
Recent Perspectives on Cardiovascular Toxicity Associated with Colorectal Cancer Drug Therapy

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Cardiovascular Complications Associated with CRC and mCRC Treatment
2.1. Monoclonal Antibodies
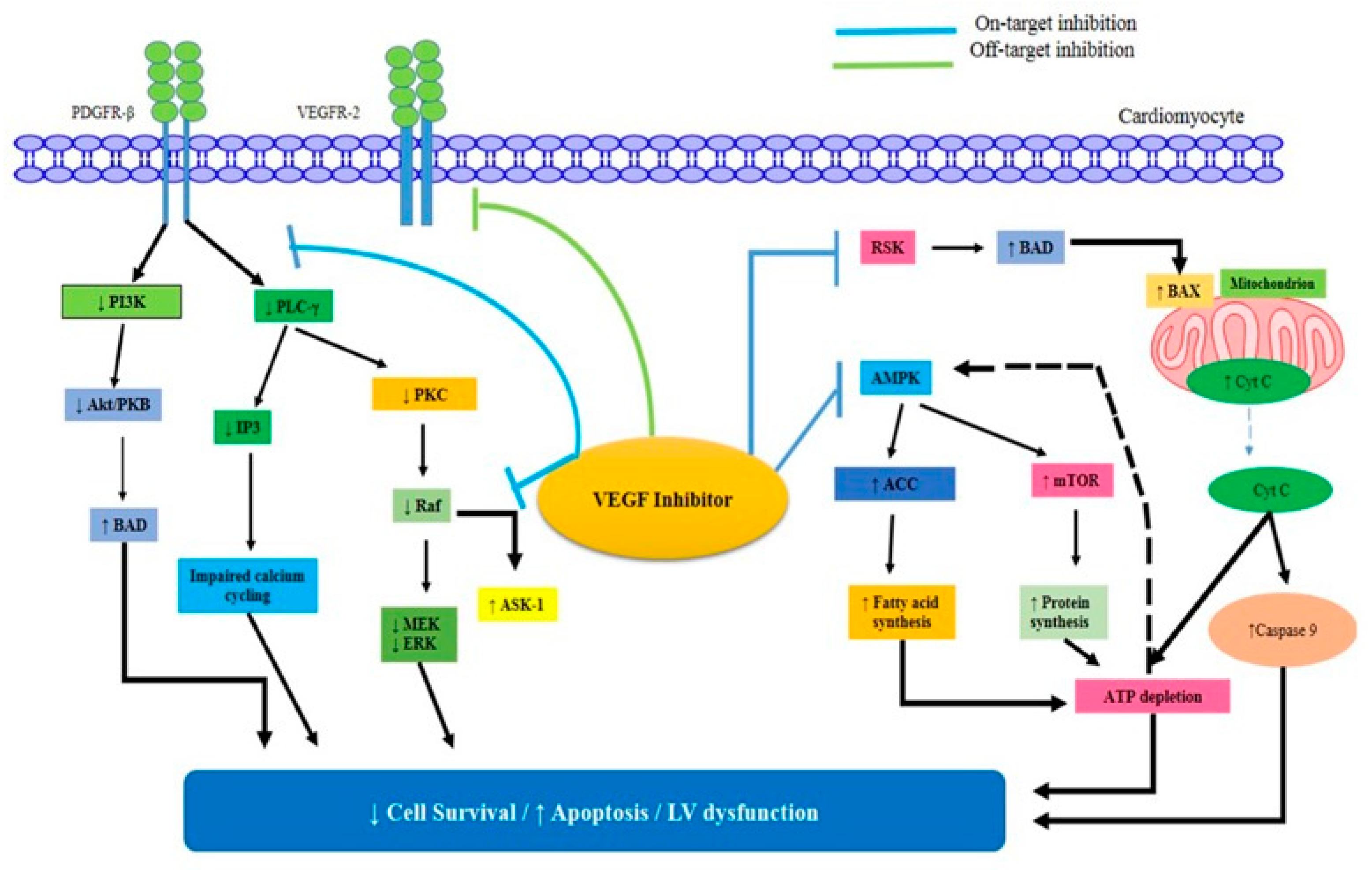
2.1.1. Bevacizumab
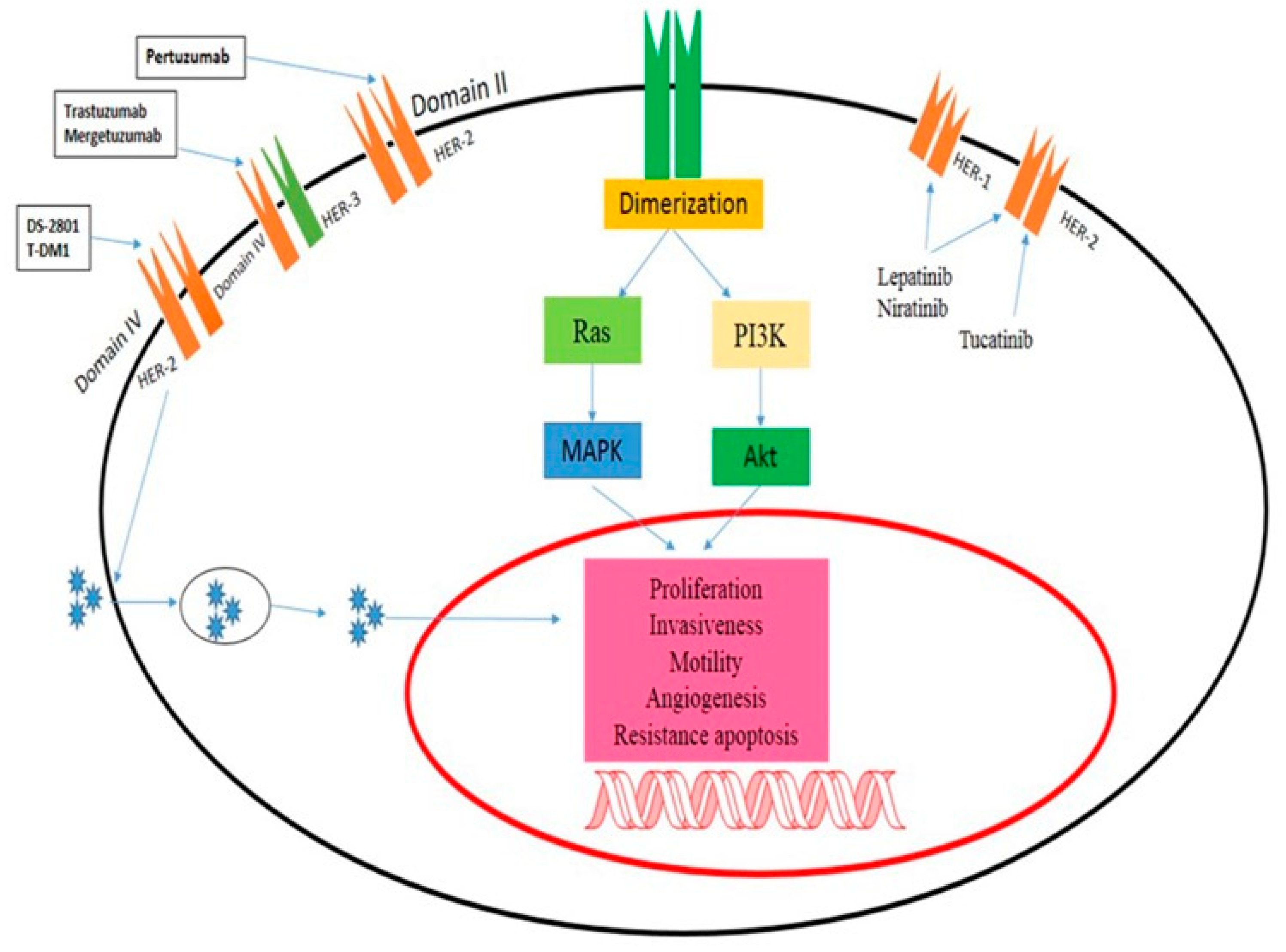
2.1.2. Cetuximab and Panitumumab
2.2. Tyrosine Kinase Inhibitors (TKIs)
2.2.1. Regorafenib
2.2.2. Novel Agents
3. Molecular Signaling and Biomarkers Influencing Cardiovascular Toxicity in CRC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Drug | Category | Mechanism of Anticancer Action | Type of Cardiotoxicity | Cancers Treated |
|---|---|---|---|---|---|
| 1 | Trastuzumab, Pertuzumab | Monoclonal Antibodies | HER2 inhibitors | LVSD, heart failure, orthostatic hypotension | Metastatic colorectal cancer, lung cancer, malignancies, myeloma [22] |
| 2 | Lapatinib, Sunitinib, Pazopanib, Sorafenib | Tyrosine kinase inhibitor | VEGF Inhibitors | Myocardial ischemia, LVSD, QT prolongation, arterial thromboembolic | Lymphoblastic leukemia, renal cell carcinoma, imatinib-resistant gastrointestinal stromal tumors, all types of chronic myeloid leukemia [32] |
| 3 | Imatinib, Dasatinib, Nilotinib, Bosutinib, Ponatinib | Tyrosine kinase inhibitor/ DNA damage | BCR-ABL Kinase Inhibitors | Accelerated atherosclerosis, peripheral artery disease, acute coronary syndrome, stroke, hypertension | Lymphoblastic leukemia, renal cell carcinoma, imatinib-resistant gastrointestinal stromal tumors, all types of chronic myeloid leukemia [32] |
| 4 | Carfilzomib, Bortezomib, Ixazomib, Doxorubicin, Epirubicin, Daunorubicin, Idarubicin, Mitoxantrone | Anthracyclines | Proteasome inhibitors, Oxidative stress, Topoisomerase II inhibitor | Myocardial ischemia, arterial hypertension, LVSD, heart failure, arrhythmias, QT changes, ventricular repolarization abnormalities | Gastrointestinal stromal tumors [28], acute leukemia’s, Hodgkin’s disease, non-Hodgkin’s lymphomas, breast and colorectal cancer [39] |
| 5 | Cyclophosphamide, Cisplatin, 5-Fluorouracil, Capecitabine | Alkylating agents, fluoropyrimines, fluoropyrimines | ROS production, DNA damage, Inhibit growth and metastasis | Acute heart failure (reversible), pericardial effusion, arrhythmias, myocardial ischemia, myocardial infraction | Blood, lung, breast, ovarian, endometrial, and bladder cancer [34], breast, colon, and other solid tumors [40] |
| 6 | Paclitaxel, Decetaxel | Taxane | Microtubule inhibition | Cardiomyocyte toxicity, bradycardia, LVSD, ventricular arrhythmias, myocardial ischemia | Cancers of the head and neck, prostatic, breast, bladder, and ovarian as well as Kaposi’s sarcoma, non-small-cell lung, and gastric adenocarcinoma [45] |
| 7 | Vincristine, Vinblastine, Vinorelbine | Vinca alkaloids | Microtubule inhibition | Myocardial ischemia | Lymphomas and leukemias [46] |
| 8 | Gemcitabine | Antimetabolite | Pyrimidine nucleoside antimetabolite | Pericardial effusion | Breast, bladder, pancreatic, and non-small-cell lung cancer [27] |
| 9 | Retinoic acid | Nutrient | Inhibiting the cell proliferation | Pericardial effusion, LVSD | Acute promyelocytic leukemia [40] |
| 10 | Arsenic Trioxide | Angiogenic | Interaction with ion channels | Prolonged QT Interval, Torsades De Pointes | Relapsing acute promyelocytic, leukemia [42] |
| 11 | Thalidomide, Lenalidomide | Angiogenic | Immunomodulator | Edema and sinus bradycardia, deep vein thrombosis | Multiple myeloma [45] |
| 12 | Interleukin 2 | Cytokine | Block reproduction and spread of cancer cells | Hypotension, arrhythmias, myocardial ischemia, cardiomyopathy, myocarditis | Metastatic renal cell carcinoma and melanoma [47] |
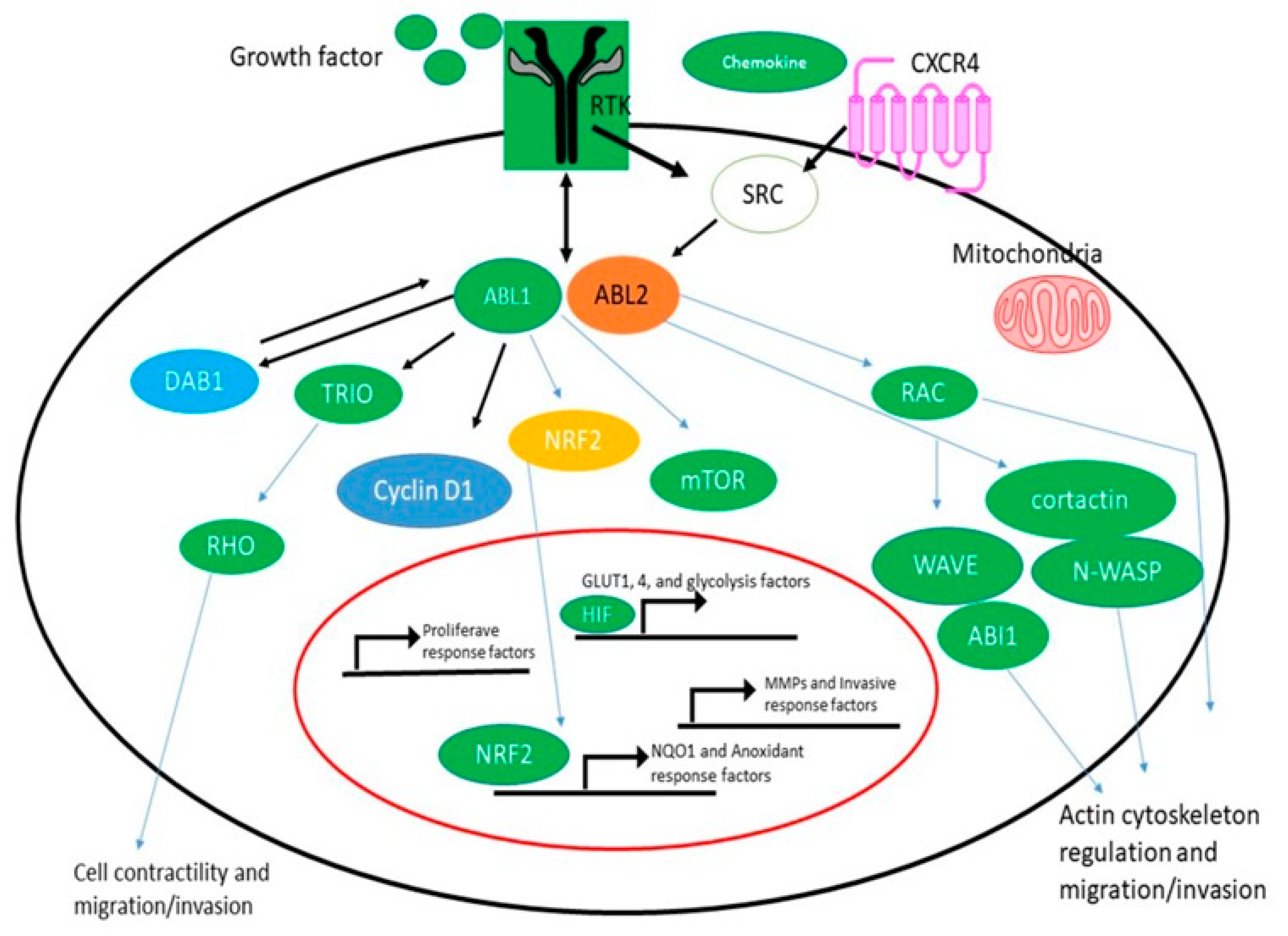
3.1. Cardiotoxicity of the BCR-ABL1
3.2. Colorectal Tumors and Epidermal Growth Factor Receptor Signaling
3.3. VEGFRs
3.4. PDGF
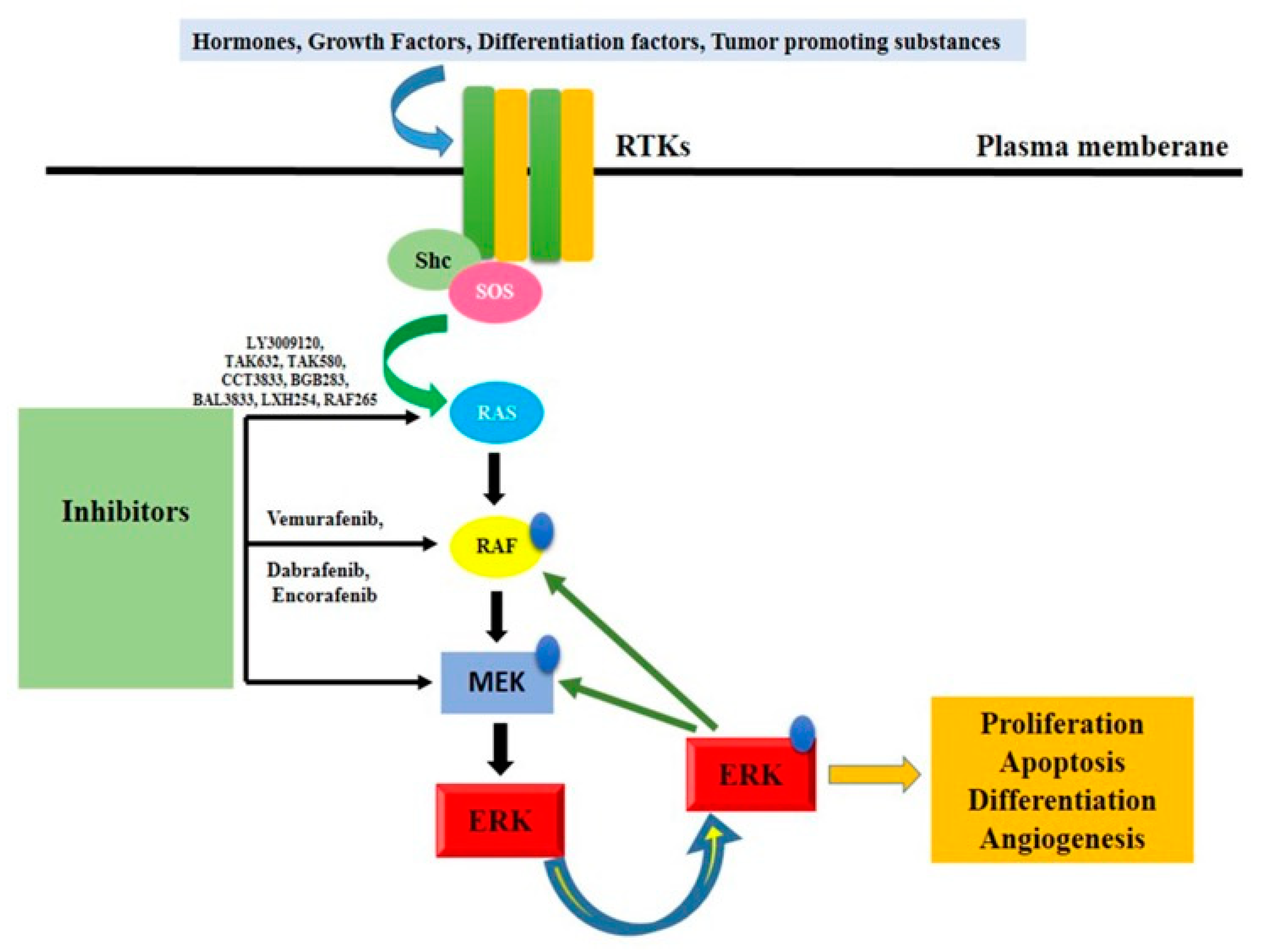
3.5. BRAF/Ras/Raf/MEK/ERK Pathway
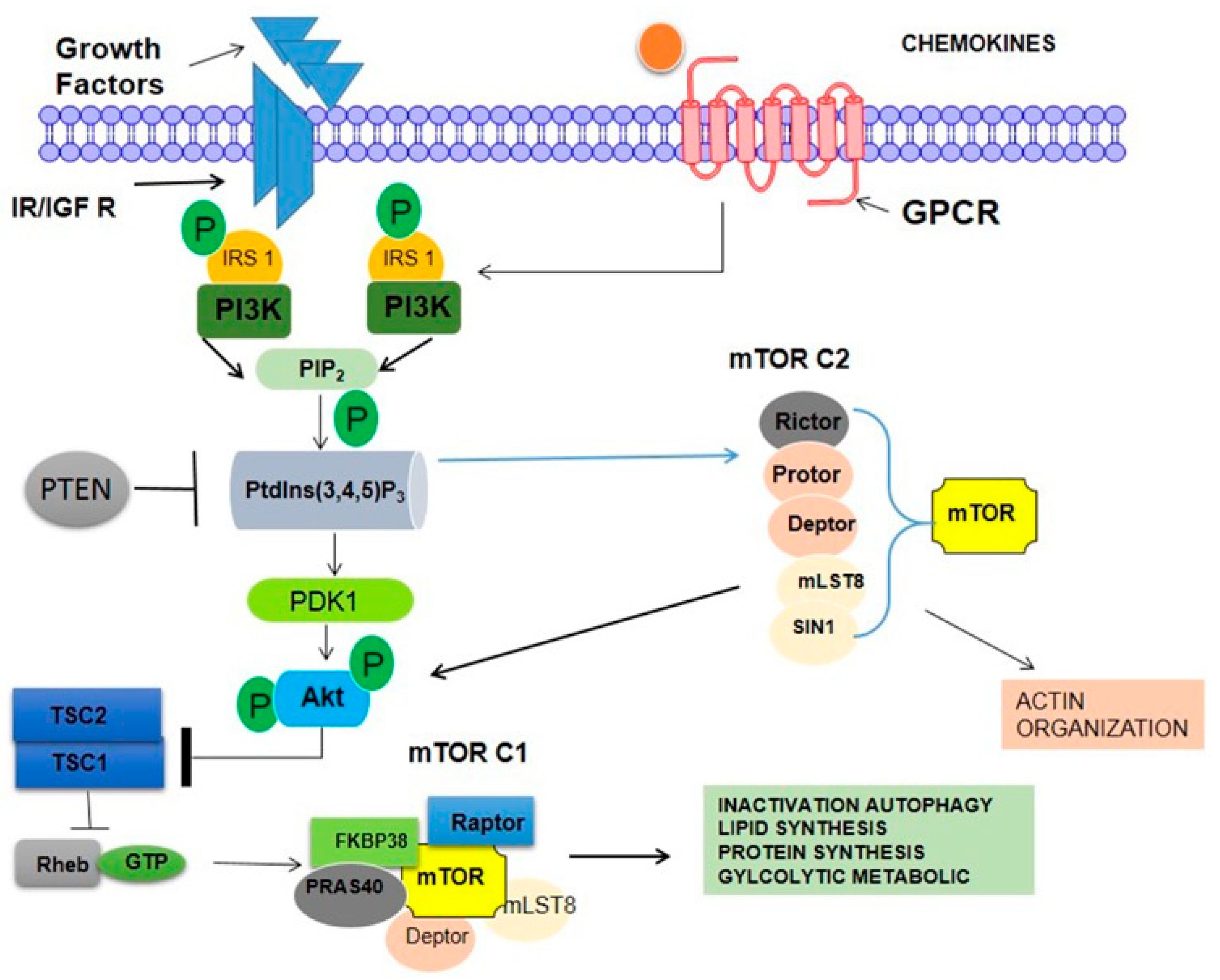
3.6. mTOR
3.7. PI3K Pathway
3.8. p38 Mitogen-Activated Protein Kinase
4. Principle Techniques Used for Monitoring Cardiotoxicity in Clinical Practice
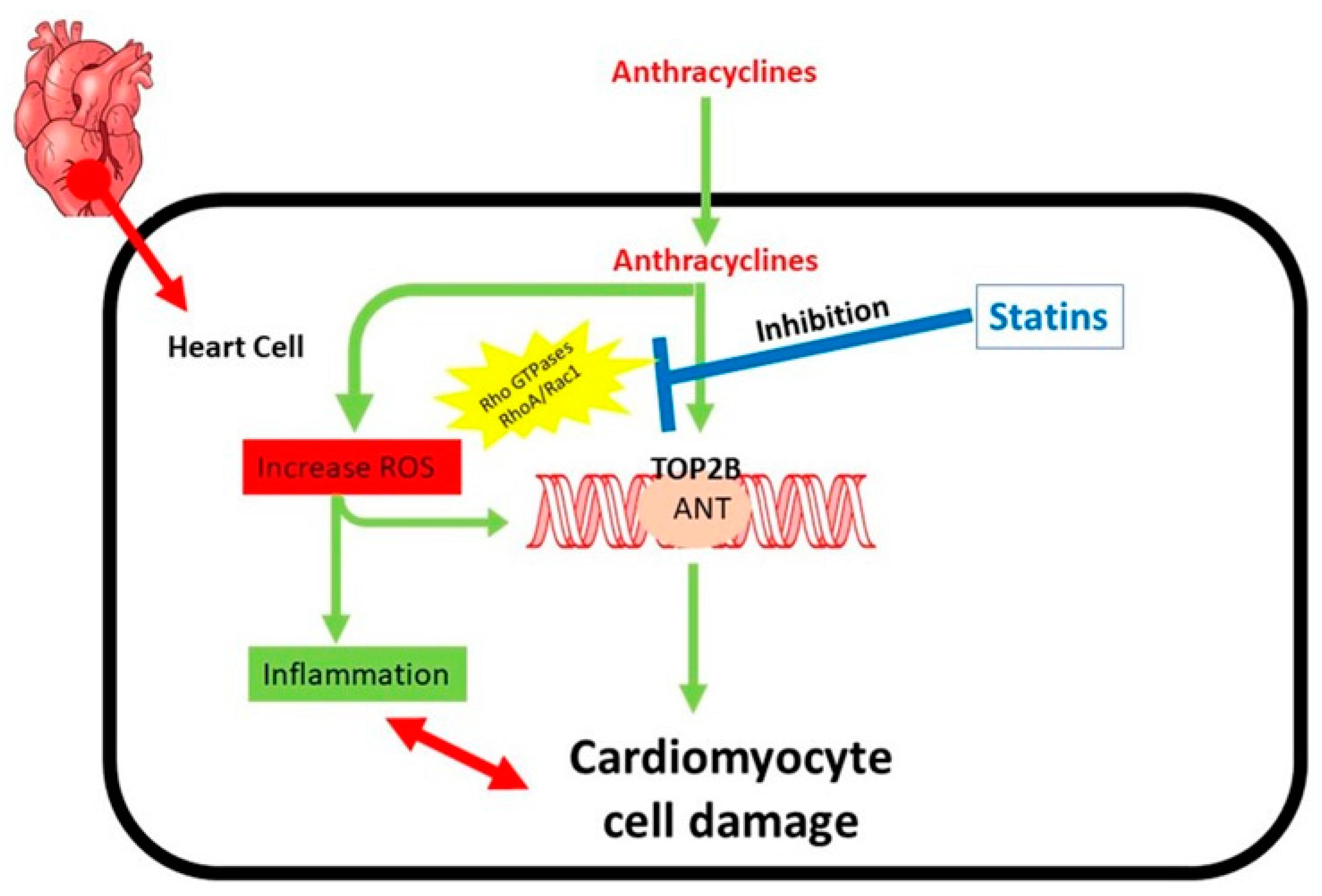
Statins Reduce Anthracycline-Induced Heart Failure and Cell Damage
5. Management of CRC Anticancer Drug-Related Cardiotoxicity
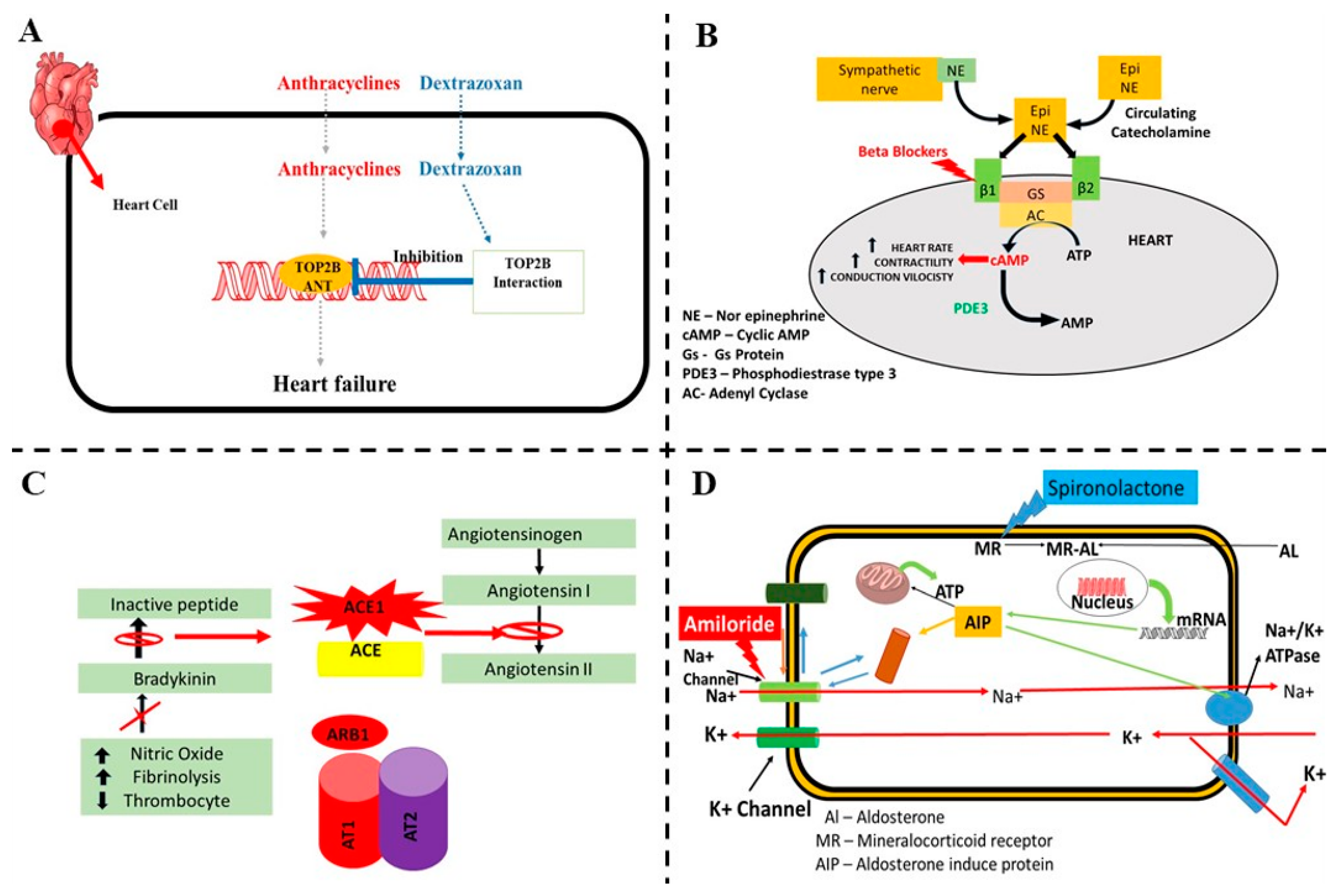
5.1. Dexrazoxane
5.2. Beta-Blockers
5.3. Angiotensin-Converting Enzyme Inhibitors (ACE-I) and Angiotensin Receptor Blockers
5.4. Statins
5.5. Aldosterone Antagonists
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.-B.; Ewer, M.; et al. Prevention and Monitoring of Cardiac Dysfunction in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Pignone, M.P.; Flitcroft, K.L.; Howard, K.; Trevena, L.J.; Salkeld, G.P.; St. John, D.J.B. Costs and Cost-effectiveness of Full Implementation of a Biennial Faecal Occult Blood Test Screening Program for Bowel Cancer in Australia. Med. J. Aust. 2011, 194, 180–185. [Google Scholar] [CrossRef]
- Alexandre, J.; Cautela, J.; Ederhy, S.; Damaj, G.L.; Salem, J.; Barlesi, F.; Farnault, L.; Charbonnier, A.; Mirabel, M.; Champiat, S.; et al. Cardiovascular Toxicity Related to Cancer Treatment: A Pragmatic Approach to the American and European Cardio-Oncology Guidelines. J. Am. Heart Assoc. 2020, 9, e018403. [Google Scholar] [CrossRef]
- Bohdan, M.; Kowalczys, A.; Mickiewicz, A.; Gruchała, M.; Lewicka, E. Cancer Therapy-Related Cardiovascular Complications in Clinical Practice: Current Perspectives. J. Clin. Med. 2021, 10, 1647. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of Anticancer Treatments: Epidemiology, Detection, and Management. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.A.C.; Brito, H.R.d.A.; Mitidieri, G.G.; de Souza, E.P.; Sobral, A.C.G.; Melo, H.H.M.A.; Vasconcelos, G.B.; de Almeida, B.B.D.; Figueiredo, T.d.A.D.; Filho, M.A.A.S.; et al. Cardiotoxicity in Cancer Patients Treated with Chemotherapy: A Systematic Review. Int. J. Health Sci. 2022, 16, 39–46. [Google Scholar]
- Dobbin, S.J.H.; Petrie, M.C.; Myles, R.C.; Touyz, R.M.; Lang, N.N. Cardiotoxic Effects of Angiogenesis Inhibitors. Clin. Sci. 2021, 135, 71–100. [Google Scholar] [CrossRef]
- Wittayanukorn, S.; Qian, J.; Westrick, S.C.; Billor, N.; Johnson, B.; Hansen, R.A. Risk of Cardiotoxicity and All-Cause Mortality in Breast Cancer Patients after Adjuvant Chemotherapy or Hormonal Therapy. Value Health 2015, 18, A190. [Google Scholar] [CrossRef][Green Version]
- Demissei, B.G.; Vedage, N.A.; Hubbard, R.A.; Smith, A.M.; Chung, J.; Lefebvre, B.; Getz, K.D.; Thavendiranathan, P.; Narayan, H.K.; Ky, B. Longitudinal Right Ventricular Systolic Function Changes in Breast Cancer Patients Treated with Cardiotoxic Cancer Therapy. JACC CardioOncol. 2022, 4, 552–554. [Google Scholar] [CrossRef]
- Ansari, A.; Hussain, A.; Wadekar, R.; Altamimi, M.A.; Malik, A.; Mujtaba, M.A.; Ansari, M.Y.; Siddique, M.U.M.; Goyal, S.N. Nanovesicles Based Drug Targeting to Control Tumor Growth and Metastasis. Adv. Cancer Biol.-Metastasis 2023, 7, 100083. [Google Scholar] [CrossRef]
- Arangalage, D.; Degrauwe, N.; Michielin, O.; Monney, P.; Özdemir, B.C. Pathophysiology, Diagnosis and Management of Cardiac Toxicity Induced by Immune Checkpoint Inhibitors and BRAF and MEK Inhibitors. Cancer Treat. Rev. 2021, 100, 102282. [Google Scholar] [CrossRef] [PubMed]
- Bronte, E.; Bronte, G.; Novo, G.; Rinaldi, G.; Bronte, F.; Passiglia, F.; Russo, A. Cardiotoxicity Mechanisms of the Combination of BRAF-Inhibitors and MEK-Inhibitors. Pharmacol. Ther. 2018, 192, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Jiang, J.; Yao, W.; Hu, Y.; Kong, S.; Fan, Q.; Yan, X.; Li, F.; Shi, Z. Pirfenidone Inhibits Stromal Collagen Deposition and Improves Intra-Tumoral Delivery and Antitumor Efficacy of Pegylated Liposomal Doxorubicin. Biomed. Pharmacother. 2023, 157, 114015. [Google Scholar] [CrossRef]
- Ganatra, S.; Dani, S.S.; Yang, E.H.; Zaha, V.G.; Nohria, A. Cardiotoxicity of T-Cell Antineoplastic Therapies. JACC CardioOncol. 2022, 4, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, A.; Adam, R.; Roselló, S.; Arnold, D.; Normanno, N.; Taïeb, J.; Seligmann, J.; De Baere, T.; Osterlund, P.; Yoshino, T.; et al. Metastatic Colorectal Cancer: ESMO Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2023, 34, 10–32. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Berry, G.J.; Witteles, R.M.; Le, D.T.; Wu, S.M.; Fisher, G.A.; Zhu, H. Late-Onset Immunotherapy-Induced Myocarditis 2 Years after Checkpoint Inhibitor Initiation. JACC CardioOncol. 2022, 4, 727–730. [Google Scholar] [CrossRef]
- Chen, W.; Shi, K.; Liu, J.; Yang, P.; Han, R.; Pan, M.; Yuan, L.; Fang, C.; Yu, Y.; Qian, Z. Sustained Co-Delivery of 5-Fluorouracil and Cis-Platinum via Biodegradable Thermo-Sensitive Hydrogel for Intraoperative Synergistic Combination Chemotherapy of Gastric Cancer. Bioact. Mater. 2023, 23, 1–15. [Google Scholar] [CrossRef]
- Cheng, H.; Force, T. Molecular Mechanisms of Cardiovascular Toxicity of Targeted Cancer Therapeutics. Circ. Res. 2010, 106, 21–34. [Google Scholar] [CrossRef]
- Choksey, A.; Timm, K.N. Cancer Therapy-Induced Cardiotoxicity—A Metabolic Perspective on Pathogenesis, Diagnosis and Therapy. Int. J. Mol. Sci. 2021, 23, 441. [Google Scholar] [CrossRef]
- Ciardiello, D.; Maiorano, B.A.; Martinelli, E. Targeting KRASG12C in Colorectal Cancer: The Beginning of a New Era. ESMO Open 2023, 8, 100745. [Google Scholar] [CrossRef]
- Di Bella, S.; Venturini, F.; Marrapese, G.; Tarenzi, E.; Siena, S. Cardiotoxicity of Novel Molecular Targeted Therapies of Cancer. J. Cardiovasc. Echogr. 2011, 21, 78–85. [Google Scholar] [CrossRef]
- El-Tanani, M.; Al Khatib, A.O.; Al-Najjar, B.O.; Shakya, A.K.; El-Tanani, Y.; Lee, Y.-F.; Serrano-Aroca, Á.; Mishra, V.; Mishra, Y.; Aljabali, A.A.; et al. Cellular and Molecular Basis of Therapeutic Approaches to Breast Cancer. Cell Signal 2023, 101, 110492. [Google Scholar] [CrossRef] [PubMed]
- Volkova, M.; Russell, R. Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment. Curr. Cardiol. Rev. 2012, 7, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, B. Doxorubicin Induces Cardiotoxicity through Upregulation of Death Receptors Mediated Apoptosis in Cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated Cell Death Pathways in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Glen, C.; Tan, Y.Y.; Waterston, A.; Evans, T.R.J.; Jones, R.J.; Petrie, M.C.; Lang, N.N. Mechanistic and Clinical Overview Cardiovascular Toxicity of BRAF and MEK Inhibitors. JACC CardioOncol. 2022, 4, 1–18. [Google Scholar] [CrossRef]
- Grela-Wojewoda, A.; Pacholczak-Madej, R.; Adamczyk, A.; Korman, M.; Püsküllüoğlu, M. Cardiotoxicity Induced by Protein Kinase Inhibitors in Patients with Cancer. Int. J. Mol. Sci. 2022, 23, 2815. [Google Scholar] [CrossRef]
- Haider, M.; Zaki, K.Z.; El Hamshary, M.R.; Hussain, Z.; Orive, G.; Ibrahim, H.O. Polymeric Nanocarriers: A Promising Tool for Early Diagnosis and Efficient Treatment of Colorectal Cancer. J. Adv. Res. 2022, 39, 237–255. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Y.; Liu, W. Precision Cardio-Oncology: Understanding the Cardiotoxicity of Cancer Therapy. NPJ Precis. Oncol. 2017, 1, 31. [Google Scholar] [CrossRef]
- Harris, I.S.; Zhang, S.; Treskov, I.; Kovacs, A.; Weinheimer, C.; Muslin, A.J. Raf-1 Kinase Is Required for Cardiac Hypertrophy and Cardiomyocyte Survival in Response to Pressure Overload. Circulation 2004, 110, 718–723. [Google Scholar] [CrossRef]
- Tang, X.; Chen, H.; Li, Q.; Song, Y.; Zhang, S.; Xu, X.-S.; Xu, Y.; Chen, S. Assessment of the Cardiac Safety between Cetuximab and Panitumumab as Single Therapy in Chinese Chemotherapy-Refractory MCRC. OncoTargets Ther. 2017, 11, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Bendell, J.C.; Smith, D.C.; Diefenbach, K.; Lettieri, J.; Boix, O.; Lockhart, A.C.; O’Bryant, C.; Moore, K.N. A Phase I Open-Label Trial Evaluating the Cardiovascular Safety of Regorafenib in Patients with Advanced Cancer. Cancer Chemother. Pharmacol. 2015, 76, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Vošmik, M.; Hodek, M.; Buka, D.; Sýkorová, P.; Grepl, J.; Paluska, P.; Paulíková, S.; Sirák, I. Cardiotoxicity of Radiation Therapy in Esophageal Cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 318–322. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J. Risk of Regorafenib-Induced Cardiovascular Events in Patients with Solid Tumors. Medicine 2018, 97, e12705. [Google Scholar] [CrossRef]
- Merhi, M.; Ahmad, F.; Taib, N.; Inchakalody, V.; Uddin, S.; Shablak, A.; Dermime, S. The Complex Network of Transcription Factors, Immune Checkpoint Inhibitors and Stemness Features in Colorectal Cancer: A Recent Update. Semin. Cancer Biol. 2023, 89, 1–17. [Google Scholar] [CrossRef]
- Jacobs, A.T.; Martinez Castaneda-Cruz, D.; Rose, M.M.; Connelly, L. Targeted Therapy for Breast Cancer: An Overview of Drug Classes and Outcomes. Biochem. Pharmacol. 2022, 204, 115209. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, G.; Choi, S.; Kim, K.H.; Chang, J.; Kim, S.M.; Kim, K.; Son, J.S.; Cho, Y.; Park, S.M. Estimating Risk of Cardiovascular Disease among Long-Term Colorectal Cancer Survivors: A Nationwide Cohort Study. Front. Cardiovasc. Med. 2022, 8, 721107. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, Y.; Zhang, J.; Zhang, S.; Zha, Y.; Wang, J. RRM2 Alleviates Doxorubicin-Induced Cardiotoxicity through the AKT/MTOR Signaling Pathway. Biomolecules 2022, 12, 299. [Google Scholar] [CrossRef]
- Kang, Y.J. Molecular and Cellular Mechanisms of Cardiotoxicity. Environ. Health Perspect. 2001, 109, 27–34. [Google Scholar] [CrossRef]
- Keramida, K.; Charalampopoulos, G.; Filippiadis, D.; Tsougos, E.; Farmakis, D. Cardiovascular Complications of Metastatic Colorectal Cancer Treatment. J. Gastrointest. Oncol. 2019, 10, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Adão, R.; de Keulenaer, G.; Leite-Moreira, A.; Brás-Silva, C. Cardiotoxicity Associated with Cancer Therapy: Pathophysiology and Prevention. Rev. Port. Cardiol. 2013, 32, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Montrasio, G.; Scherr, B.F.; Schimmer, R.; Matter, C.M.; Bühler, K.P.; Manz, M.G.; Müller, A.M.S. Fulminant Cardiotoxicity in a Patient with Cardiac Lymphoma Treated with CAR-T Cells. JACC CardioOncol. 2022, 4, 708–712. [Google Scholar] [CrossRef]
- Aghel, N.; Delgado, D.H.; Lipton, J.H. Cardiovascular Toxicities of BCR-ABL Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia: Preventive Strategies and Cardiovascular Surveillance. Vasc. Health Risk Manag. 2017, 13, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Kourek, C.; Touloupaki, M.; Rempakos, A.; Loritis, K.; Tsougkos, E.; Paraskevaidis, I.; Briasoulis, A. Cardioprotective Strategies from Cardiotoxicity in Cancer Patients: A Comprehensive Review. J. Cardiovasc. Dev. Dis. 2022, 9, 259. [Google Scholar] [CrossRef]
- Latifi, Y.; Moccetti, F.; Wu, M.; Xie, A.; Packwood, W.; Qi, Y.; Ozawa, K.; Shentu, W.; Brown, E.; Shirai, T.; et al. Thrombotic Microangiopathy as a Cause of Cardiovascular Toxicity from the BCR-ABL1 Tyrosine Kinase Inhibitor Ponatinib. Blood 2019, 133, 1597–1606. [Google Scholar] [CrossRef]
- Kwakman, J.J.M.; Simkens, L.H.J.; Mol, L.; Kok, W.E.M.; Koopman, M.; Punt, C.J.A. Incidence of Capecitabine-Related Cardiotoxicity in Different Treatment Schedules of Metastatic Colorectal Cancer: A Retrospective Analysis of the CAIRO Studies of the Dutch Colorectal Cancer Group. Eur. J. Cancer 2017, 76, 93–99. [Google Scholar] [CrossRef]
- Liu, C.; Wu, K.; Li, J.; Mu, X.; Gao, H.; Xu, X. Nanoparticle-Mediated Therapeutic Management in Cholangiocarcinoma Drug Targeting: Current Progress and Future Prospects. Biomed. Pharmacother. 2023, 158, 114135. [Google Scholar] [CrossRef]
- López-Sendón, J.; Álvarez-Ortega, C.; Zamora Auñon, P.; Buño Soto, A.; Lyon, A.R.; Farmakis, D.; Cardinale, D.; Canales Albendea, M.; Feliu Batlle, J.; Rodríguez Rodríguez, I.; et al. Classification, Prevalence, and Outcomes of Anticancer Therapy-Induced Cardiotoxicity: The CARDIOTOX Registry. Eur. Heart J. 2020, 41, 1720–1729. [Google Scholar] [CrossRef]
- Manzat Saplacan, R.M.; Balacescu, L.; Gherman, C.; Chira, R.I.; Craiu, A.; Mircea, P.A.; Lisencu, C.; Balacescu, O. The Role of PDGFs and PDGFRs in Colorectal Cancer. Mediat. Inflamm. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Muraine, L.; Bensalah, M.; Butler-Browne, G.; Bigot, A.; Trollet, C.; Mouly, V.; Negroni, E. Update on Anti-Fibrotic Pharmacotherapies in Skeletal Muscle Disease. Curr. Opin. Pharmacol. 2023, 68, 102332. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Kulkarni, K.; MacDonald, T. Incidence and Risk Factors of Anthracycline-Induced Cardiotoxicity in Long-Term Survivors of Pediatric Cancer: A Population Based Cohort Study. Pediatr. Hematol. Oncol. J. 2022, 7, 136–141. [Google Scholar] [CrossRef]
- Negishi, T.; Thavendiranathan, P.; Penicka, M.; Lemieux, J.; Murbraech, K.; Miyazaki, S.; Shirazi, M.; Santoro, C.; Cho, G.-Y.; Popescu, B.A.; et al. Cardioprotection Using Strain-Guided Management of Potentially Cardiotoxic Cancer Therapy. JACC Cardiovasc. Imaging 2023, 16, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Al-Sohaily, S.; Biankin, A.; Leong, R.; Kohonen-Corish, M.; Warusavitarne, J. Molecular Pathways in Colorectal Cancer. J. Gastroenterol. Hepatol. 2012, 27, 1423–1431. [Google Scholar] [CrossRef]
- Okem, A.; Henstra, C.; Lambert, M.; Hayeshi, R. A Review of the Pharmacodynamic Effect of Chemo-Herbal Drug Combinations Therapy for Cancer Treatment. Med. Drug Discov. 2023, 17, 100147. [Google Scholar] [CrossRef]
- Okpara, E.S.; Adedara, I.A.; Guo, X.; Klos, M.L.; Farombi, E.O.; Han, S. Molecular Mechanisms Associated with the Chemoprotective Role of Protocatechuic Acid and Its Potential Benefits in the Amelioration of Doxorubicin-Induced Cardiotoxicity: A Review. Toxicol. Rep. 2022, 9, 1713–1724. [Google Scholar] [CrossRef]
- Osterlund, P.; Kinos, S.; Pfeiffer, P.; Salminen, T.; Kwakman, J.J.M.; Frödin, J.-E.; Shah, C.H.; Sorbye, H.; Ristamäki, R.; Halonen, P.; et al. Continuation of Fluoropyrimidine Treatment with S-1 after Cardiotoxicity on Capecitabine- or 5-Fluorouracil-Based Therapy in Patients with Solid Tumours: A Multicentre Retrospective Observational Cohort Study. ESMO Open 2022, 7, 100427. [Google Scholar] [CrossRef]
- Papadatos-Pastos, D.; Rabbie, R.; Ross, P.; Sarker, D. The Role of the PI3K Pathway in Colorectal Cancer. Crit. Rev. Oncol. Hematol. 2015, 94, 18–30. [Google Scholar] [CrossRef]
- Platt, J.R.; Todd, O.M.; Hall, P.; Craig, Z.; Quyn, A.; Seymour, M.; Braun, M.; Roodhart, J.; Punt, C.; Christou, N.; et al. FOxTROT2: Innovative Trial Design to Evaluate the Role of Neoadjuvant Chemotherapy for Treating Locally Advanced Colon Cancer in Older Adults or Those with Frailty. ESMO Open 2023, 8, 100642. [Google Scholar] [CrossRef]
- Bersell, K.; Arab, S.; Haring, B.; Kühn, B. Neuregulin1/ErbB4 Signaling Induces Cardiomyocyte Proliferation and Repair of Heart Injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef]
- Polomski, E.-A.S.; Antoni, M.L.; Jukema, J.W.; Kroep, J.R.; Dibbets-Schneider, P.; Sattler, M.G.A.; de Geus-Oei, L.-F. Nuclear Medicine Imaging Methods of Radiation-Induced Cardiotoxicity. Semin. Nucl. Med. 2022, 52, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Azam, T.U.; Mohananey, D.; Kumar, R.; Chu, J.; Lenihan, D.; Dent, S.; Ganatra, S.; Beasley, G.S.; Okwuosa, T. Permissive Cardiotoxicity. JACC CardioOncol. 2022, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Purkayastha, K.; Seth, R.; Seth, S.; Lyon, A. Cancer Therapy-Induced Cardiotoxicity: Review and Algorithmic Approach toward Evaluation. J. Pract. Cardiovasc. Sci. 2017, 3, 82. [Google Scholar] [CrossRef]
- Foersch, S.; Sperka, T.; Lindner, C.; Taut, A.; Rudolph, K.L.; Breier, G.; Boxberger, F.; Rau, T.T.; Hartmann, A.; Stürzl, M.; et al. VEGFR2 Signaling Prevents Colorectal Cancer Cell Senescence to Promote Tumorigenesis in Mice with Colitis. Gastroenterology 2015, 149, 177–189.e10. [Google Scholar] [CrossRef]
- Qiao, C.; Wang, H.; Guan, Q.; Wei, M.; Li, Z. Ferroptosis-Based Nano Delivery Systems Targeted Therapy for Colorectal Cancer: Insights and Future Perspectives. Asian J. Pharm. Sci. 2022, 17, 613–629. [Google Scholar] [CrossRef]
- Brandão, S.R.; Carvalho, F.; Amado, F.; Ferreira, R.; Costa, V.M. Insights on the Molecular Targets of Cardiotoxicity Induced by Anticancer Drugs: A Systematic Review Based on Proteomic Findings. Metabolism 2022, 134, 155250. [Google Scholar] [CrossRef]
- Welty, N.E.; Gill, S.I. Cancer Immunotherapy beyond Checkpoint Blockade. JACC CardioOncol. 2022, 4, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Omland, T.; Heck, S.L.; Gulati, G. The Role of Cardioprotection in Cancer Therapy Cardiotoxicity. JACC CardioOncol. 2022, 4, 19–37. [Google Scholar] [CrossRef]
- Saif, M.W. Alternative Treatment Options in Patients with Colorectal Cancer Who Encounter Fluoropyrimidine-Induced Cardiotoxicity. OncoTargets Ther. 2020, 13, 10197–10206. [Google Scholar] [CrossRef]
- Taeger, J.; Moser, C.; Hellerbrand, C.; Mycielska, M.E.; Glockzin, G.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O.; Lang, S.A. Targeting FGFR/PDGFR/VEGFR Impairs Tumor Growth, Angiogenesis, and Metastasis by Effects on Tumor Cells, Endothelial Cells, and Pericytes in Pancreatic Cancer. Mol. Cancer Ther. 2011, 10, 2157–2167. [Google Scholar] [CrossRef]
- Singh, A.P.; Umbarkar, P.; Tousif, S.; Lal, H. Cardiotoxicity of the BCR-ABL1 Tyrosine Kinase Inhibitors: Emphasis on Ponatinib. Int. J. Cardiol. 2020, 316, 214–221. [Google Scholar] [CrossRef]
- Chan, J.S.K.; Lakhani, I.; Lee, T.T.L.; Chou, O.H.I.; Lee, Y.H.A.; Cheung, Y.M.; Yeung, H.W.; Tang, P.; Ng, K.; Dee, E.C.; et al. Cardiovascular Outcomes and Hospitalizations in Asian Patients Receiving Immune Checkpoint Inhibitors: A Population-Based Study. Curr. Probl. Cardiol. 2023, 48, 101380. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Bi, Z.; Liu, Y.; Qin, F.; Wei, Y.; Wei, X. Targeting RAS-RAF-MEK-ERK Signaling Pathway in Human Cancer: Current Status in Clinical Trials. Genes Dis. 2023, 10, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Spano, J.P.; Milano, G.; Vignot, S.; Khayat, D. Potential Predictive Markers of Response to EGFR-Targeted Therapies in Colorectal Cancer. Crit. Rev. Oncol. Hematol. 2008, 66, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, G. Targeting the Ras/Raf/MEK/ERK Pathway in Hepatocellular Carcinoma. Oncol. Lett. 2017, 13, 1041–1047. [Google Scholar] [CrossRef]
- Nel, J.; Elkhoury, K.; Velot, É.; Bianchi, A.; Acherar, S.; Francius, G.; Tamayol, A.; Grandemange, S.; Arab-Tehrany, E. Functionalized Liposomes for Targeted Breast Cancer Drug Delivery. Bioact. Mater. 2023, 24, 401–437. [Google Scholar] [CrossRef]
- Zheng, P.-P.; Li, J.; Kros, J.M. Breakthroughs in Modern Cancer Therapy and Elusive Cardiotoxicity: Critical Research-Practice Gaps, Challenges, and Insights. Med. Res. Rev. 2018, 38, 325–376. [Google Scholar] [CrossRef]
- Wang, X.-W. Targeting MTOR Network in Colorectal Cancer Therapy. World J. Gastroenterol. 2014, 20, 4178. [Google Scholar] [CrossRef]
- Timm, K.N.; Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar] [CrossRef]
- Lyon, A.R.; López-Fernández, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on Cardio-Oncology Developed in Collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef]
- Dent, S.F.; Moore, H.; Raval, P.; Alder, L.; Guha, A. How to Manage and Monitor Cardiac Dysfunction in Patients with Metastatic HER2-Positive Breast Cancer. JACC CardioOncol. 2022, 4, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Trapani, D.; Zagami, P.; Nicolò, E.; Pravettoni, G.; Curigliano, G. Management of Cardiac Toxicity Induced by Chemotherapy. J. Clin. Med. 2020, 9, 2885. [Google Scholar] [CrossRef] [PubMed]
- Vuong, J.T.; Stein-Merlob, A.F.; Cheng, R.K.; Yang, E.H. Novel Therapeutics for Anthracycline Induced Cardiotoxicity. Front. Cardiovasc. Med. 2022, 9, 863314. [Google Scholar] [CrossRef] [PubMed]
- Kubin, T.; Cetinkaya, A.; Kubin, N.; Bramlage, P.; Sen-Hild, B.; Gajawada, P.; Akintürk, H.; Schönburg, M.; Schaper, W.; Choi, Y.-H.; et al. The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes. Int. J. Mol. Sci. 2020, 21, 6348. [Google Scholar] [CrossRef]
- Walls, G.M.; O’Kane, R.; Ghita, M.; Kuburas, R.; McGarry, C.K.; Cole, A.J.; Jain, S.; Butterworth, K.T. Murine Models of Radiation Cardiotoxicity: A Systematic Review and Recommendations for Future Studies. Radiother. Oncol. 2022, 173, 19–31. [Google Scholar] [CrossRef]
- Wang, J.; Pendergast, A.M. The Emerging Role of ABL Kinases in Solid Tumors. Trends Cancer 2015, 1, 110–123. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, A.-J.; Wu, Y.-J.; Wu, L.-M.; Zhang, L. Silymarin in Cancer Therapy: Mechanisms of Action, Protective Roles in Chemotherapy-Induced Toxicity, and Nanoformulations. J. Funct. Foods 2023, 100, 105384. [Google Scholar] [CrossRef]
- Deng, S.; Yan, T.; Jendrny, C.; Nemecek, A.; Vincetic, M.; Gödtel-Armbrust, U.; Wojnowski, L. Dexrazoxane May Prevent Doxorubicin-Induced DNA Damage via Depleting Both Topoisomerase II Isoforms. BMC Cancer 2014, 14, 842. [Google Scholar] [CrossRef]
- Wu, L.; Wang, L.; Du, Y.; Zhang, Y.; Ren, J. Mitochondrial Quality Control Mechanisms as Therapeutic Targets in Doxorubicin-Induced Cardiotoxicity. Trends Pharmacol. Sci. 2023, 44, 34–49. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, Z.; Cheng, K.; Bi, H.; Chen, J. Small Molecule-Based Immunomodulators for Cancer Therapy. Acta Pharm. Sin. B 2022, 12, 4287–4308. [Google Scholar] [CrossRef]
- Xiao, J.J.; Chen, J.S.; Lum, B.L.; Graham, R.A. A Survey of Renal Impairment Pharmacokinetic Studies for New Oncology Drug Approvals in the USA from 2010 to Early 2015. Anticancer Drugs 2017, 28, 677–701. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Yang, Y.; Luo, Z.; Li, X.; Liu, J.; Zhang, B.; Li, W. Role of Non-Cardiomyocytes in Anticancer Drug-Induced Cardiotoxicity: A Systematic Review. iScience 2022, 25, 105283. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Meng, Q.H.; Gilchrist, S.C.; Lin, S.H.; Lin, R.; Xu, T.; Milgrom, S.A.; Gandhi, S.J.; Wu, H.; Zhao, Y.; et al. Assessment of Prognostic Value of High-Sensitivity Cardiac Troponin T for Early Prediction of Chemoradiation Therapy-Induced Cardiotoxicity in Patients with Non-Small Cell Lung Cancer: A Secondary Analysis of a Prospective Randomized Trial. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Gelblum, D.; Jones, L.; Yu, A.; Zhigang, Z.; Cahlon, O.; McCormick, B.; Powell, S.N.; Ho, A.Y. HEART (Heart Evaluation after Radiation Therapy): Novel Detection of Cardiotoxicity in Breast Cancer Patients. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, E58–E59. [Google Scholar] [CrossRef][Green Version]
- Yang, Z.; Wang, W.; Wang, X.; Qin, Z. Cardiotoxicity of Epidermal Growth Factor Receptor 2-Targeted Drugs for Breast Cancer. Front. Pharmacol. 2021, 12, 741451. [Google Scholar] [CrossRef]







| Symptomatic CTRCD (HF) | Very severe | HF requiring inotropic support, mechanical circulatory support, or consideration of transplantation. |
| Severe | HF hospitalization. | |
| Mild | Mild HF symptoms, no intensification of therapy required. | |
| Asymptomatic CTRCD | Severe | New LVEF reduction to 40%. |
| Moderate | New LVEF reduction by ≥10 percentage points to an LVEF of 40–49% OR New LVEF reduction by 10 percentage points to an LVEF of 40–49% AND either new relative decline in GLS by 0.15% from baseline OR new rise in cardiac biomarkers. | |
| Mild | LVEF ≥50% AND new relative decline in GLS by 0.15% from baseline AND/OR new rise in cardiac biomarkers. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashyap, M.K.; Mangrulkar, S.V.; Kushwaha, S.; Ved, A.; Kale, M.B.; Wankhede, N.L.; Taksande, B.G.; Upaganlawar, A.B.; Umekar, M.J.; Koppula, S.; et al. Recent Perspectives on Cardiovascular Toxicity Associated with Colorectal Cancer Drug Therapy. Pharmaceuticals 2023, 16, 1441. https://doi.org/10.3390/ph16101441
Kashyap MK, Mangrulkar SV, Kushwaha S, Ved A, Kale MB, Wankhede NL, Taksande BG, Upaganlawar AB, Umekar MJ, Koppula S, et al. Recent Perspectives on Cardiovascular Toxicity Associated with Colorectal Cancer Drug Therapy. Pharmaceuticals. 2023; 16(10):1441. https://doi.org/10.3390/ph16101441
Chicago/Turabian StyleKashyap, Monu Kumar, Shubhada V. Mangrulkar, Sapana Kushwaha, Akash Ved, Mayur B. Kale, Nitu L. Wankhede, Brijesh G. Taksande, Aman B. Upaganlawar, Milind J. Umekar, Sushruta Koppula, and et al. 2023. "Recent Perspectives on Cardiovascular Toxicity Associated with Colorectal Cancer Drug Therapy" Pharmaceuticals 16, no. 10: 1441. https://doi.org/10.3390/ph16101441
APA StyleKashyap, M. K., Mangrulkar, S. V., Kushwaha, S., Ved, A., Kale, M. B., Wankhede, N. L., Taksande, B. G., Upaganlawar, A. B., Umekar, M. J., Koppula, S., & Kopalli, S. R. (2023). Recent Perspectives on Cardiovascular Toxicity Associated with Colorectal Cancer Drug Therapy. Pharmaceuticals, 16(10), 1441. https://doi.org/10.3390/ph16101441