbanic acid and docetaxel to prostate cancer cells. J. Cell. Physiol. 2020, 235, 4618–4630. [Google Scholar] [CrossRef]
- Cheng, J.; Teply, B.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.; Levy-Nissenbaum, E.; Radovic-Moreno, A.; Langer, R.; Farokhzad, O. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef]
- Gu, F.; Zhang, L.; Teply, B.; Mann, N.; Wang, A.; Radovic-Moreno, A.; Langer, R.; Farokhzad, O. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc. Natl. Acad. Sci. USA 2008, 105, 2586–2591. [Google Scholar] [CrossRef]
- Banerjee, S.; Foss, C.; Horhota, A.; Pullambhatla, M.; McDonnell, K.; Zale, S.; Pomper, M. In-111- and IRDye800CW-Labeled PLA-PEG Nanoparticle for Imaging Prostate-Specific Membrane Antigen-Expressing Tissues. Biomacromolecules 2017, 18, 201–209. [Google Scholar] [CrossRef]
- Hu, K.; Yang, Z.; Zhang, L.; Xie, L.; Wang, L.; Xu, H.; Josephson, L.; Liang, S.; Zhang, M. Boron agents for neutron capture therapy. Coord. Chem. Rev. 2020, 405, 213139. [Google Scholar] [CrossRef]
- Barth, R.; Mi, P.; Yang, W. Boron delivery agents for neutron capture therapy of cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef] [PubMed]
- Xuan, S.; Vicente, M.d.G.H. Recent Advances in Boron Delivery Agents for Boron Neutron Capture Therapy (BNCT). In Boron-Based Compounds; Wiley: Hoboken, NJ, USA, 2018; pp. 298–342. [Google Scholar]
- Haapaniemi, A.; Kankaanranta, L.; Saat, R.; Koivunoro, H.; Saarilahti, K.; Makitie, A.; Atula, T.; Joensuu, H. Boron Neutron Capture Therapy in the Treatment of Recurrent Laryngeal Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Blaha, C.; Santos, R.; Huynh, T.; Hayes, T.; Beckford-Vera, D.; Blecha, J.; Hong, A.; Fogarty, M.; Hope, T.; et al. Synthesis and Initial Biological Evaluation of Boron-Containing Prostate-Specific Membrane Antigen Ligands for Treatment of Prostate Cancer Using Boron Neutron Capture Therapy. Mol. Pharm. 2019, 16, 3831–3841. [Google Scholar] [CrossRef]
- Meher, N.; Seo, K.; Wang, S.; Bidkar, A.; Fogarty, M.; Dhrona, S.; Huang, X.; Tang, R.; Blaha, C.; Evans, M.; et al. Synthesis and Preliminary Biological Assessment of Carborane-Loaded Theranostic Nanoparticles to Target Prostate-Specific Membrane Antigen. Acs Appl. Mater. Interfaces 2021, 13, 54739–54752. [Google Scholar] [CrossRef]
- Vera, D.; Fontaine, S.; VanBrocklin, H.; Hearn, B.; Reid, R.; Ashley, G.; Santi, D. PET Imaging of the EPR Effect in Tumor Xenografts Using Small 15 nm Diameter Polyethylene Glycols Labeled with Zirconium-89. Mol. Cancer Ther. 2020, 19, 673–679. [Google Scholar] [CrossRef]
- Chen, Z.; Tai, Z.; Gu, F.; Hu, C.; Zhu, Q.; Gao, S. Aptamer-mediated delivery of docetaxel to prostate cancer through polymeric nanoparticles for enhancement of antitumor efficacy. Eur. J. Pharm. Biopharm. 2016, 107, 130–141. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.; Sood, A.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef]
- Cheng, M.; Overchuk, M.; Rajora, M.; Lou, J.; Chen, Y.; Pomper, M.; Chen, J.; Zheng, G. Targeted Theranostic 111In/Lu-Nanotexaphyrin for SPECT Imaging and Photodynamic Therapy. Mol. Pharm. 2022, 19, 1803–1813. [Google Scholar] [CrossRef]
- Vaughan, L.; Glanzel, W.; Korch, C.; Capes-Davis, A. Widespread Use of Misidentified Cell Line KB (HeLa): Incorrect Attribution and Its Impact Revealed through Mining the Scientific Literature. Cancer Res. 2017, 77, 2784–2788. [Google Scholar] [CrossRef]
- Zhu, C.; Bandekar, A.; Sempkowski, M.; Banerjee, S.; Pomper, M.; Bruchertseifer, F.; Morgenstern, A.; Sofou, S. Nanoconjugation of PSMA-Targeting Ligands Enhances Perinuclear Localization and Improves Efficacy of Delivered Alpha-Particle Emitters against Tumor Endothelial Analogues. Mol. Cancer Ther. 2016, 15, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Li, L.; Chea, J.; Delgado, M.; Crow, D.; Poku, E.; Szpikowska, B.; Bowles, N.; Channappa, D.; Colcher, D.; et al. PET imaging of Cu-64-DOTA-scFv-anti-PSMA lipid nanoparticles (LNPs): Enhanced tumor targeting over anti-PSMA scFv or untargeted LNPs. Nucl. Med. Biol. 2017, 47, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Li, L.; Chea, J.; Delgado, M.; Poku, E.; Szpikowska, B.; Bowles, N.; Minnix, M.; Colcher, D.; Wong, J.; et al. Synthesis, Positron Emission Tomography Imaging, and Therapy of Diabody Targeted Drug Lipid Nanoparticles in a Prostate Cancer Murine Model. Cancer Biother. Radiopharm. 2017, 32, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Zuckerman, J.; Choi, C.; Seligson, D.; Tolcher, A.; Alabi, C.; Yen, Y.; Heidel, J.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef]
- Chen, Z.; Penet, M.; Nimmagadda, S.; Li, C.; Banerjee, S.; Winnard, P.; Artemov, D.; Glunde, K.; Pomper, M.; Bhujwalla, Z. PSMA-Targeted Theranostic Nanoplex for Prostate Cancer Therapy. Acs Nano 2012, 6, 7752–7762. [Google Scholar] [CrossRef]
- Lesniak, W.; Boinapally, S.; Banerjee, S.; Azad, B.; Foss, C.; Shen, C.; Lisok, A.; Wharram, B.; Nimmagadda, S.; Pomper, M. Evaluation of PSMA-Targeted PAMAM Dendrimer Nanoparticles in a Murine Model of Prostate Cancer. Mol. Pharm. 2019, 16, 2590–2604. [Google Scholar] [CrossRef]
- Meher, N.; Ashley, G.; Bidkar, A.; Dhrona, S.; Fong, C.; Fontaine, S.; Vera, D.; Wilson, D.; Seo, Y.; Santi, D.; et al. Prostate-Specific Membrane Antigen Targeted Deep Tumor Penetration of Polymer Nanocarriers. Acs Appl. Mater. Interfaces 2022, 14, 50569–50582. [Google Scholar] [CrossRef]
- Gratton, S.; Ropp, P.; Pohlhaus, P.; Luft, J.; Madden, V.; Napier, M.; DeSimone, J. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef]
- Safari, H.; Kelley, W.; Saito, E.; Kaczorowski, N.; Carethers, L.; Shea, L.; Eniola-Adefeso, O. Neutrophils preferentially phagocytose elongated particles-An opportunity for selective targeting in acute inflammatory diseases. Sci. Adv. 2020, 6, eaba1474. [Google Scholar] [CrossRef]
- Elci, S.; Jiang, Y.; Yan, B.; Kim, S.; Saha, K.; Moyano, D.; Tonga, G.; Jackson, L.; Rotello, V.; Vachet, R. Surface Charge Controls the Suborgan Biodistributions of Gold Nanoparticles. Acs Nano 2016, 10, 5536–5542. [Google Scholar] [CrossRef]
- Pijeira, M.; Viltres, H.; Kozempel, J.; Sakmar, M.; Vlk, M.; Ilem-Ozdemir, D.; Ekinci, M.; Srinivasan, S.; Rajabzadeh, A.; Ricci-Junior, E.; et al. Radiolabeled nanomaterials for biomedical applications: Radiopharmacy in the era of nanotechnology. Ejnmmi Radiopharm. Chem. 2022, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Androvic, L.; Woldrichova, L.; Jozefjakova, K.; Pechar, M.; Lynn, G.; Kankova, D.; Malinova, L.; Laga, R. Cyclotriphosphazene-Based Star Copolymers as Structurally Tunable Nanocarriers with Programmable Biodegradability. Macromolecules 2021, 54, 3139–3157. [Google Scholar] [CrossRef]
- Ernsting, M.; Murakami, M.; Roy, A.; Li, S. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Maecke, H.; Borner, A.; Weckesser, E.; Schoffski, P.; Oei, M.; Schumacher, J.; Henze, M.; Heppeler, A.; Meyer, G.; et al. Biokinetics and imaging with the somatostatin receptor PET radioligand Ga-68-DOTATOC: Preliminary data. Eur. J. Nucl. Med. 2001, 28, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Debela, D.; Muzazu, S.; Heraro, K.; Ndalama, M.; Mesele, B.; Haile, D.; Kitui, S.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. Sage Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chemical Structures | CA | CN | Radiometals |
|---|---|---|---|---|
| DFO; desferrioxamine B |  | O6 | 6 | 89Zr4+ |
| DTPA; diethylenetriaminepentaacetic acid |  | N3O5 | 8 | 111In3+, 177Lu3+, 86/90Y3+ |
| Pa Family; H2dedpa; 1,2-[[6-(carboxy)-pyridin-2-yl]- methylamino]ethane |  | N4O2 | 6 | 67/68Ga3+,111In3+, 177Lu3+ |
| HOPO; 3,4,3-(LI-1,2-HOPO) |  | O8 | 8 | 89Zr4+, 227Th4+ |
| NOTA; 1,4,7-triazacyclononane-1,4,7- triacetic acid |  | N3O3 | 6 | 177Lu3+, 64Cu2+, 67/68Ga3+, 86/90Y3+, 212/213Bi3+ |
| DOTA; 1,4,7,10-tetraazacyclododecane- 1,4,7,10-tetraacetic acid, maximum |  | N4O4 | 8 | 64Cu2+, 212Pb2+, 212/213Bi3+, 177Lu3+, 225Ac3+, 111In3+, 44/47Sc3+, 86/90Y3+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meher, N.; VanBrocklin, H.F.; Wilson, D.M.; Flavell, R.R. PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer. Pharmaceuticals 2023, 16, 315. https://doi.org/10.3390/ph16020315
Meher N, VanBrocklin HF, Wilson DM, Flavell RR. PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer. Pharmaceuticals. 2023; 16(2):315. https://doi.org/10.3390/ph16020315
Chicago/Turabian StyleMeher, Niranjan, Henry F. VanBrocklin, David M. Wilson, and Robert R. Flavell. 2023. "PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer" Pharmaceuticals 16, no. 2: 315. https://doi.org/10.3390/ph16020315
APA StyleMeher, N., VanBrocklin, H. F., Wilson, D. M., & Flavell, R. R. (2023). PSMA-Targeted Nanotheranostics for Imaging and Radiotherapy of Prostate Cancer. Pharmaceuticals, 16(2), 315. https://doi.org/10.3390/ph16020315