
3D-Printing of Capsule Devices as Compartmentalization Tools for Supported Reagents in the Search of Antiproliferative Isatins
 , , ,
, , ,  and
and 
Abstract
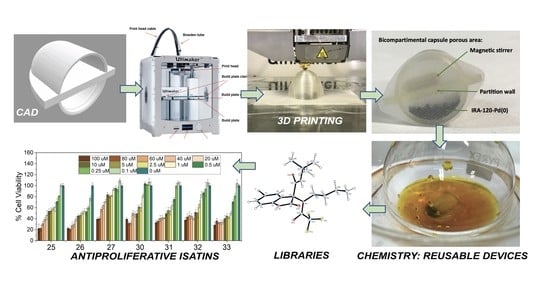
1. Introduction
2. Results and Discussion
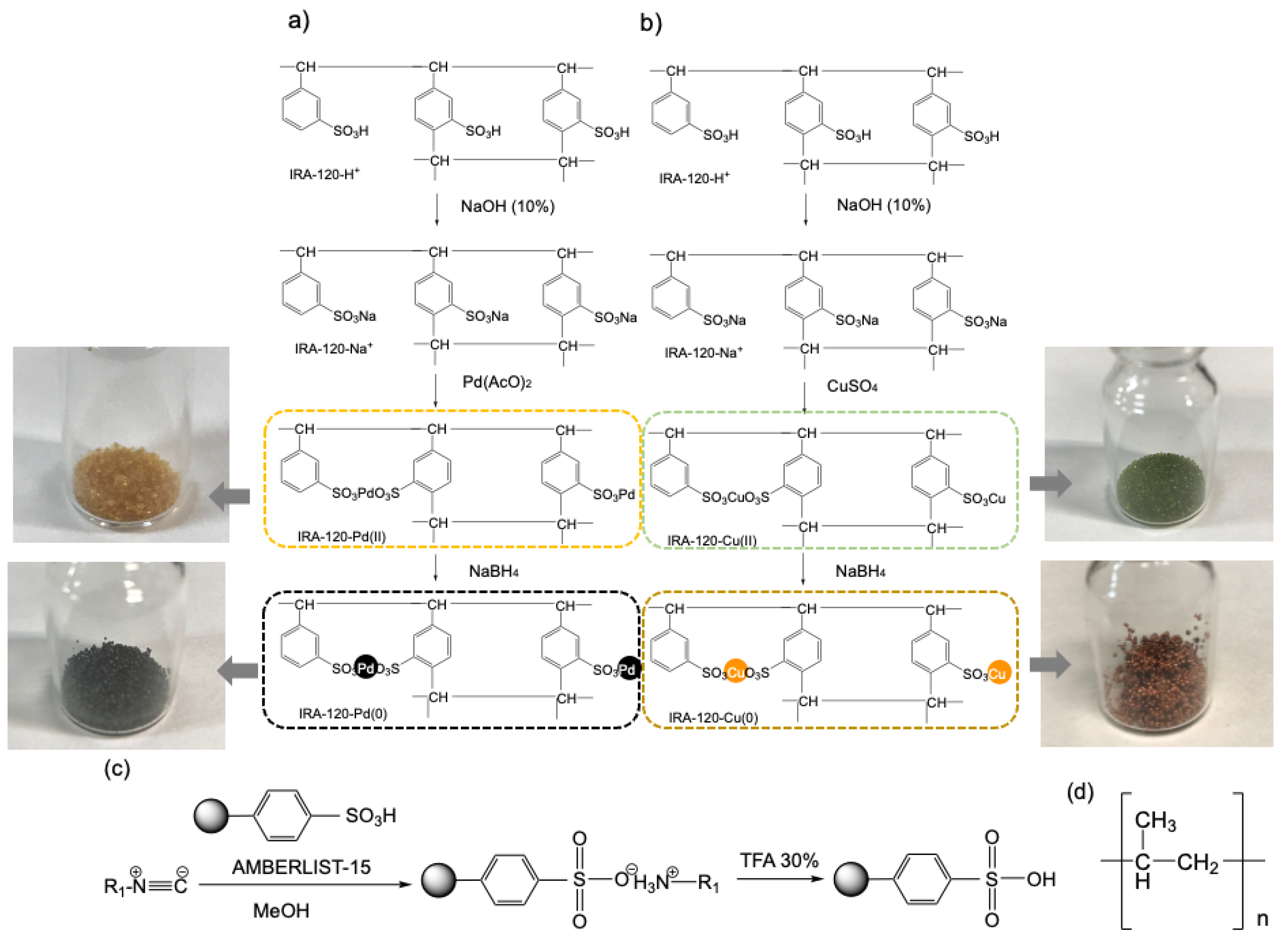
- Preparation of the catalytic systems to be compartmentalized (Scheme 1). Synthesis of efficient and robust resin–metal heterogeneous catalysts IRA-120-Cu and IRA-120-Pd (Section 2.1).
- Computer-aided design of novel capsule prototypes using the editing programs Tinkercad® and Cura® (Section 2.2).
- Manufacture of PP capsule prototypes using 3D printing (fused deposition modeling) with a semipermeable membrane, enabling the compartmentation of the metal–resin type catalysts of the first point, providing Capsule@IRA-120-Cu(0) and Capsule@IRA-120-Pd(0). In addition, enabling the compartmentation of the isocyanide scavenger Amberlyst-15 to create Capsule@Amberlyst-15 [53]. This supported reagent is a brown-gray granule reticular polystyrene based on ion exchange resins with strong acidic sulfonic groups (Scheme 1c). It is used as a strongly acidic heterogeneous acid catalysis and is suitable for nonaqueous catalysis.
- Study of the chemical stability of the capsule as a resin container, according to the reaction conditions (solvent, base, and temperature), to carry out the CuAAC or Heck reactions as well as the scavenging of isocyanide in Ugi reactions. (Section 2.2).
- Study of the behavior of the capsule–catalyst pairing. Application to heterogeneous catalysis as CuAAC reactions or PCCCRs (Heck) or as isocyanide scavenger (Ugi). Study of the recyclability of the resins and the capsules. Customization of capsule designs (of different sizes), aiming to carry out reactions at different scales (from milligrams to grams) (Section 2.3 and Section 2.4).
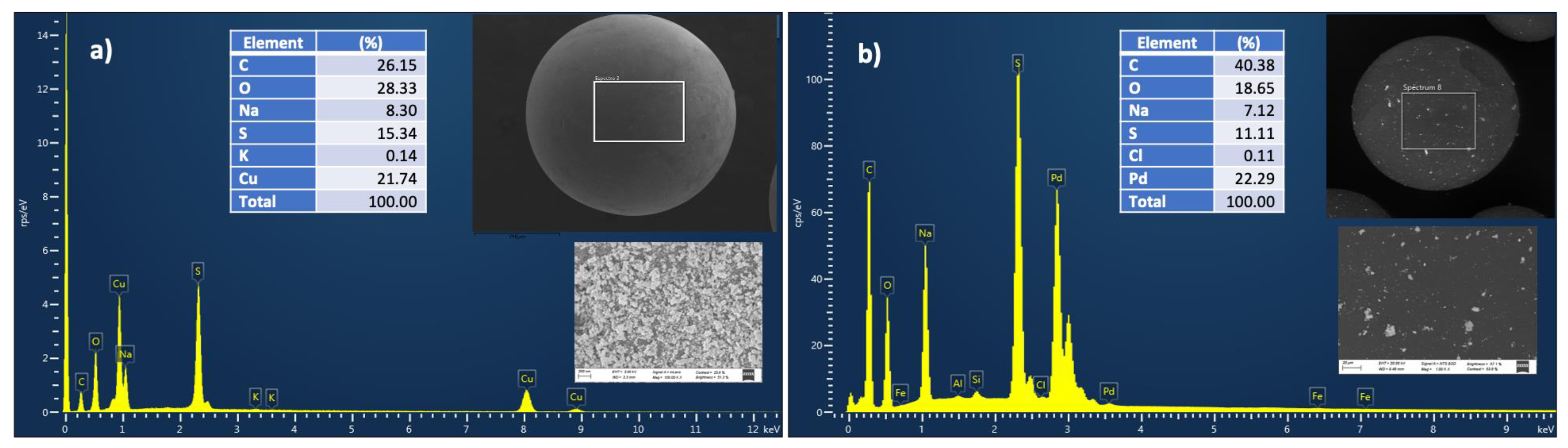
2.1. Synthesis of the Catalytic Materials IRA-120-Cu(0) and IRA-120-Pd(0)
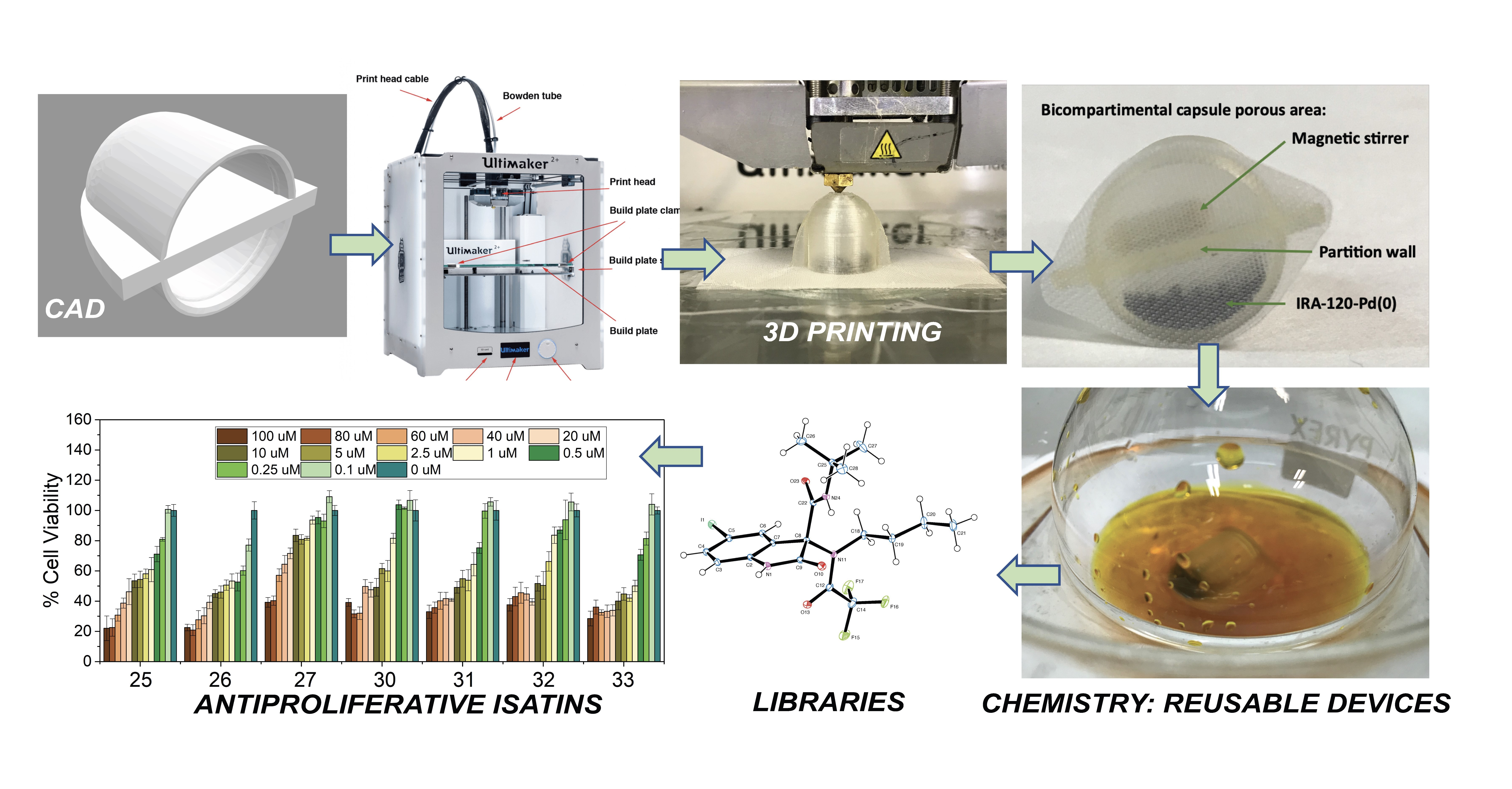
2.2. Fabrication of the Polypropylene Capsules and Compartmentation of Amberlyst 15 and the Catalytic Materials IRA-120-Cu(0) and IRA-120-Pd(0)
2.2.1. Computer-Aided Design (CAD)
2.2.2. 3D Printing
2.2.3. Chemical and Mechanical Resistance Tests under SPOS Conditions
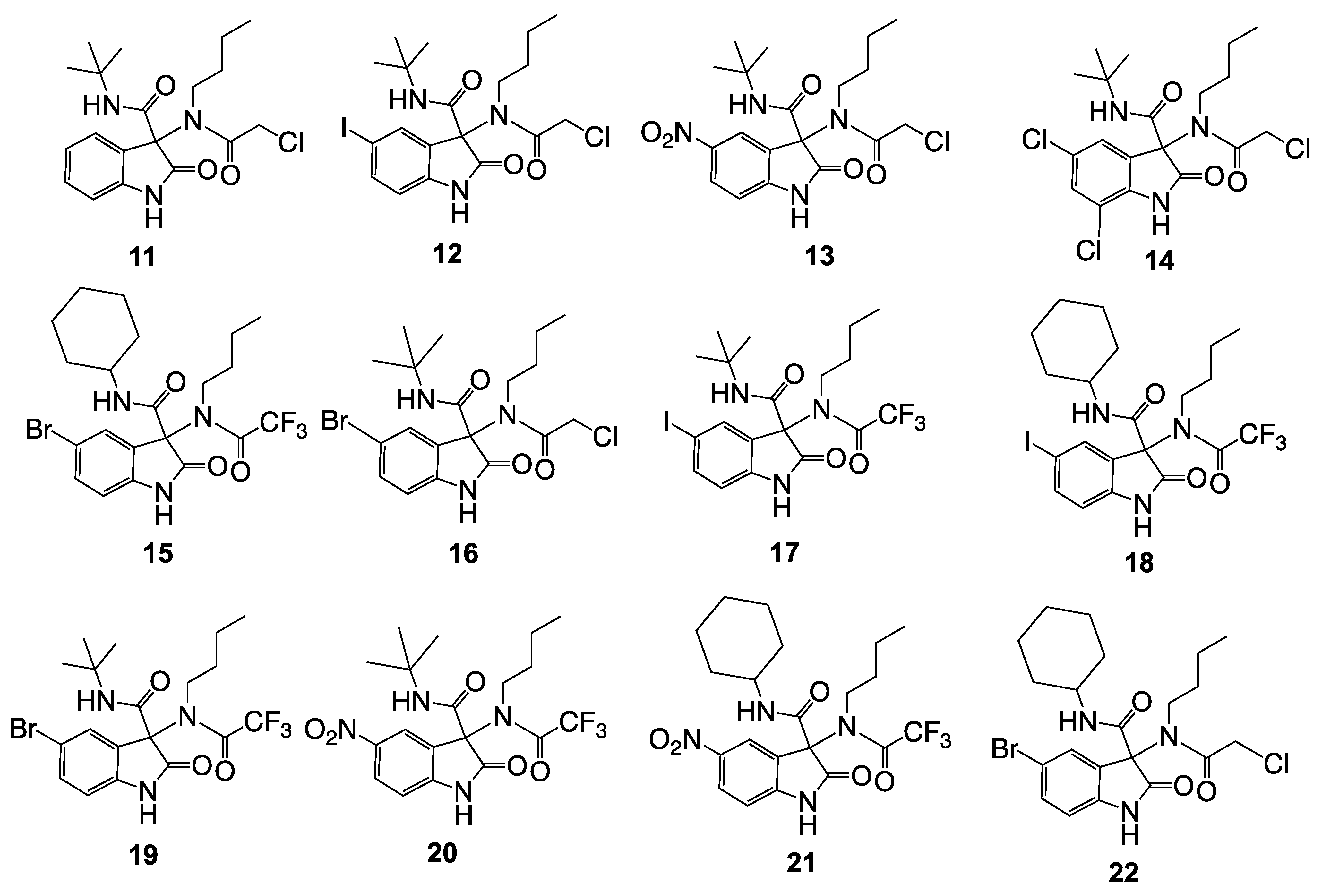
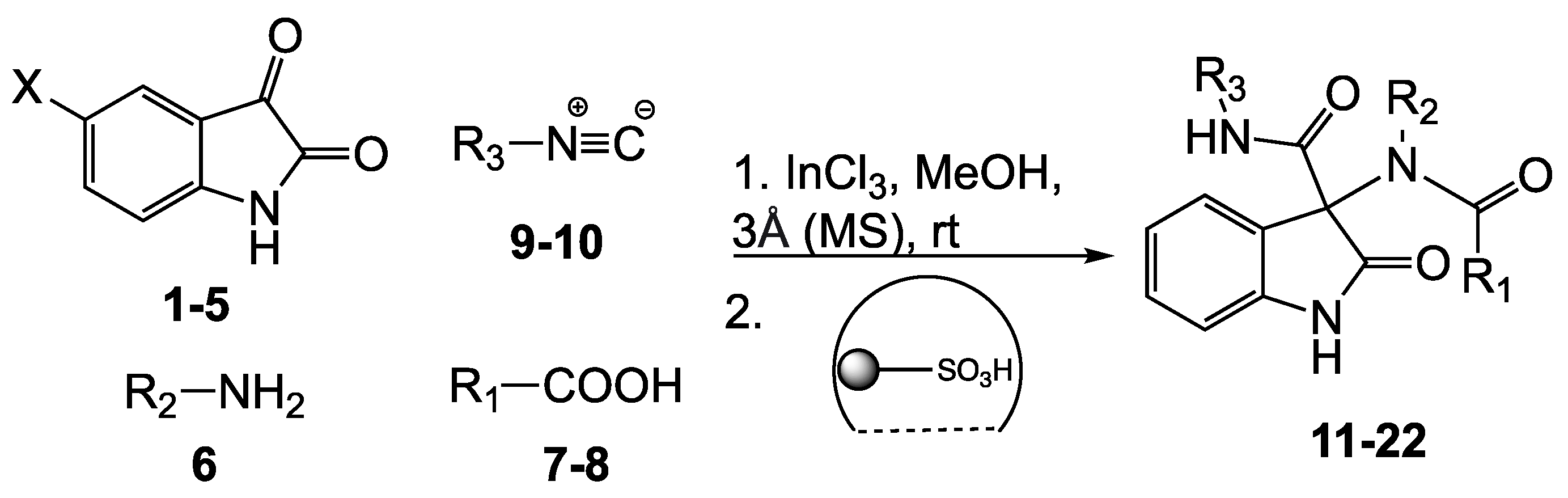
2.3. Preparation of Series 1 (Compounds 11–22): Ugi Reaction
2.4. Capsule@Amberlyst-15 Behavior as Scavenger of Isocyanides, Post-Ugi Isocyanide Removal, Recycling of the Capsule after Scavenging
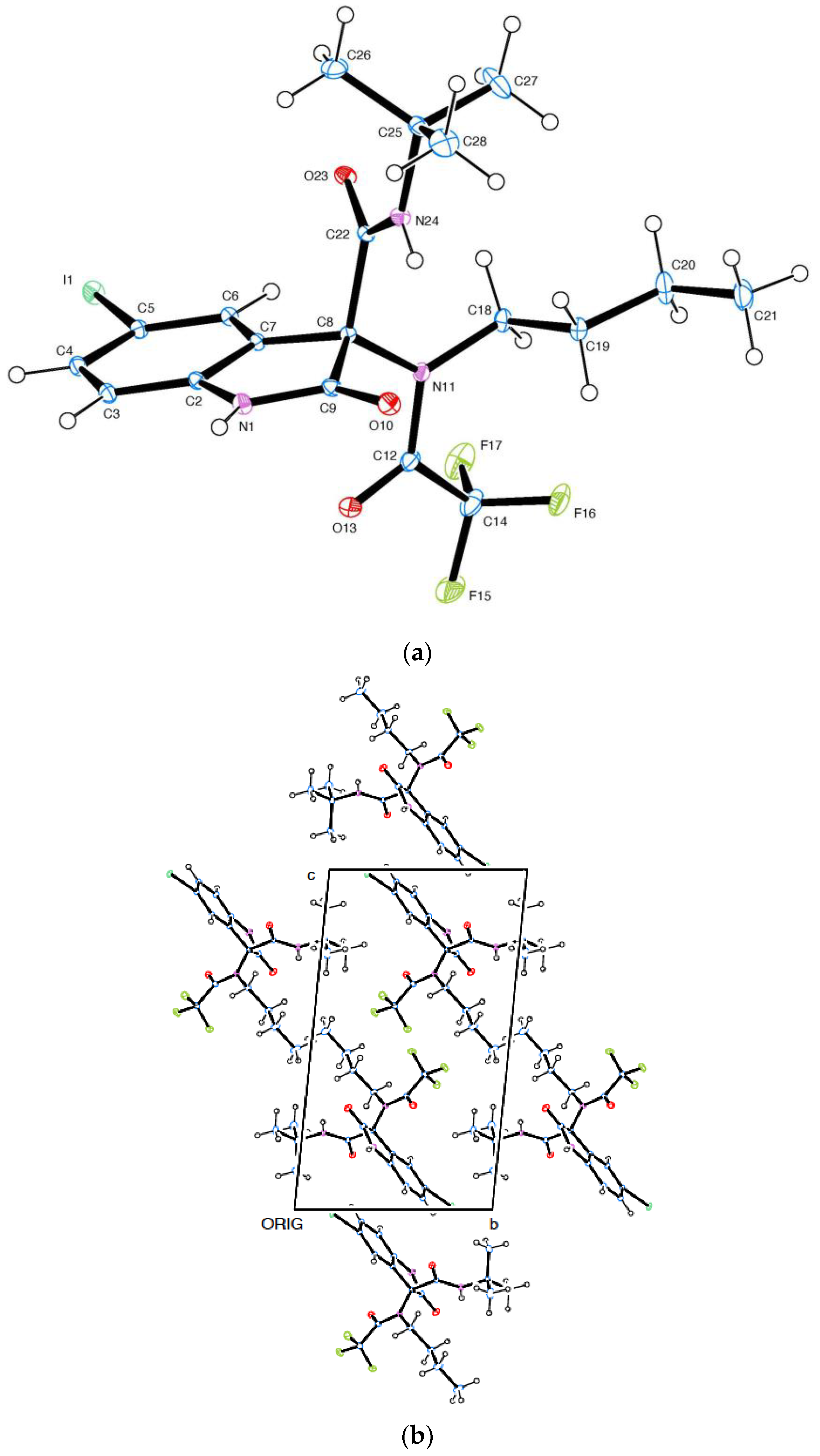
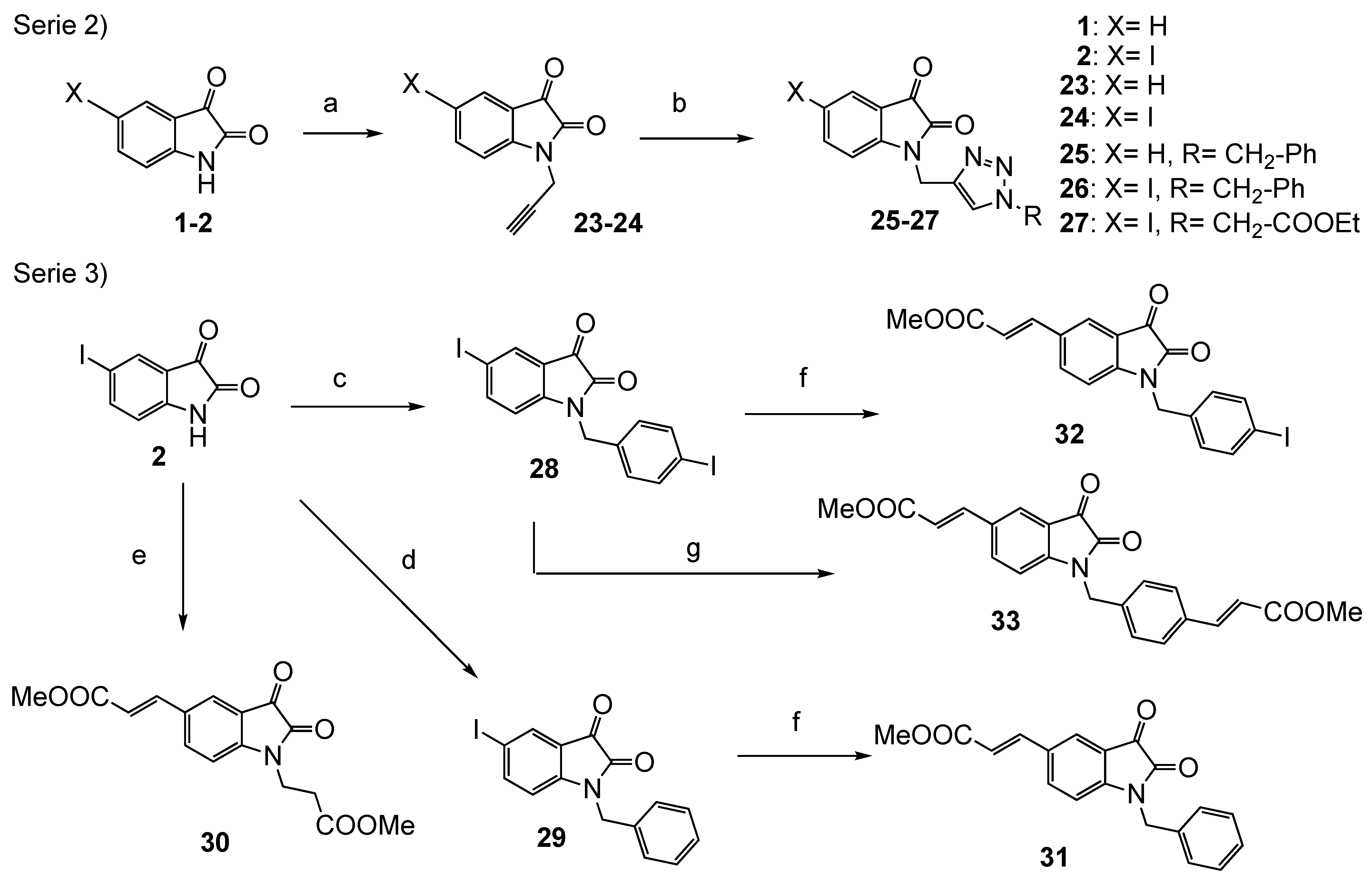
2.5. Preparation of Series 2 (Compounds 25–27) and 3 (Compounds 30–33)
2.6. Capsule Behavior and Reuse, Leaching Studies
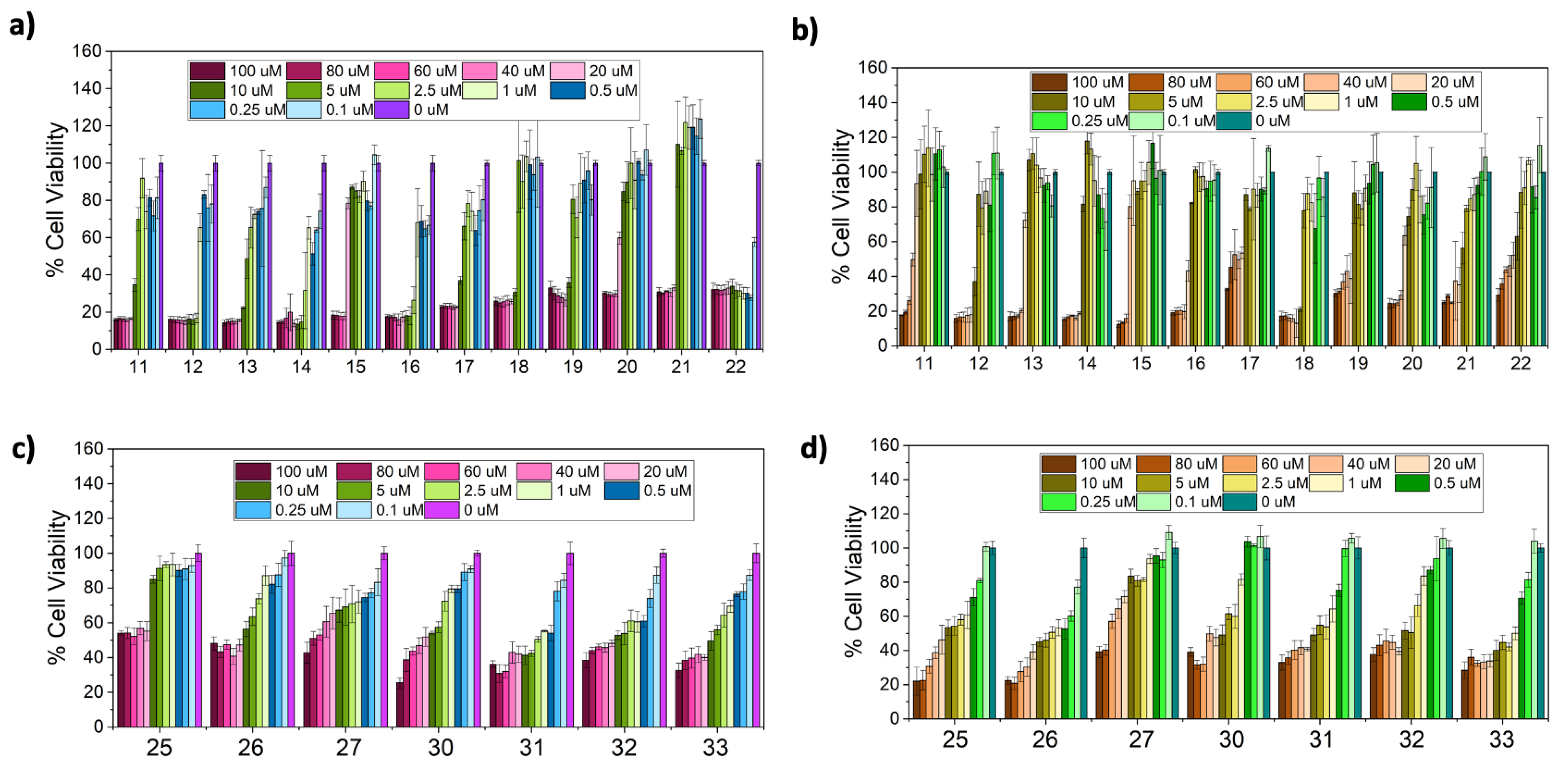
2.7. Biological Activity, Evaluation of Antiproliferative Activity
3. Materials and Methods
3.1. Chemistry: General Procedures for the Syntheses of the Catalytic Materials, Programs, Reagents, and Materials
3.2. Manufacture of Polypropylene Capsules by 3D Printing
3.3. Chemistry, General Procedures for the Syntheses of the Series 1, 2, and 3
3.4. General Procedure for the Syntheses of Series 1 (Compounds 11–22)
3.5. General Procedures for the Syntheses of the Intermediates for Series 2 and 3
3.6. General Procedures for the Syntheses of the Serie 2 (CuAAC Reaction)
3.7. Syntheses of the Series 3 (Heck Reaction)
3.8. Procedure for the Syntheses of Compound 30
3.9. Cell Cultures
3.10. In Vitro Cell Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bozkurt, Y.; Karayel, E. 3D printing technology; Methods, biomedical applications, future opportunities and trends. J. Mater. Res. Technol. 2021, 14, 1430–1450. [Google Scholar] [CrossRef]
- Paul, G.M.; Rezaienia, A.; Wen, P.; Condoor, S.; Parkar, N.; King, W.; Korakianitis, T. Medical applications for 3D printing: Recent developments. Mol. Med. 2018, 115, 75. [Google Scholar]
- Kitson, P.J.; Glatzel, S.; Chen, W.; Lin, C.G.; Song, Y.F.; Cronin, L. 3D printing of versatile reactionware for chemical synthesis. Nat. Protoc. 2016, 11, 920–936. [Google Scholar] [CrossRef] [PubMed]
- Symes, M.D.; Kitson, P.J.; Yan, J.; Richmond, C.J.; Cooper, G.J.T.; Bowman, R.W.; Vilbrandt, T.; Cronin, L. Integrated 3D-printed reactionware for chemical synthesis and analysis. Nat. Chem. 2012, 4, 349–354. [Google Scholar] [CrossRef]
- Kitson, P.J.; Marie, G.; Francoia, J.P.; Zalesskiy, S.S.; Sigerson, R.C.; Mathieson, J.S.; Cronin, L. Digitization of multistep organic synthesis in reactionware for on-demand pharmaceuticals. Science 2018, 359, 314–319. [Google Scholar] [CrossRef]
- Bubliauskas, A.; Blair, D.J.; Powell-Davies, H.; Kitson, P.J.; Burke, M.D.; Cronin, L. Digitizing chemical synthesis in 3D printed reactionware. Angew. Chem. Int. Ed. 2022, 61, e202116108. [Google Scholar] [CrossRef]
- Ludwig, T.; Boden, A.; Pipek, V. 3D printers as sociable technologies. ACM Trans. Comput. Interact. 2017, 24, 1–28. [Google Scholar] [CrossRef]
- Noorani, R. 3D Printing: Technology, Applications, and Selection, 1st ed.; CRC Press: Boca Raton, FL, USA, 2017; Chapter 1. [Google Scholar]
- Climent, M.J.; Corma, A.; Iborra, S. Heterogeneous catalysts for the one-pot synthesis of chemicals and fine chemicals. Chem. Rev. 2011, 111, 1072–1133. [Google Scholar] [CrossRef]
- Lucarelli, C.; Vaccari, A. Examples of heterogeneous catalytic processes for fine chemistry. Green Chem. 2011, 13, 1941–1949. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Marco, M. Preparation of polymer-supported ligands and metal complexes for use in catalysis. Chem. Rev. 2002, 102, 3217–3274. [Google Scholar] [CrossRef]
- Sun, Q.; Dai, Z.; Meng, X.; Xiao, F.S. Porous polymer catalysts with hierarchical structures. Chem. Soc. Rev. 2015, 44, 6018–6034. [Google Scholar] [CrossRef] [PubMed]
- Wassel, A.R.; El-Naggar, M.E.; Shoueir, K. Recent advances in polymer/metal/metal oxide hybrid nanostructures for catalytic applications: A review. J. Environ. Chem. Eng. 2020, 8, 104175. [Google Scholar] [CrossRef]
- Sarkar, S.; Guibal, E.; Quignard, F.; SenGupta, A.K. Polymer-supported metals and metal oxide nanoparticles: Synthesis, characterization, and applications. J. Nanoparticle Res. 2012, 14, 715. [Google Scholar] [CrossRef]
- Meldal, M.; Diness, F. Recent fascinating aspects of the CuAAC click reaction. Trends Chem. 2020, 2, 569–584. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Xu, M.; Wang, B.; Zhou, Z.; Wang, L. Sensitive detection of Au(III) using regenerative rhodamine B-functionalized chitosan nanoparticles. Sens. Actuators B Chem. 2016, 233, 361–368. [Google Scholar] [CrossRef]
- McGlacken, G.P.; Fairiamb, I.J.S. Palladium-catalysed cross-coupling and related processes: Some interesting observations that have been exploited in synthetic chemistry. Eur. J. Org. Chem. 2009, 2009, 4011–4029. [Google Scholar] [CrossRef]
- Saldan, I.; Semenyuk, Y.; Marchuk, I.; Reshetnyak, O. Chemical synthesis and application of palladium nanoparticles. J. Mater. Sci. 2015, 50, 2337–2354. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Len, C.; Fihri, A. Silica-supported palladium: Sustainable catalysts for cross-coupling reactions. Coord. Chem. Rev. 2009, 21–22, 2599–2626. [Google Scholar] [CrossRef]
- Mikhaylov, V.N.; Sorokoumov, V.N.; Liakhov, D.M.; Tskhovrebov, A.G.; Balova, I.A. Polystyrene-Supported Acyclic Diaminocarbene Palladium Complexes in Sonogashira Cross-Coupling: Stability vs. Catalytic Activity. Catalysts 2018, 8, 141. [Google Scholar] [CrossRef]
- Lu, J.; Dimroth, J.; Weck, M. Compartmentalization of incompatible catalytic transformations for tandem catalysis. J. Am. Chem. Soc. 2015, 137, 12984–12989. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, J.L.; Kang, M.K.; Gong, J.U.H.; Han, S.Y.; Shim, J.H.; Lim, S.S.; Kang, Y.H. Sage weed (Salvia Plebeia) extract antagonizes foam cell formation and promotes cholesterol efflux in murine macrophages. Int. J. Mol. Med. 2012, 30, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Shibasaki, M. Recent advances in cooperative bimetallic asymmetric catalysis: Dinuclear schiff base complexes. Chem. Commun. 2013, 50, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sonaglia, L.; Stacey, J.; Lautens, M. Multicomponent multicatalyst reactions (MC)2R: One-pot synthesis of 3,4-dihydroquinolinones. Org. Lett. 2013, 15, 2128–2131. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.J.R.W.; Marguet, M.; Marais, S.; Fraaije, M.W.; van Hest, J.C.M.; Lecommandoux, S. Cascade reactions in multicompartmentalized polymersomes. Angew. Chem. Int. Ed. Engl. 2014, 53, 146–150. [Google Scholar] [CrossRef]
- Qu, P.; Cleveland, J.W.; Ahmed, E.; Liu, F.; Dubrawski, S.; Jones, C.W.; Weck, M. Compartmentalisation of molecular catalysts for nonorthogonal tandem catalysis. Chem. Soc. Rev. 2022, 51, 57–70. [Google Scholar] [CrossRef]
- Houghten, R.A. General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. USA 1985, 82, 5131–5135. [Google Scholar] [CrossRef]
- Phan, N.T.S.; Gill, C.S.; Nguyen, J.V.; Zhang, Z.J.; Jones, C.W. Expanding the utility of one-pot multistep reaction networks through compartmentation and recovery of the catalyst. Angew. Chem. Int. Ed. 2006, 45, 2209–2212. [Google Scholar] [CrossRef]
- Cleveland, J.W.; Choi, J., II; Sekiya, R.S.; Cho, J.; Moon, H.J.; Jang, S.S.; Jones, C.W. Cooperativity in the aldol condensation using bifunctional mesoporous Silica-Poly(Styrene) MCM-41 organic/inorganic hybrid catalysts. ACS Appl. Mater. Interfaces 2022, 14, 11235–11247. [Google Scholar] [CrossRef]
- Helms, B.; Guillaudeu, S.J.; Xie, Y.; McMurdo, M.; Hawker, C.J.; Fréchet, J.M.J. One-pot reaction cascades using star polymers with core-confined catalysts. Angew. Chem. Int. Ed. 2005, 44, 6384–6387. [Google Scholar] [CrossRef]
- Díaz-Marta, A.S.; Tubío, C.R.; Carbajales, C.; Fernández, C.; Escalante, L.; Sotelo, E.; Guitián, F.; Barrio, V.L.; Gil, A.; Coelho, A. Three-dimensional printing in catalysis: Combining 3D heterogeneous copper and palladium catalysts for multicatalytic multicomponent reactions. ACS Catal. 2018, 8, 392–404. [Google Scholar] [CrossRef]
- Ciriminna, R.; Carà, P.D.; Sciortino, M.; Pagliaro, M. Catalysis with doped sol-gel silicates. Adv. Synth. Catal. 2011, 353, 677–687. [Google Scholar] [CrossRef]
- Cane, A.; Tournaire, M.C.; Barritault, D.; Crumeyrolle-Arias, M. The endogenous oxindoles 5-hydroxyoxindole and isatin are antiproliferative and proapoptotic. Biochem. Biophys. Res. Commun. 2000, 276, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Benkendorff, K.; Pyne, S.G.; Bremner, J.B. In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorg. Med. Chem. 2007, 15, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.S.; Singh, T.; Lakhan, R. Synthesis, C-13 NMR and anticonvulsant activity of new isatin-based spiroazetidi-nones. Indian J. Chem. 1997, 36, 951–954. [Google Scholar]
- Verma, M.; Pandeya, S.N.; Singh, K.N.; Stables, J.P. Anticonvulsant activity of schiff bases of isatin derivatives. Acta Pharm. 2004, 54, 49–56. [Google Scholar] [PubMed]
- Pandeya, S.N.; Yogeeshwari, P.; Sriram, D.; Nath, G. Synthesis, antibacterial and antifungal activities of N-Mannich bases of 3-(N2-Pyrimethaminylimino) isatin. Indian J. Pharm. Sci. 2002, 64, 209–212. [Google Scholar]
- Selvam, P.; Murugesh, N.; Chandramohan, M.; Debyser, Z.; Witvrouw, M. Design, synthesis and AntiHIV activity of novel isatine-sulphonamides. Indian J. Pharm. Sci. 2008, 70, 779–782. [Google Scholar] [CrossRef]
- Sridhar, S.K.; Ramesh, A. Synthesis and pharmacological activities of hydrazones, schiff and mannich bases of isatin derivatives. Biol. Pharm. Bull. 2001, 24, 1149–1152. [Google Scholar] [CrossRef]
- Ferraz de Paiva, R.E.; Vieira, E.G.; Rodrigues da Silva, D.; Wegermann, C.A.; Costa Ferreira, A.M. Anticancer compounds based on isatin-derivatives: Strategies to ameliorate selectivity and efficiency. Front. Mol. Biosci. 2021, 7, 627272. [Google Scholar] [CrossRef]
- Dhokne, P.; Sakla, A.P.; Shankaraiah, N. Structural insights of oxindole based kinase inhibitors as anticancer agents: Recent advances. Eur. J. Med. Chem. 2021, 216, 113334. [Google Scholar] [CrossRef]
- Brandão, P.; Marques, C.S.; Carreiro, E.P.; Pineiro, M.; Burke, A.J. Engaging isatins in Multicomponent Reactions (MCRs)—Easy access to structural diversity. Chem. Rec. 2021, 21, 924–1037. [Google Scholar] [CrossRef] [PubMed]
- Brandão, P.; Puerta, A.; Padrón, J.M.; Kuznetsov, M.L.; Burke, A.J.; Pineiro, M. Ugi adducts of isatin as promising antiproliferative agents with druglike properties. Asian J. Org. Chem. 2021, 10, 3434–3455. [Google Scholar] [CrossRef]
- Kudo, M.; Cheng, A.L.; Park, J.W.; Park, J.H.; Liang, P.C.; Hidaka, H.; Izumi, N.; Heo, J.; Lee, Y.J.; Sheen, I.S.; et al. Orantinib versus placebo combined with transcatheter arterial chemoembolisation in patients with unresectable hepatocellular carcinoma (ORIENTAL): A randomised, double-blind, placebo-controlled, multicentre, phase 3 study. Lancet Gastroenterol. Hepatol. 2018, 3, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef]
- Roth, G.J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, S.L.; Kaiser, R. Nintedanib: From discovery to the clinic. J. Med. Chem. 2015, 58, 1053–1063. [Google Scholar] [CrossRef]
- Han, K.; Zhou, Y.; Liu, F.; Guo, Q.; Wang, P.; Yang, Y.; Song, B.; Liu, W.; Yao, Q.; Teng, Y.; et al. Design, synthesis and in vitro cytotoxicity evaluation of 5-(2-Carboxyethenyl) isatin derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 591–594. [Google Scholar] [CrossRef]
- Teng, Y.O.; Zhao, H.Y.; Wang, J.; Liu, H.; Le Gao, M.; Zhou, Y.; Han, K.L.; Fan, Z.C.; Zhang, Y.M.; Sun, H.; et al. Synthesis and anti-cancer activity evaluation of 5-(2-Carboxyethenyl)-isatin derivatives. Eur. J. Med. Chem. 2016, 112, 145–156. [Google Scholar] [CrossRef]
- Chang, Y.; Yuan, Y.; Zhang, Q.; Rong, Y.; Yang, Y.; Chi, M.; Liu, Z.; Zhang, Y.; Yu, P.; Teng, Y. Effects of an isatin derivative on tumor cell migration and angiogenesis. RSC Adv. 2020, 10, 1191–1197. [Google Scholar] [CrossRef]
- Firoozpour, L.; Gao, L.; Moghimi, S.; Pasalar, P.; Davoodi, J.; Wang, M.W.; Rezaei, Z.; Dadgar, A.; Yahyavi, H.; Amanlou, M.; et al. Efficient synthesis, biological evaluation, and docking study of isatin based derivatives as caspase inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 1674–1684. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. The heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef]
- Fouad, M.A.; Abdel-Hamid, H.; Ayoup, M.S. Two decades of recent advances of Ugi reactions: Synthetic and pharmaceutical applications. RSC Adv. 2020, 10, 42644–42681. [Google Scholar] [CrossRef]
- Pal, R. Amberlyst-15 in organic synthesis amberlyst-15 in organic synthesis. Arch. Org. Chem. 2015, 2012, 570–609. [Google Scholar]
- Gautier, A. Ueber die einwirkung des chlorwasserstoffs u. a. auf das aethyl- und methylcyanür. Justus Liebigs Ann. Chem. 1867, 142, 289–294. [Google Scholar] [CrossRef]
- Hofmann, A.W. Ueber eine neue reihe von homologen der cyanwasserstoffsäure. Justus Liebigs Ann. Chem. 1867, 144, 114–120. [Google Scholar] [CrossRef]
- Azuaje, J.; Coelho, A.; El Maatougui, A.; Blanco, J.M.; Sotelo, E. Supported P-toluenesulfonic acid as a highly robust and eco-friendly isocyanide scavenger. ACS Comb. Sci. 2011, 13, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Sotelo-Perez, E.; Azuaje-Guerrero, J.A.; Coelho-Coton, A.J. Method for Capturing Isocyanides. WO2012049345A1, 19 April 2012. [Google Scholar]
- Biswas, K.; Ghosh, S.; Basu, B. Ion-exchange resins and polypeptide supported catalysts: A critical review. Curr. Green Chem. 2020, 7, 40–52. [Google Scholar]
- Silva, T.R.; de Oliveira, D.C.; Pal, T.; Domingos, J.B. The catalytic evaluation of bimetallic pd-based nanocatalysts supported on ion exchange resin in nitro and alkyne reduction reactions. New J. Chem. 2019, 43, 7083–7092. [Google Scholar] [CrossRef]
- Friedrich, K. Structure and Properties of Additive Manufactured Polymer Components; Woodhead Publishing: Cambridge, UK, 2020. [Google Scholar]
- Maddah, H.A. Polypropylene as a promising plastic: A review. Am. J. Polym. Sci. 2016, 6, 1–11. [Google Scholar]
- Available online: https://www.hmcpolymers.com/storage/download/hmc-pp-chemical-resistance.pdf (accessed on 9 February 2023).
- Tri, N.M.; Thanh, N.D.; Ha, L.N.; Anh, D.T.T.; Toan, V.N.; Giang, N.T.K. Study on synthesis of some substituted N-Propargyl isatins by propargylation reaction of corresponding isatins using potassium carbonate as base under ultrasound- and microwave-assisted conditions. Chem. Pap. 2021, 75, 4793–4801. [Google Scholar] [CrossRef]
- Alonso, F.; Moglie, Y.; Radivoy, G. Copper nanoparticles in click chemistry. Acc. Chem. Res. 2015, 48, 2516–2528. [Google Scholar] [CrossRef]
- Koraneekit, A.; Limpaiboon, T.; Sangka, A.; Boonsiri, P.; Daduang, S.; Daduang, J. Synergistic effects of cisplatin-caffeic acid induces apoptosis in human cervical cancer cells via the mitochondrial pathways. Oncol. Lett. 2018, 15, 7397–7402. [Google Scholar] [CrossRef] [PubMed]
- Robledo-Cadena, D.X.; Gallardo-Pérez, J.C.; Dávila-Borja, V.; Pacheco-Velázquez, S.C.; Belmont-Díaz, J.A.; Ralph, S.J.; Blanco-Carpintero, B.A.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S. Non-steroidal anti-inflammatory drugs increase cisplatin, paclitaxel, and doxorubicin efficacy against human cervix cancer cells. Pharmaceuticals 2020, 13, 463. [Google Scholar] [CrossRef] [PubMed]
- Bayoumi, H.M.; Alkhatib, M.H.; Al-Seeni, M.N. Carvacrol effect on toptecan cytotoxicity in various human cancer cells in vitro. Pharmacia 2021, 68, 353–363. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | X5 | Isocyanide | Acid | Time (h) | Yield (%) a,b,c |
|---|---|---|---|---|---|
| 11 | H | tert-butyl | ClCH2COOH | 48 | 65 |
| 12 | I | tert-butyl | ClCH2COOH | 72 | 28 |
| 13 | NO2 | tert-butyl | ClCH2COOH | 72 | 52 |
| 14 | Cl (Cl = X7) | tert-butyl | ClCH2COOH | 72 | 30 |
| 15 | Br | Cyclohexyl | CF3COOH | 48 | 58 |
| 16 | Br | tert-butyl | ClCH2COOH | 72 | 21 |
| 17 | I | tert-butyl | CF3COOH | 48 | 60 |
| 18 | I | Cyclohexyl | CF3COOH | 48 | 55 |
| 19 | Br | tert-butyl | CF3COOH | 48 | 58 |
| 20 | NO2 | tert-butyl | CF3COOH | 48 | 55 |
| 21 | NO2 | Cyclohexyl | CF3COOH | 48 | 57 |
| 22 | Br | Cyclohexyl | ClCH2COOH | 72 | 42 |
| Compound | Method | Catalyst | Time (h) | Yield (%) a |
|---|---|---|---|---|
| 25 | CuAAC b | IRA-120-Cu(0) | 10 | 95 |
| 26 | CuAAC b | IRA-120-Cu(0) | 10 | 90 |
| 27 | CuAAC b | IRA-120-Cu(0) | 10 | 93 |
| 30 | Aza-Michae/Heck c,d | IRA-120-Pd(0) | 24 | 85 |
| 31 | Heck c | IRA-120-Pd(0) | 24 | 88 |
| 32 | Heck c | IRA-120-Pd(0) | 24 | 60 e |
| 33 | Heck d | IRA-120-Pd(0) | 12 | 85 |
| Comp. | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 Balb (μM) | 7.3 ± 0.7 | 1.3 ± 0.2 | 4.8 ± 0.9 | 1.5 ± 0.3 | 28.3 ± 1.7 | 1.5 ± 0.8 | 7.2 ± 0.5 | 8.5 ± 0.7 | 7.8 ± 0.4 | 25.0 ± 1.9 | 17.0 ± 0.7 | 0.12 ± 0.01 |
| IC50 HeLa (μM) | 40.1 ± 1.5 | 8.3 ± 0.8 | 26.9 ± 1.0 | 14.1 ± 0.3 | 47.7 ± 1.2 | 17.9 ± 1.3 | 33.0 ± 1.4 | 6.8 ± 0.5 | 16.8 ± 1.7 | 26.0 ± 1.9 | 12.4 ± 0.7 | 27.0 ± 0.8 |
| Comp. | 25 | 26 | 27 | 30 | 31 | 32 | 33 |
|---|---|---|---|---|---|---|---|
| IC50 Balb (μM) | 100 ± 3.1 | 21.5 ± 2.0 | 7.3 ± 0.7 | 42.1 ± 2.7 | 3.5 ± 0.8 | 32.1 ± 3.4 | 12.6 ± 0.9 |
| IC50 HeLa (μM) | 13.9 ± 4.5 | 2.5 ± 0.4 | 66.9 ± 8.9 | 9.3 ± 0.8 | 8.8 ± 1.1 | 10.8 ± 0.9 | 1.0 ± 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malatini, C.; Carbajales, C.; Luna, M.; Beltrán, O.; Amorín, M.; Masaguer, C.F.; Blanco, J.M.; Barbosa, S.; Taboada, P.; Coelho, A. 3D-Printing of Capsule Devices as Compartmentalization Tools for Supported Reagents in the Search of Antiproliferative Isatins. Pharmaceuticals 2023, 16, 310. https://doi.org/10.3390/ph16020310
Malatini C, Carbajales C, Luna M, Beltrán O, Amorín M, Masaguer CF, Blanco JM, Barbosa S, Taboada P, Coelho A. 3D-Printing of Capsule Devices as Compartmentalization Tools for Supported Reagents in the Search of Antiproliferative Isatins. Pharmaceuticals. 2023; 16(2):310. https://doi.org/10.3390/ph16020310
Chicago/Turabian StyleMalatini, Camilla, Carlos Carbajales, Mariángel Luna, Osvaldo Beltrán, Manuel Amorín, Christian F. Masaguer, José M. Blanco, Silvia Barbosa, Pablo Taboada, and Alberto Coelho. 2023. "3D-Printing of Capsule Devices as Compartmentalization Tools for Supported Reagents in the Search of Antiproliferative Isatins" Pharmaceuticals 16, no. 2: 310. https://doi.org/10.3390/ph16020310
APA StyleMalatini, C., Carbajales, C., Luna, M., Beltrán, O., Amorín, M., Masaguer, C. F., Blanco, J. M., Barbosa, S., Taboada, P., & Coelho, A. (2023). 3D-Printing of Capsule Devices as Compartmentalization Tools for Supported Reagents in the Search of Antiproliferative Isatins. Pharmaceuticals, 16(2), 310. https://doi.org/10.3390/ph16020310