

Novel Derivatives of Quinoxaline-2-carboxylic Acid 1,4-Dioxides as Antimycobacterial Agents: Mechanistic Studies and Therapeutic Potential

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
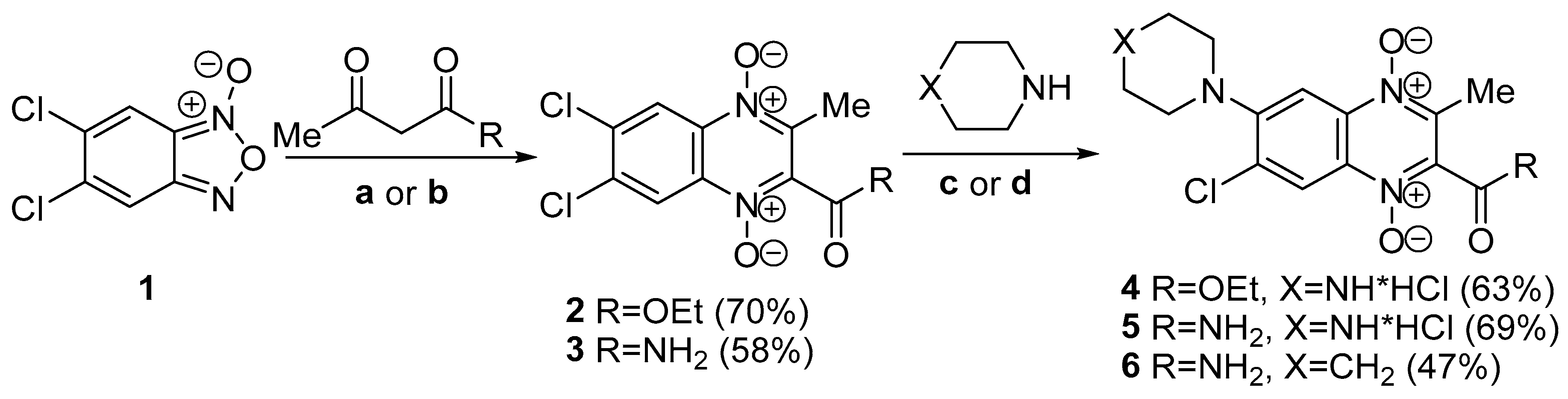
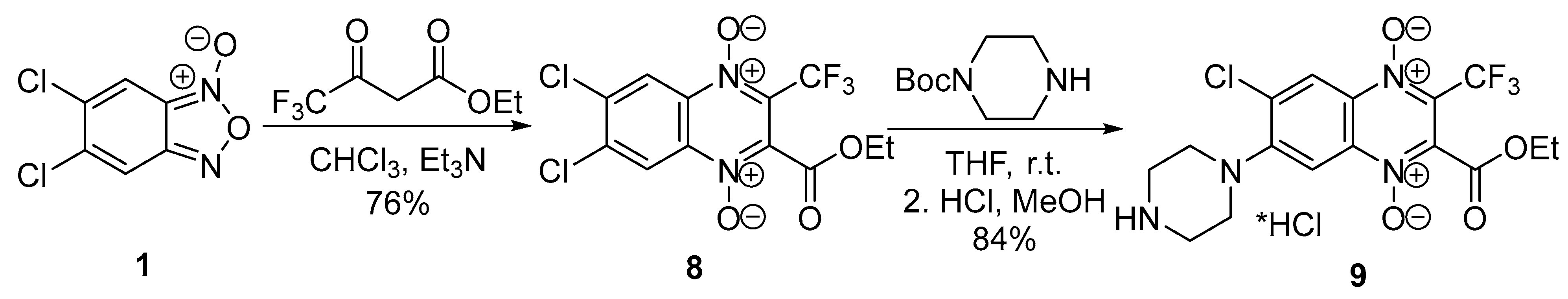
2.1. Chemistry
2.2. Biology
2.2.1. Biological Screening of Quinoxaline 1,4-Dioxides Derivates on M. smegmatis and M. tuberculosis
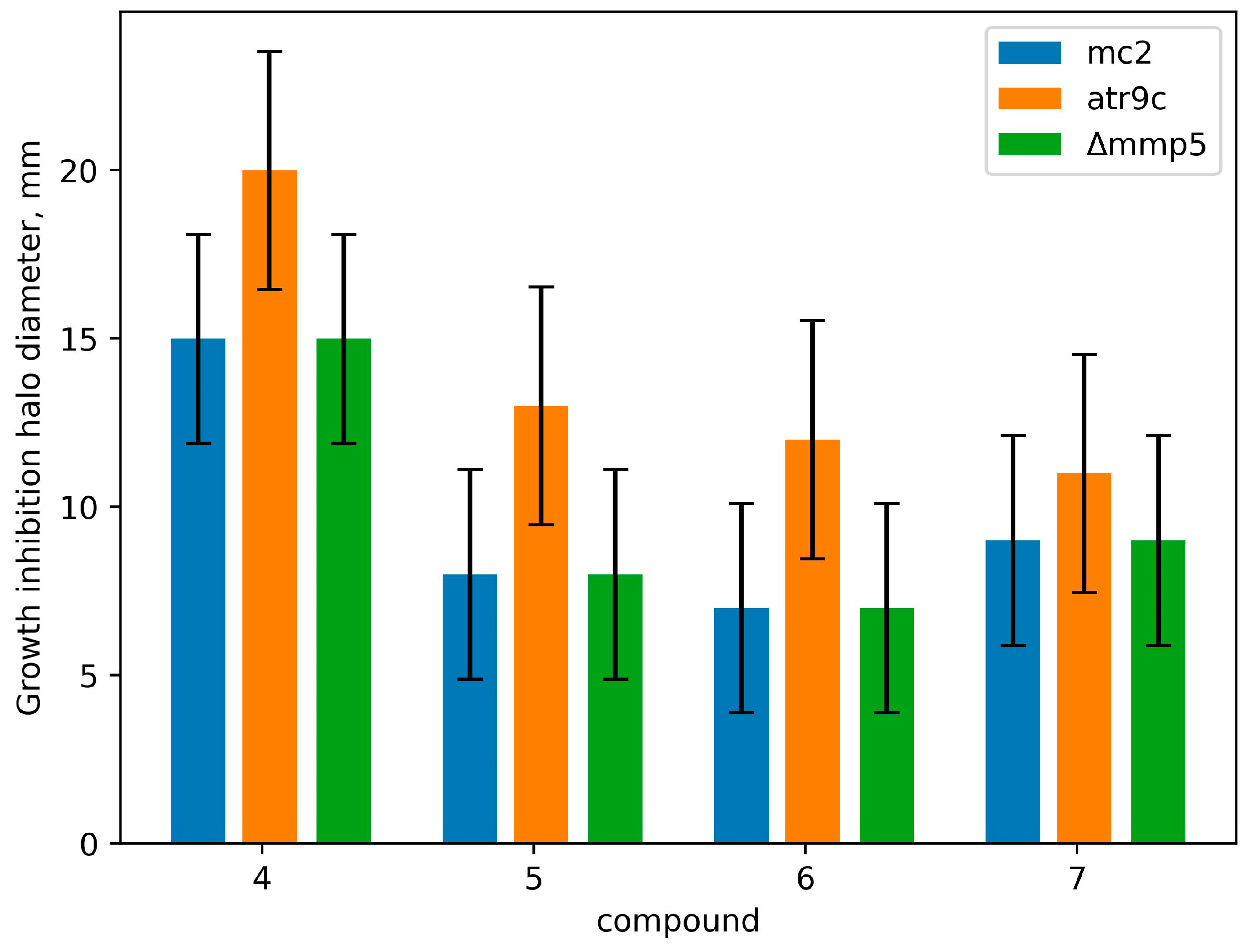
2.2.2. M. smegmatis Drug-Resistant Mutants and Their Genomic Analysis
2.2.3. Investigating the Effect of Individual Gene Mutations on Sensitivity to Quinoxalin-1,4-dioxide
- M. smegmatis 4646c (carrying mutation AAC95CAC in MSMEG_4646, analogous to M. smegmatis qdr4);
- M. smegmatis 4648c (with mutation CAG49CCG in MSMEG_4648, matching M. smegmatis qdR1);
- M. smegmatis 5122c (harboring mutation AT(-72-71)GA in the MSMEG_5122 promoter, akin to M. smegmatis qdR1).
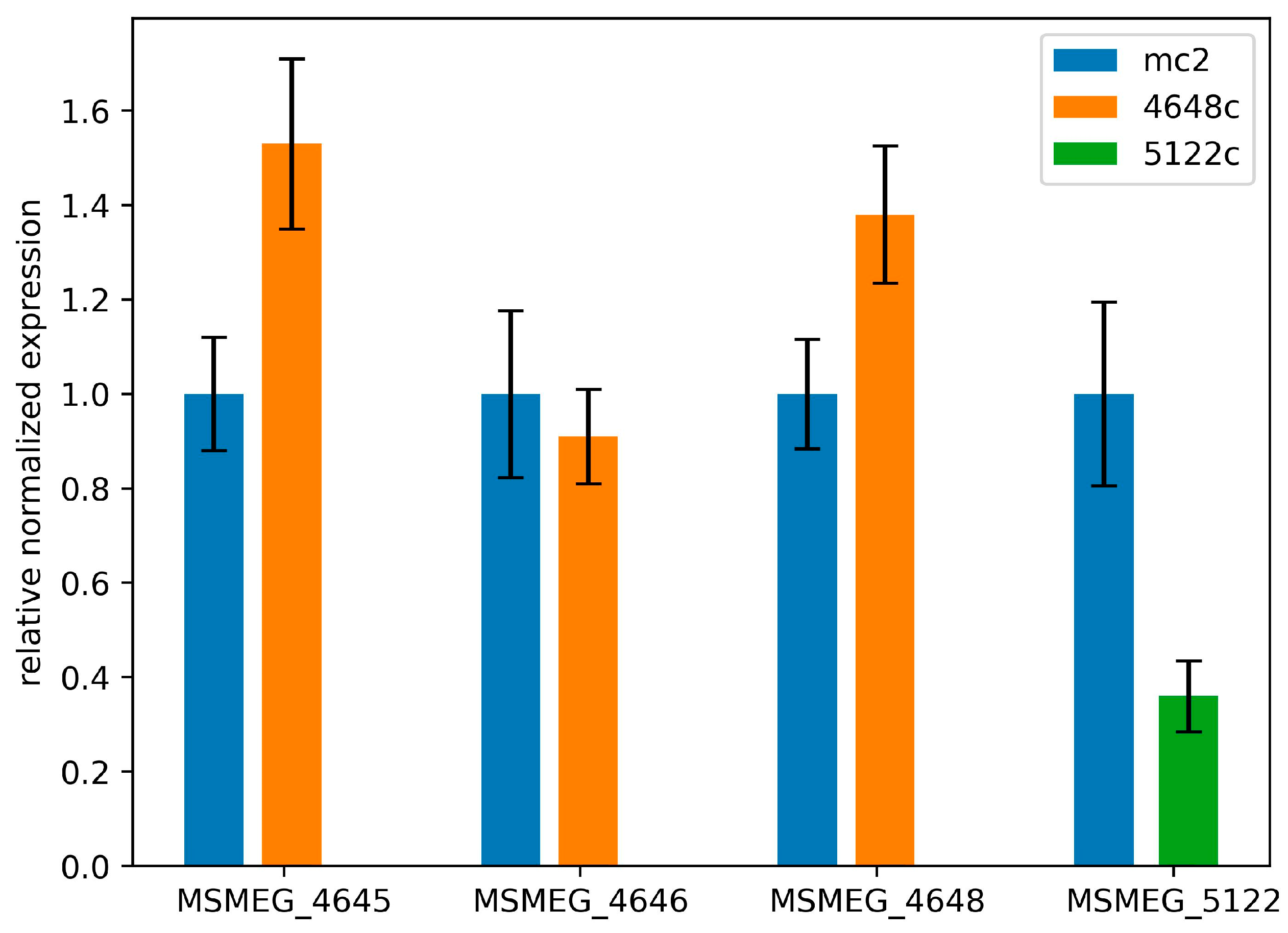
2.2.4. Mutations in Genes MSMEG_5122 and MSMEG_4648 Change Their Expression
2.2.5. Derivative 4 Displays Minimal Cytotoxicity on Human Fibroblasts
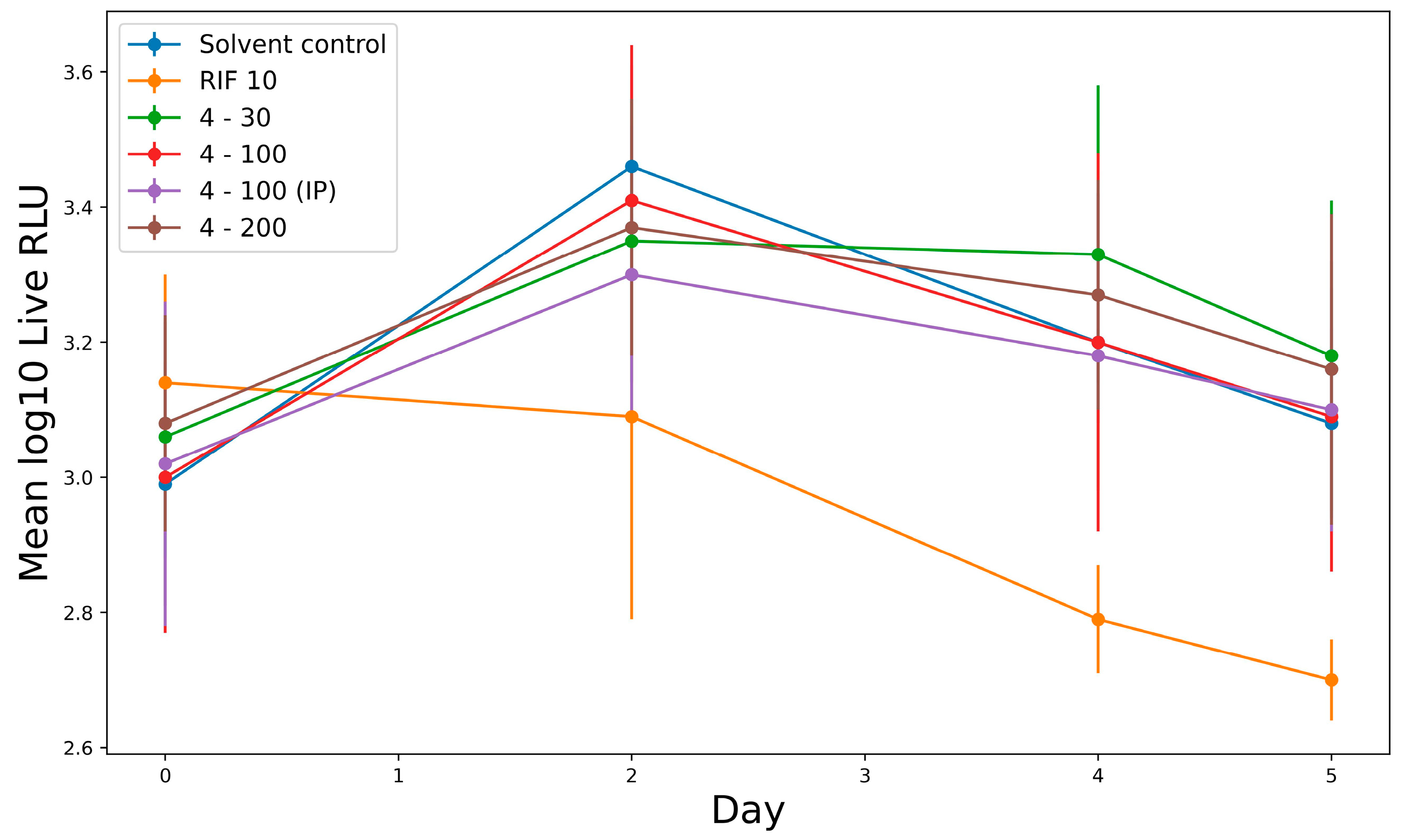
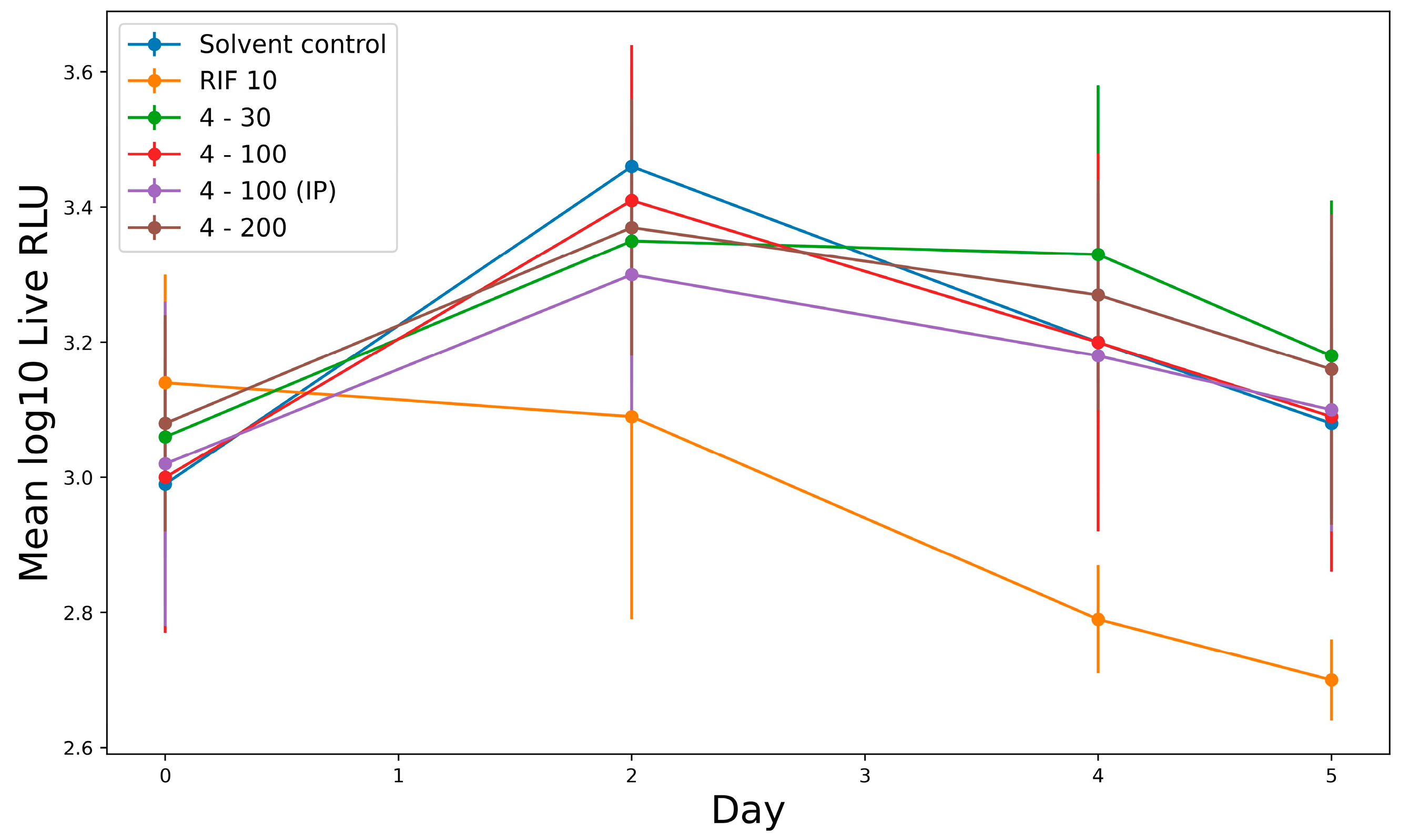
2.2.6. In Vivo Study Activity of Derivative 4 in Murine Tuberculosis Model
3. Discussion
4. Materials and Methods
4.1. Materials and General Methods
4.2. Synthesis
4.2.1. General Procedure for Synthesis of Compounds 2–9
4.2.2. 2-(Ethoxycarbonyl)-3-methylquinoxaline 1,4-dioxide
4.2.3. 6,7-Dichloro-2-(ethoxycarbonyl)-3-methylquinoxaline 1,4-dioxide (2)
4.2.4. 2-Carbamoyl-6,7-dichloro-3-methylquinoxaline 1,4-dioxide (3)
4.2.5. 7-Chloro-2-(ethoxycarbonyl)-3-methyl-6-(piperazin-1-yl)quinoxaline 1,4-dioxide hydrochloride (4)
4.2.6. 2-Carbamoyl-7-chloro-3-methyl-6-(piperazin-1-yl)quinoxaline 1,4-dioxide hydrochloride (5)
4.2.7. 2-Carbamoyl-7-chloro-3-methyl-6-(piperidin-1-yl)quinoxaline 1,4-dioxide (6)
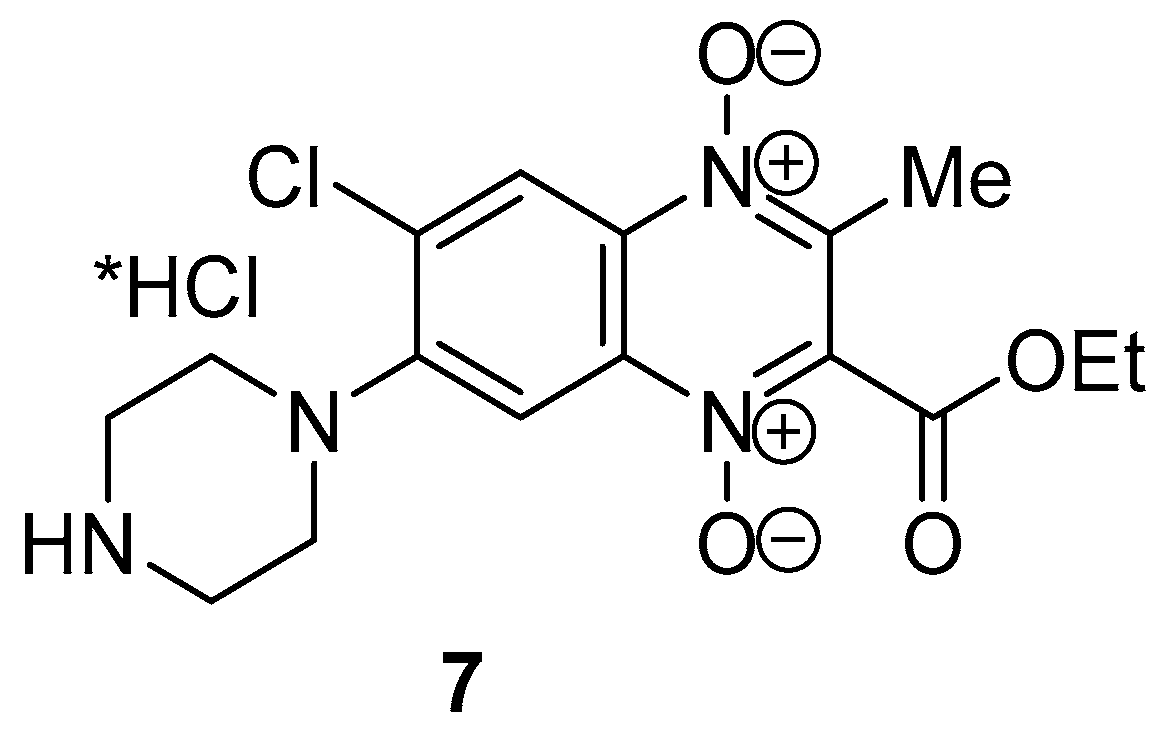
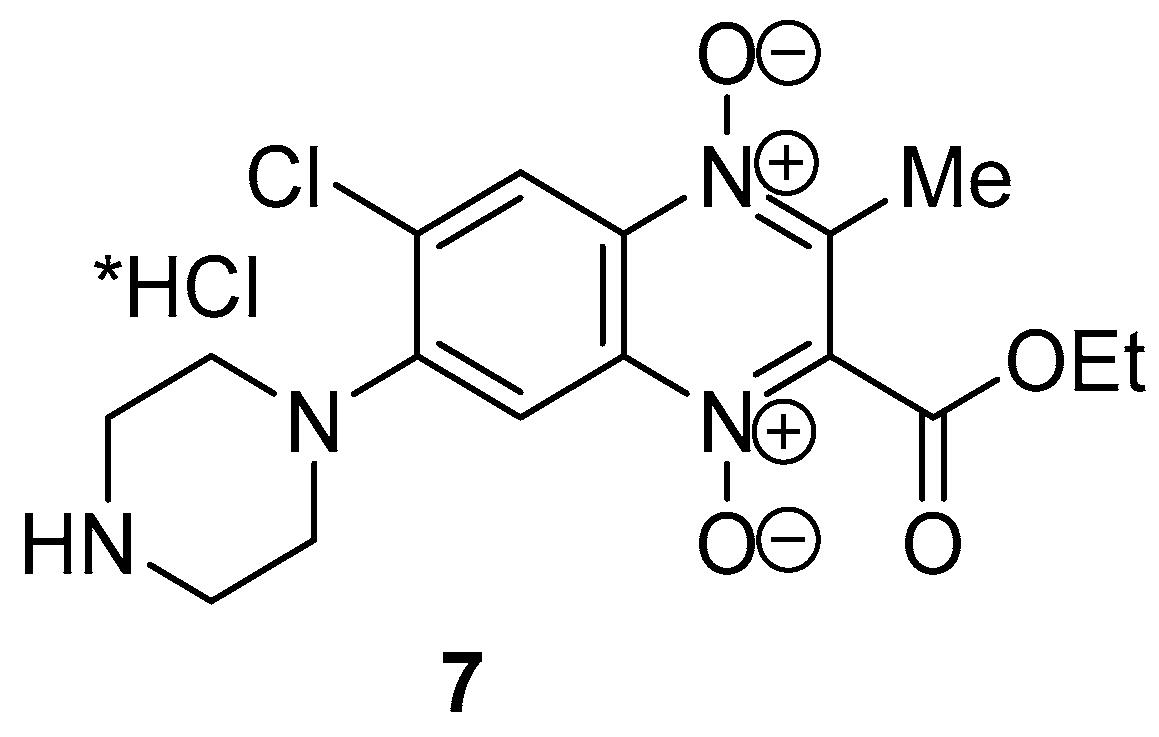
4.2.8. 6-Chloro-2-(ethoxycarbonyl)-3-methyl-7-(piperazin-1-yl)quinoxaline 1,4-dioxide hydrochloride (7)
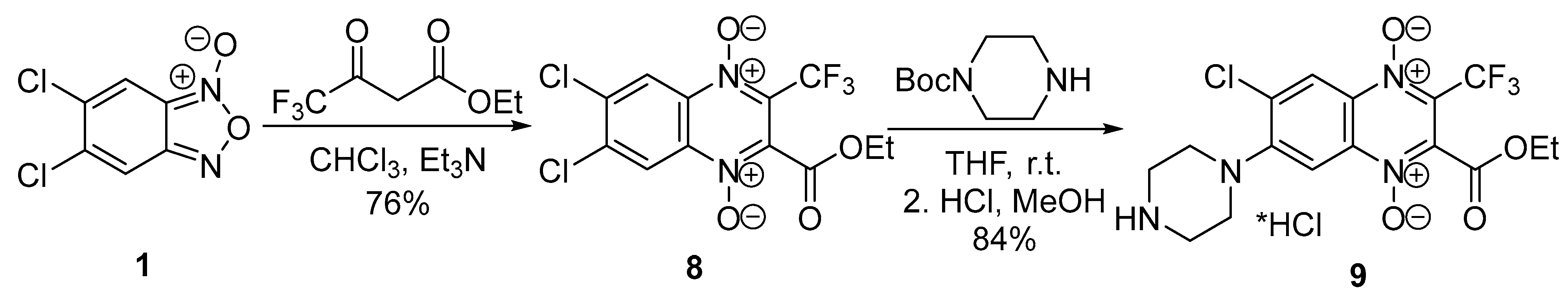
4.2.9. 6,7-Dichloro-2-(ethoxycarbonyl)-3-trifluoromethylquinoxaline 1,4-dioxide (8)
4.2.10. 6-Chloro-2-(ethoxycarbonyl)-3-trifluoromethyl-7-(piperazin-1-yl)quinoxaline 1,4-dioxide hydrochloride (9)
4.3. Study of the Stability of Compounds 4–7
4.4. Biology
4.4.1. Microbial Cultures and Growth Conditions
4.4.2. Minimal Inhibitory Concentration Determinations
MIC Determination on M. smegmatis Strains
MIC Determination on M. tuberculosis Strains
4.4.3. Paper Disk Assay
4.4.4. Generation of Resistant Mutants and Their Phenotype Characterizations
4.4.5. M. smegmatis Whole-Genomic Sequencing and Analysis
4.4.6. Construction of the M. smegmatis Recombinant Strains
4.4.7. RNA Extraction and Real-Time PCR
4.4.8. Cytotoxic Measurement Assay
4.4.9. In Vivo Activity against M. tuberculosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Bagcchi, S. WHO’s Global Tuberculosis Report 2022. Lancet Microbe 2023, 4, e20. [Google Scholar] [CrossRef]
- Caminero, J.A. Multidrug-Resistant Tuberculosis: Epidemiology, Risk Factors and Case Finding. Int. J. Tuberc. Lung Dis. 2010, 14, 382–390. [Google Scholar] [PubMed]
- Gómez-González, P.J.; Perdigao, J.; Gomes, P.; Puyen, Z.M.; Santos-Lazaro, D.; Napier, G.; Hibberd, M.L.; Viveiros, M.; Portugal, I.; Campino, S.; et al. Genetic Diversity of Candidate Loci Linked to Mycobacterium tuberculosis Resistance to Bedaquiline, Delamanid and Pretomanid. Sci. Rep. 2021, 11, 19431. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-P.; Yang, L.; Chai, X.; Lei, Y.-X.; Alam, M.S.; Liu, L.; Shen, C.; Jiang, D.-J.; Wang, Z.; Liu, Z.-Y.; et al. Discovery of Novel DprE1 Inhibitors via Computational Bioactivity Fingerprints and Structure-Based Virtual Screening. Acta Pharmacol. Sin. 2022, 43, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Buravchenko, G.I.; Shchekotikhin, A.E. Quinoxaline 1,4-Dioxides: Advances in Chemistry and Chemotherapeutic Drug Development. Pharmaceuticals 2023, 16, 1174. [Google Scholar] [CrossRef] [PubMed]
- Drumev, D. Current animal feeds with antimicrobial activity. Vet. Med. Nauki 1981, 18, 10–25. [Google Scholar] [PubMed]
- Rainier, R.H.; Chalquest, R.R.; Babcock, W.E.; Thrasher, G.W. Efficacy of Carbadox in Prevention of Field Outbreaks of Swine Dysentery. Vet. Med. Small Anim. Clin. 1973, 68, 171–175. [Google Scholar] [PubMed]
- Cheng, G.; Sa, W.; Cao, C.; Guo, L.; Hao, H.; Liu, Z.; Wang, X.; Yuan, Z. Quinoxaline 1,4-Di-N-Oxides: Biological Activities and Mechanisms of Actions. Front. Pharmacol. 2016, 7, 64. [Google Scholar] [CrossRef]
- Zanetti, S.; Sechi, L.A.; Molicotti, P.; Cannas, S.; Bua, A.; Deriu, A.; Carta, A.; Paglietti, G. In Vitro Activity of New Quinoxalin 1,4-Dioxide Derivatives against Strains of Mycobacterium tuberculosis and Other Mycobacteria. Int. J. Antimicrob. Agents 2005, 25, 179–181. [Google Scholar] [CrossRef]
- Radwan, A.A.; Abdel-Mageed, W.M. In Silico Studies of Quinoxaline-2-Carboxamide 1,4-Di-n-Oxide Derivatives as Antimycobacterial Agents. Molecules 2014, 19, 2247–2260. [Google Scholar] [CrossRef]
- Muradás, T.C.; Abbadi, B.L.; Villela, A.D.; Macchi, F.S.; Bergo, P.F.; de Freitas, T.F.; Sperotto, N.D.M.; Timmers, L.F.S.M.; Norberto de Souza, O.; Picada, J.N.; et al. Pre-Clinical Evaluation of Quinoxaline-Derived Chalcones in Tuberculosis. PLoS ONE 2018, 13, e0202568. [Google Scholar] [CrossRef] [PubMed]
- Buravchenko, G.I.; Maslov, D.A.; Alam, M.S.; Grammatikova, N.E.; Frolova, S.G.; Vatlin, A.A.; Tian, X.; Ivanov, I.V.; Bekker, O.B.; Kryakvin, M.A.; et al. Synthesis and Characterization of Novel 2-Acyl-3-Trifluoromethylquinoxaline 1,4-Dioxides as Potential Antimicrobial Agents. Pharmaceuticals 2022, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Buravchenko, G.I.; Scherbakov, A.M.; Dezhenkova, L.G.; Monzote, L.; Shchekotikhin, A.E. Synthesis of 7-Amino-6-Halogeno-3-Phenylquinoxaline-2-Carbonitrile 1,4-Dioxides: A Way Forward for Targeting Hypoxia and Drug Resistance of Cancer Cells. RSC Adv. 2021, 11, 38782–38795. [Google Scholar] [CrossRef]
- Buravchenko, G.I.; Scherbakov, A.M.; Dezhenkova, L.G.; Bykov, E.E.; Solovieva, S.E.; Korlukov, A.A.; Sorokin, D.V.; Monzote Fidalgo, L.; Shchekotikhin, A.E. Discovery of Derivatives of 6(7)-Amino-3-Phenylquinoxaline-2-Carbonitrile 1,4-Dioxides: Novel, Hypoxia-Selective HIF-1α Inhibitors with Strong Antiestrogenic Potency. Bioorg. Chem. 2020, 104, 104324. [Google Scholar] [CrossRef] [PubMed]
- Maslov, D.A.; Shur, K.V.; Vatlin, A.A.; Danilenko, V.N. MmpS5-MmpL5 Transporters Provide Mycobacterium smegmatis Resistance to Imidazo[1,2-b][1,2,4,5]Tetrazines. Pathogens 2020, 9, 166. [Google Scholar] [CrossRef]
- Andries, K.; Villellas, C.; Coeck, N.; Thys, K.; Gevers, T.; Vranckx, L.; Lounis, N.; de Jong, B.C.; Koul, A. Acquired Resistance of Mycobacterium tuberculosis to Bedaquiline. PLoS ONE 2014, 9, e102135. [Google Scholar] [CrossRef]
- Milano, A.; Pasca, M.R.; Provvedi, R.; Lucarelli, A.P.; Manina, G.; de Jesus Lopes Ribeiro, A.L.; Manganelli, R.; Riccardi, G. Azole Resistance in Mycobacterium tuberculosis Is Mediated by the MmpS5-MmpL5 Efflux System. Tuberculosis 2009, 89, 84–90. [Google Scholar] [CrossRef]
- Li, X.-Z.; Zhang, L.; Nikaido, H. Efflux Pump-Mediated Intrinsic Drug Resistance in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 2004, 48, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Frolova, S.G.; Klimina, K.M.; Kumar, R.; Vatlin, A.A.; Salunke, D.B.; Kendrekar, P.; Danilenko, V.N.; Maslov, D.A. Identification of Mutations Conferring Tryptanthrin Resistance to Mycobacterium smegmatis. Antibiotics 2020, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Parish, T.; Stoker, N.G. Use of a Flexible Cassette Method to Generate a Double Unmarked Mycobacterium tuberculosis tlyA plcABC Mutant by Gene Replacement. Microbiology 2000, 146 Pt 8, 1969–1975. [Google Scholar] [CrossRef]
- Shur, K.; Frolova, S.; Akimova, N.; Maslov, D. A Test System for in Vitro Screening Antimycobacterial Drug Candidates for MmpS5-MmpL5 Mediated Resistance. Russ. J. Genet. 2021, 57, 114–116. [Google Scholar] [CrossRef]
- Loseva, O.V.; Lutsenko, I.A.; Rodina, T.A.; Nelyubina, Y.V.; Gerasimenko, A.V.; Bekker, O.B.; Ivanov, A.V.; Eremenko, I.L. An Ionic Gold(III)–Zinc(II) Pseudo-Polymeric Compound of [H3O][Au{S2CN(CH2)5}2]3[ZnCl4]2: Synthesis, Supramolecular Architecture and Anti-Tuberculosis Activity. Polyhedron 2022, 226, 116097. [Google Scholar] [CrossRef]
- Junnotula, V.; Sarkar, U.; Sinha, S.; Gates, K.S. Initiation of DNA Strand Cleavage by 1,2,4-Benzotriazine 1,4-Dioxide Antitumor Agents: Mechanistic Insight from Studies of 3-Methyl-1,2,4-Benzotriazine 1,4-Dioxide. J. Am. Chem. Soc. 2009, 131, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of New Quinoxaline-2-Carboxylate 1,4-Dioxide Derivatives as Anti-Mycobacterium tuberculosis Agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.F.; Yadav, P.; Shinde, S.S.; Hong, C.R.; Pullen, S.M.; Reynisson, J.; Wilson, W.R.; Hay, M.P. Radical Chemistry and Cytotoxicity of Bioreductive 3-Substituted Quinoxaline Di-N-Oxides. Chem. Res. Toxicol. 2016, 29, 1310–1324. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Z.; Sun, C.; Shao, M.; Ma, J.; Wei, X.; Zhang, T.; Li, W.; Ju, J. Discovery and Biosynthesis of Atrovimycin, an Antitubercular and Antifungal Cyclodepsipeptide Featuring Vicinal-Dihydroxylated Cinnamic Acyl Chain. Org. Lett. 2019, 21, 2634–2638. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, Y.; Liu, J.; Tan, Y.; Liu, Z.; Chhotaray, C.; Jiang, H.; Lu, Z.; Chiwala, G.; Wang, S.; et al. The Compound TB47 Is Highly Bactericidal against Mycobacterium ulcerans in a Buruli Ulcer Mouse Model. Nat. Commun. 2019, 10, 524. [Google Scholar] [CrossRef]
- Erkmen, O. Practice 18—Antibiotic Sensitivity Test Technique. In Laboratory Practices in Microbiology; Erkmen, O., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 181–186. ISBN 978-0-323-91017-0. [Google Scholar]
- Belisle, J.T.; Mahaffey, S.B.; Hill, P.J. Isolation of Mycobacterium Species Genomic DNA. In Mycobacteria Protocols, 2nd ed.; Parish, T., Brown, A.C., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; pp. 1–12. ISBN 978-1-59745-207-6. [Google Scholar]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | M. tuberculosis AlRa MIC | M. smegmatis mc2 155 MIC | ||
|---|---|---|---|---|
| μg/mL | μM | μg/mL | μM | |
| 4 | 1.25 | 3 | 4 | 10 |
| 5 | 20 | 53.5 | 16 | 3.7 |
| 6 | 5 | 15 | 32 | 95 |
| 7 | >20 | 50 | 16 | 40 |
| 9 | >20 | 44 | 4 | 9 |
| RIF | 0.03 | 0.04 | 4 | 5 |
| DIOX | 10 | 45 | 16 | 72 |
| Strain | Gene | |||||||
|---|---|---|---|---|---|---|---|---|
| MSMEG_4646 | MSMEG_4648 | MSMEG_5122 | MSMEG_1380 | |||||
| Nucleotide | Amino Acid | Nucleotide | Amino Acid | Nucleotide | Amino Acid | Nucleotide | Amino Acid | |
| qdR1 | w.t. | w.t. | CAG > CCG | Q49P | AT(72-71)GA (in promoter region) | - | insC12 | - |
| qdR2 | w.t. | w.t. | w.t. | w.t. | ATT > ACT | I5T | insG156 | - |
| qdR3 | TGA > TCA | STOP562S | w.t. | w.t. | w.t. | w.t. | TAC > TCC | Y54S |
| qdR4 | AAC > CAC | N95H | w.t. | w.t. | w.t. | w.t. | w.t. | w.t. |
| qdR5 | CCG > CTG | P274L | w.t. | w.t. | w.t. | w.t. | GGA > TGA | G108STOP |
| M. smegmatis Strains | MICs to Compound, μg/mL | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 5 | 7 | 6 | RIF | DIOX | |||||||
| μg/mL | μM | μg/mL | μM | μg/mL | μM | μg/mL | μM | μg/mL | μM | μg/mL | μM | |
| mc2 155 | 4 | 9.98 | 16 | 43 | 16 | 40 | 32 | 95 | 4 | 4.9 | 16 | 72 |
| qdr1 | 8 | 20 | 32 | 86 | 32 | 80 | >32 | >95 | 4 | 4.9 | >32 | >144 |
| qdr2 | 8 | 20 | 32 | 86 | 32 | 80 | >32 | >95 | 4 | 4.9 | >32 | >144 |
| qdr3 | 16 | 40 | 16 | 43 | 32 | 80 | 32 | 95 | 4 | 4.9 | 32 | 144 |
| qdr4 | 16 | 40 | 32 | 86 | 32 | 80 | >32 | >95 | 4 | 4.9 | >32 | >144 |
| qdr5 | 16 | 40 | 16 | 43 | 32 | 80 | 32 | 95 | 4 | 4.9 | 32 | 144 |
| 4646c | 16 | 40 | 16 | 43 | 32 | 80 | 32 | 95 | 4 | 4.9 | 32 | 144 |
| 4648c | 4 | 10 | 16 | 43 | 16 | 40 | 32 | 95 | 4 | 4.9 | 32 | 144 |
| 5122c | 16 | 40 | >32 | >86 | >32 | >80 | >32 | >95 | 4 | 4.9 | 32 | 144 |
| Strain | Description | Origin |
|---|---|---|
| M. smegmatis mc2 155 | Wild-type (w.t.) strain | |
| M. smegmatis atr9c | Recombinant strain: ins C8 (frameshift) in MSMEG_1380 | [15] |
| M. smegmatis Δmmp5 | Carries a 2828 bp deletion in the mmpS5-mmpL5 operon | [21] |
| M. smegmatis qdr1–qdr5 | Spontaneous M. smegmatis mutants resistant to derivative 4 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frolova, S.G.; Vatlin, A.A.; Maslov, D.A.; Yusuf, B.; Buravchenko, G.I.; Bekker, O.B.; Klimina, K.M.; Smirnova, S.V.; Shnakhova, L.M.; Malyants, I.K.; et al. Novel Derivatives of Quinoxaline-2-carboxylic Acid 1,4-Dioxides as Antimycobacterial Agents: Mechanistic Studies and Therapeutic Potential. Pharmaceuticals 2023, 16, 1565. https://doi.org/10.3390/ph16111565
Frolova SG, Vatlin AA, Maslov DA, Yusuf B, Buravchenko GI, Bekker OB, Klimina KM, Smirnova SV, Shnakhova LM, Malyants IK, et al. Novel Derivatives of Quinoxaline-2-carboxylic Acid 1,4-Dioxides as Antimycobacterial Agents: Mechanistic Studies and Therapeutic Potential. Pharmaceuticals. 2023; 16(11):1565. https://doi.org/10.3390/ph16111565
Chicago/Turabian StyleFrolova, Svetlana G., Aleksey A. Vatlin, Dmitry A. Maslov, Buhari Yusuf, Galina I. Buravchenko, Olga B. Bekker, Ksenia M. Klimina, Svetlana V. Smirnova, Lidia M. Shnakhova, Irina K. Malyants, and et al. 2023. "Novel Derivatives of Quinoxaline-2-carboxylic Acid 1,4-Dioxides as Antimycobacterial Agents: Mechanistic Studies and Therapeutic Potential" Pharmaceuticals 16, no. 11: 1565. https://doi.org/10.3390/ph16111565
APA StyleFrolova, S. G., Vatlin, A. A., Maslov, D. A., Yusuf, B., Buravchenko, G. I., Bekker, O. B., Klimina, K. M., Smirnova, S. V., Shnakhova, L. M., Malyants, I. K., Lashkin, A. I., Tian, X., Alam, M. S., Zatonsky, G. V., Zhang, T., Shchekotikhin, A. E., & Danilenko, V. N. (2023). Novel Derivatives of Quinoxaline-2-carboxylic Acid 1,4-Dioxides as Antimycobacterial Agents: Mechanistic Studies and Therapeutic Potential. Pharmaceuticals, 16(11), 1565. https://doi.org/10.3390/ph16111565