
Site-Specific Structural Changes in Long-Term-Stressed Monoclonal Antibody Revealed with DEPC Covalent-Labeling and Quantitative Mass Spectrometry

 , and
, and
Abstract
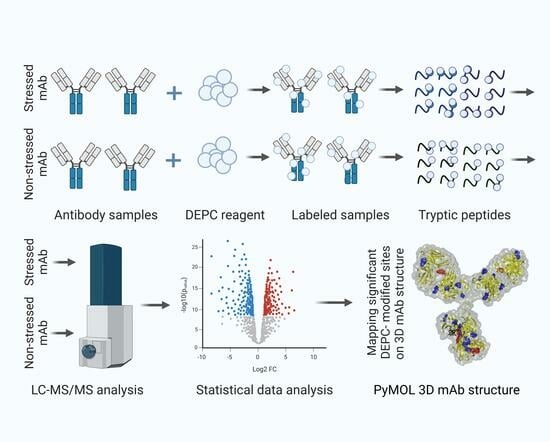
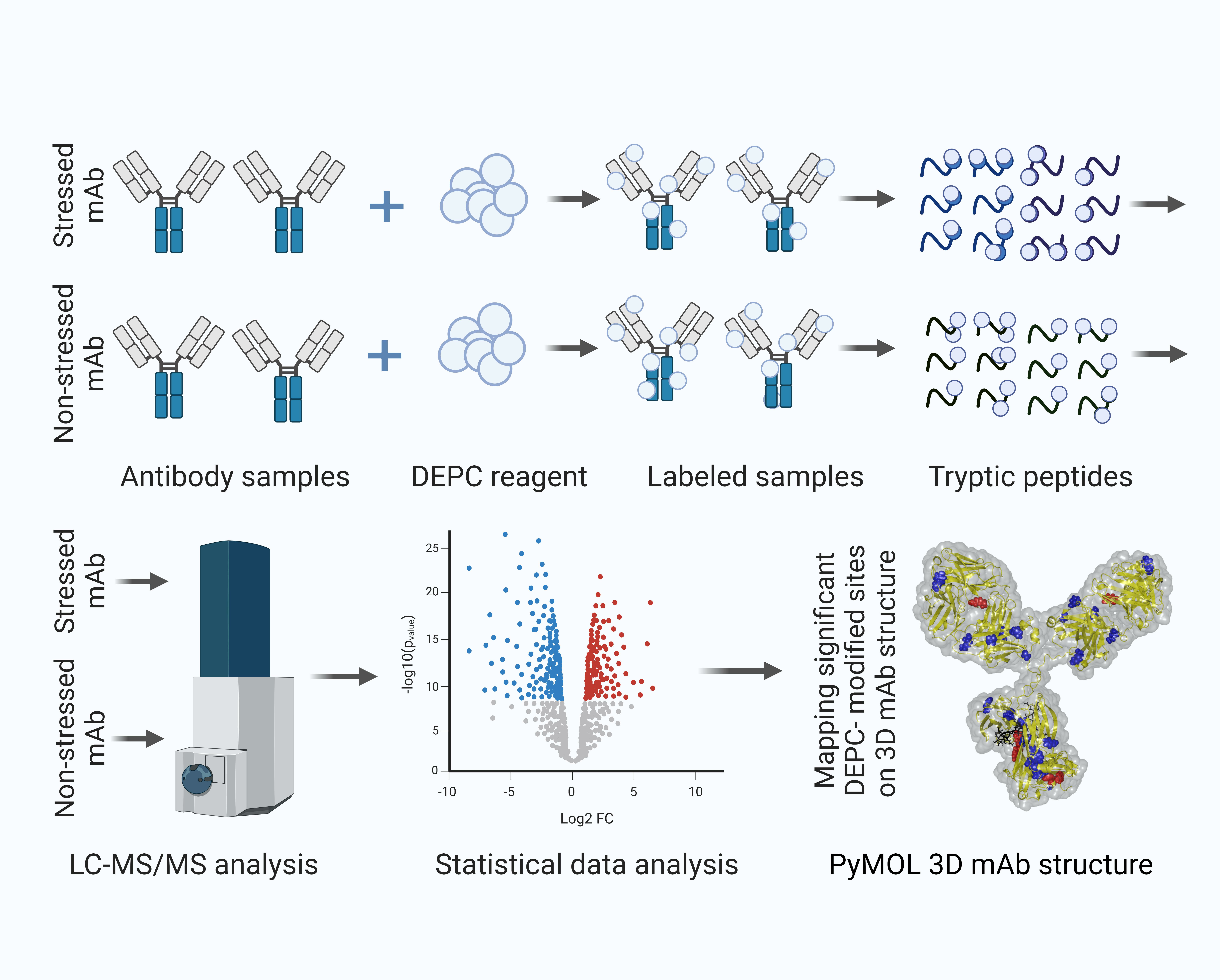{kind=link}
{kind=link}
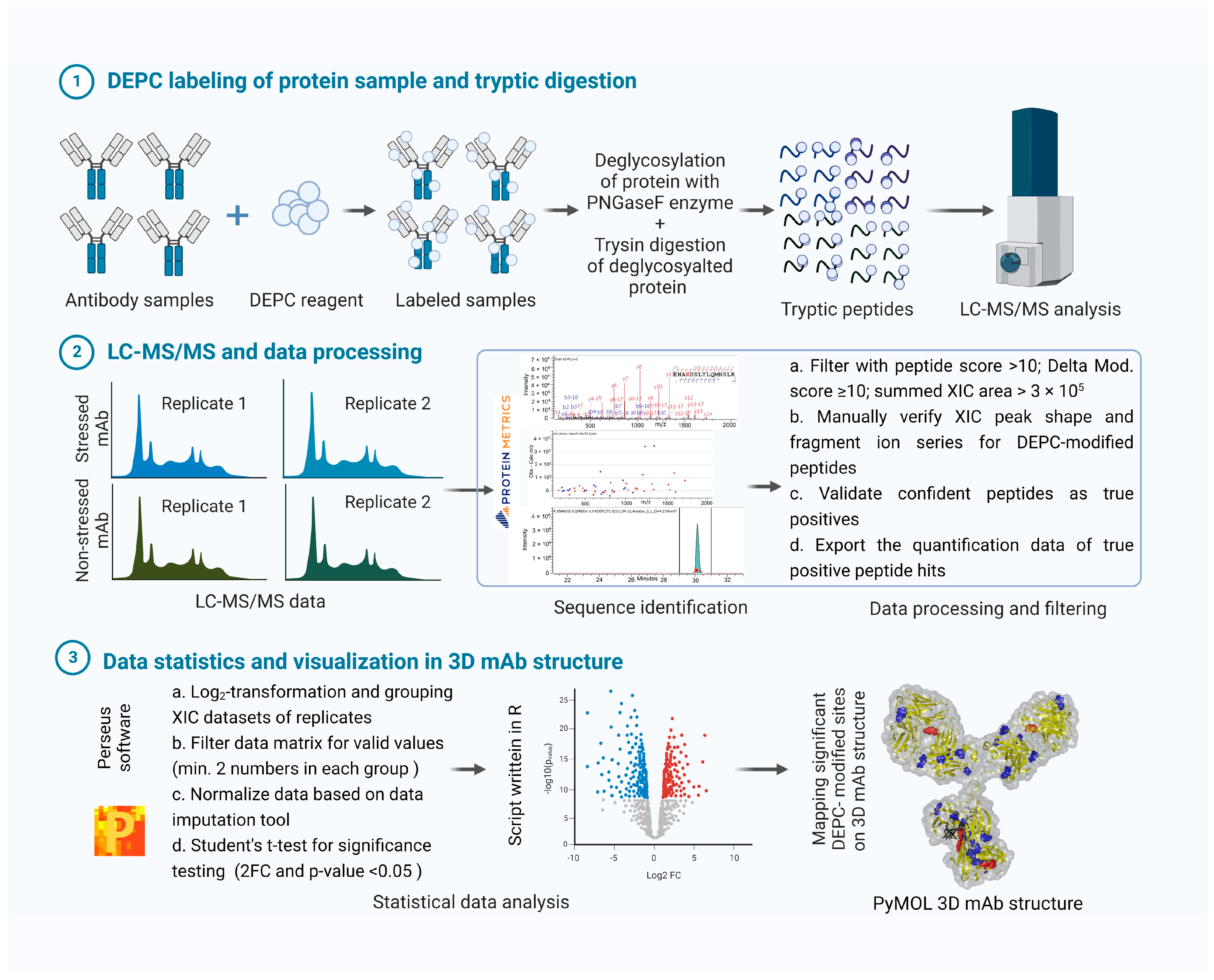
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
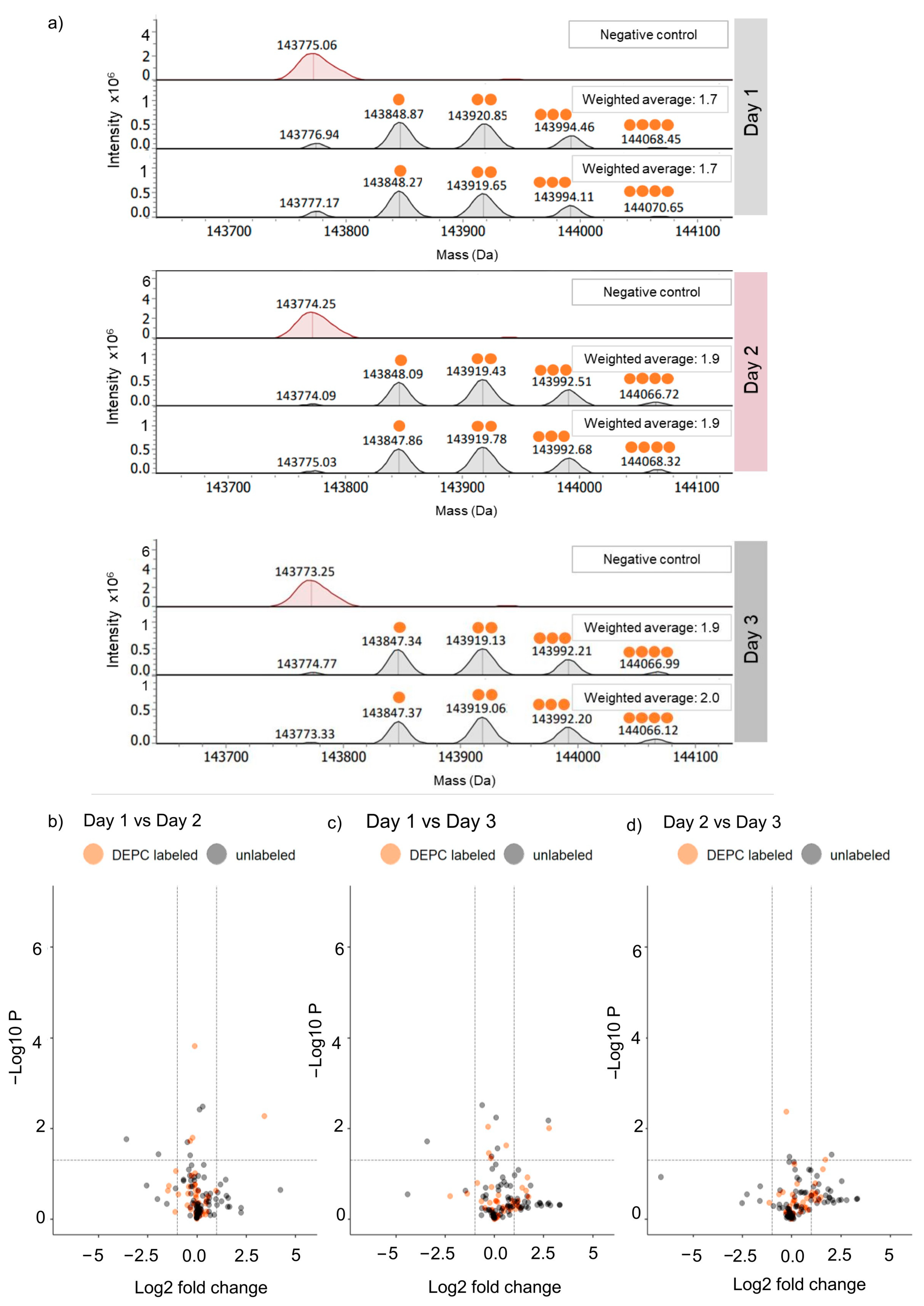
2.1. Reproducibility of DEPC Labeling
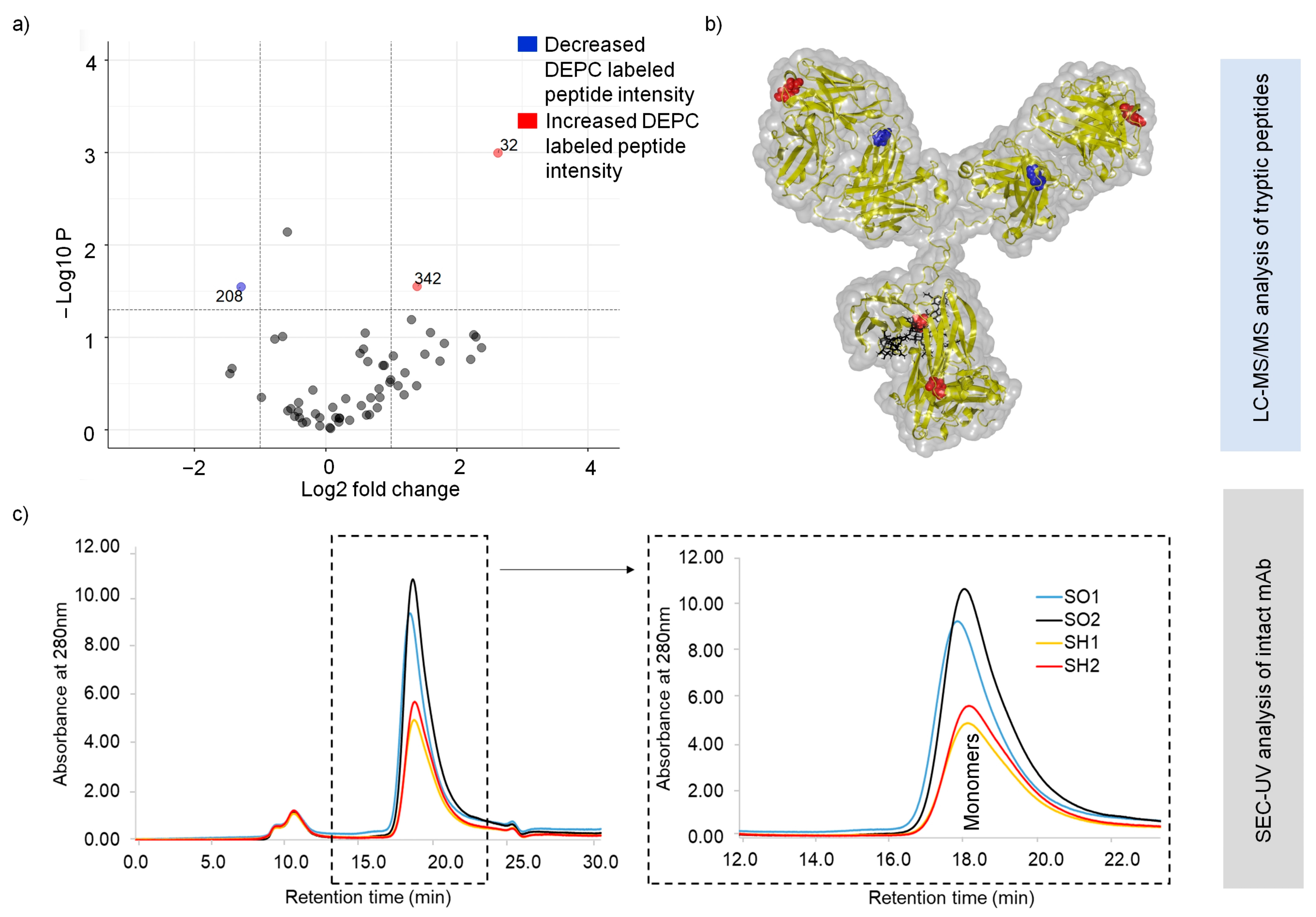
2.2. Heat-Stress-Induced Structural Changes in SILuMAb
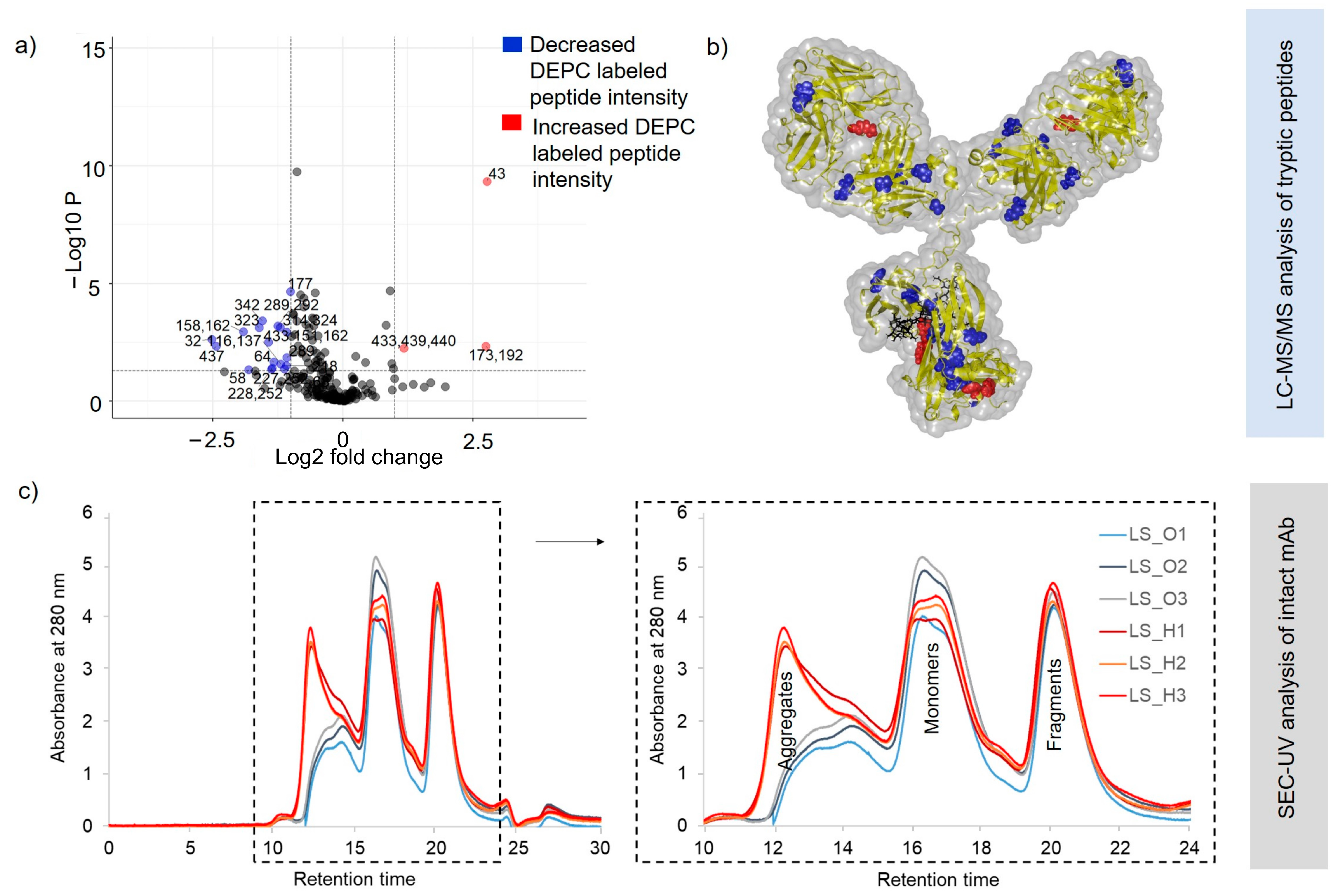
2.3. Structural Changes in Heat-Stressed, One-Year-Stored SILuMAb
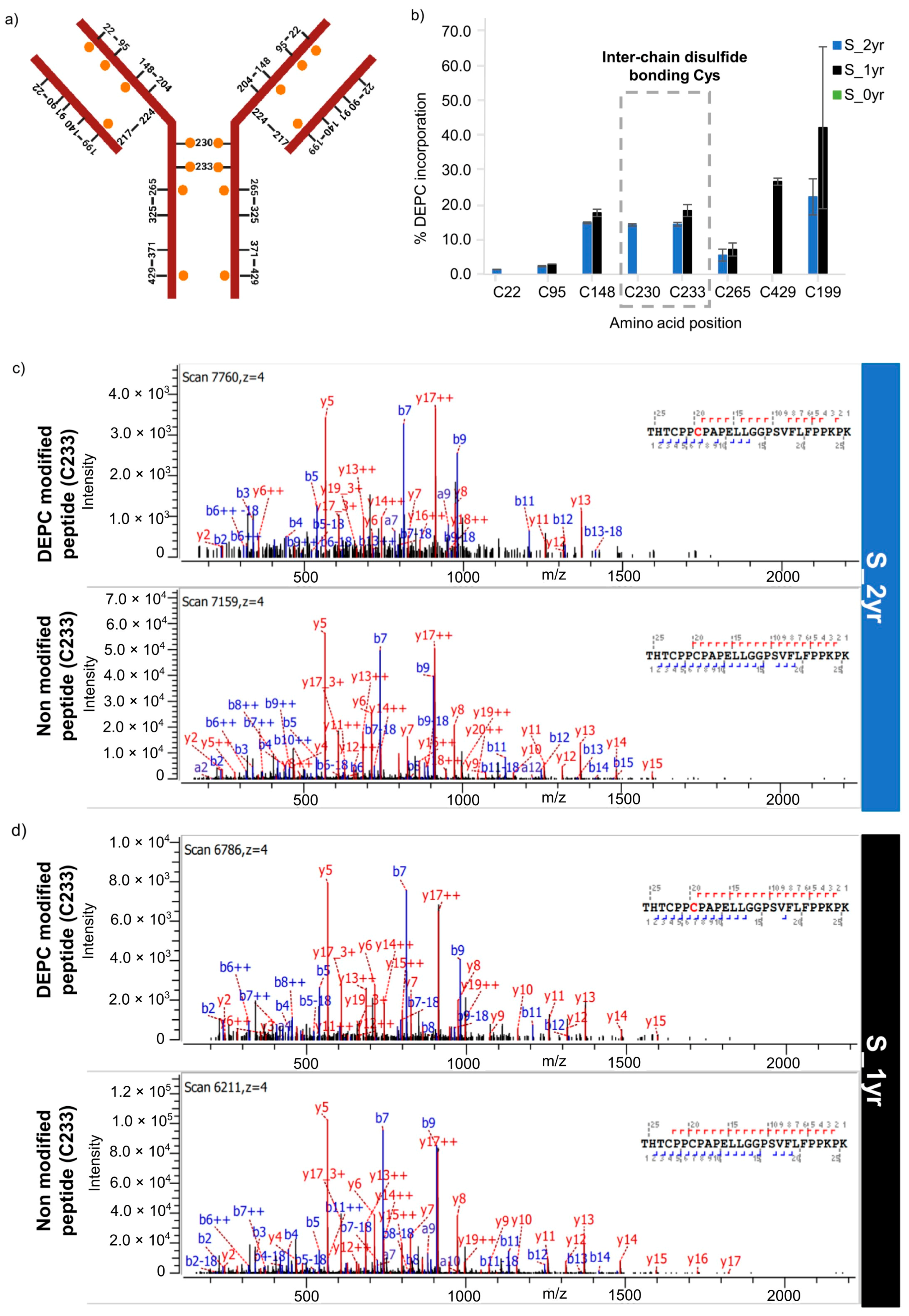
2.4. Intactness of Disulfide Bonds in Long-Term-Stored SILuMAb
3. Discussion
4. Materials and Methods
4.1. Overview of Experiments
4.2. Antibodies, Chemicals, Enzymes, and Materials
4.3. Sample Preparation for CL-MS
4.3.1. Diethyl Pyrocarbonate (DEPC) Labeling
4.3.2. Sample Preparation for LC-MS-Based Peptide Analysis
4.3.3. Sample Preparation for Intact-Protein Mass Analysis
4.4. Analytical Techniques
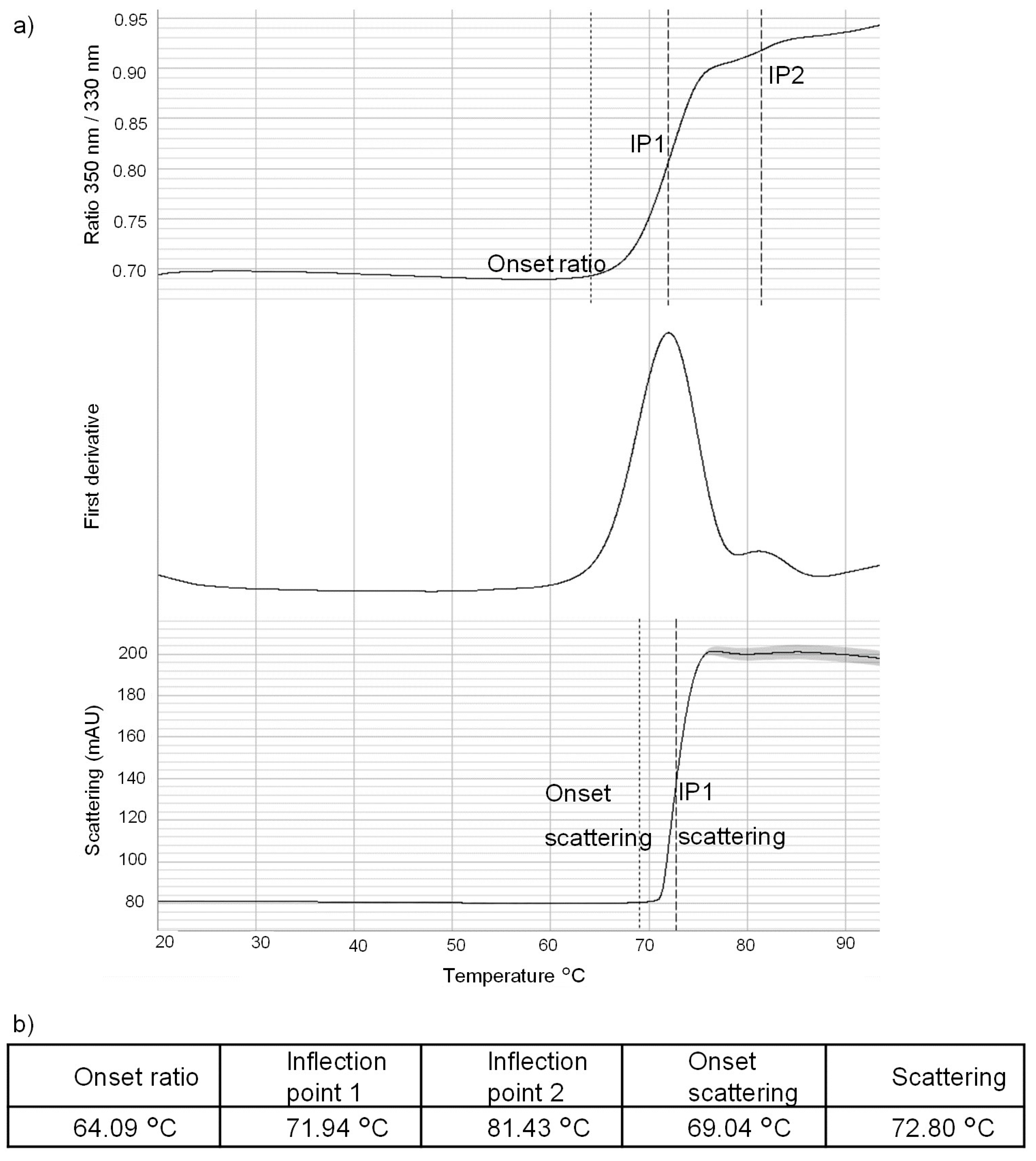
4.4.1. Nanodifferential Scanning Fluorimetry
4.4.2. LC-MS/MS-Based Tryptic Peptide Analysis
4.4.3. LC-MS-Based Intact-Protein Mass Analysis
4.4.4. SEC-UV Intact-Protein Analysis
4.5. LC-MS/MS Data Analysis and Representation
4.5.1. Peptide Quantification from LC-MS/MS Data
4.5.2. Statistical Analysis of Quantitative Peptide Data
4.5.3. Visualization of Labeled mAb as a 3D Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bunc, M.; Hadži, S.; Graf, C.; Bončina, M.; Lah, J. Aggregation Time Machine: A Platform for the Prediction and Optimization of Long-Term Antibody Stability Using Short-Term Kinetic Analysis. J. Med. Chem. 2022, 65, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. Stability Testing of New Drug Substances and Products. In Handbook of Pharmaceutical Manufacturing Formulations, 3rd ed.; CRC Press: New York, NY, USA, 2019; pp. 31–40. [Google Scholar]
- Matthews, B.R. Regulatory aspects of stability testing in Europe. Drug Dev. Ind. Pharm. 1999, 25, 831–856. [Google Scholar] [CrossRef]
- Vallejo, D.D.; Rojas Ramírez, C.; Parson, K.F.; Han, Y.; Gadkari, V.V.; Ruotolo, B.T. Mass Spectrometry Methods for Measuring Protein Stability. Chem. Rev. 2022, 122, 7690–7719. [Google Scholar] [CrossRef]
- Akbarian, M.; Chen, S.H. Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins. Pharmaceutics 2022, 14, 2533. [Google Scholar] [CrossRef]
- Campuzano, I.D.G.; Netirojjanakul, C.; Nshanian, M.; Lippens, J.L.; Kilgour, D.P.A.; Van Orden, S.; Loo, J.A. Native-MS Analysis of Monoclonal Antibody Conjugates by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2017, 90, 745–751. [Google Scholar] [CrossRef]
- Hernandez-Alba, O.; Wagner-Rousset, E.; Beck, A.; Cianférani, S. Native Mass Spectrometry, Ion Mobility, and Collision-Induced Unfolding for Conformational Characterization of IgG4 Monoclonal Antibodies. Anal. Chem. 2018, 90, 8865–8872. [Google Scholar] [CrossRef]
- Kaltashov, I.A.; Bobst, C.E.; Pawlowski, J.; Wang, G. Mass spectrometry-based methods in characterization of the higher order structure of protein therapeutics. J. Pharm. Biomed. Anal. 2020, 184, 113169. [Google Scholar] [CrossRef]
- Iacob, R.E.; Engen, J.R. Hydrogen exchange mass spectrometry: Are we out of the quicksand? J. Am. Soc. Mass Spectrom. 2012, 23, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Hamuro, Y.; Coales, S.J. Optimization of Feasibility Stage for Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 623–629. [Google Scholar] [CrossRef]
- Liu, X.R.; Huang, R.Y.C.; Zhao, F.; Chen, G.; Tao, L. Advances in mass spectrometry-based epitope mapping of protein therapeutics. J. Pharm. Biomed. Anal. 2022, 215, 114754. [Google Scholar] [CrossRef] [PubMed]
- Limpikirati, P.; Pan, X.; Vachet, R.W. Covalent Labeling with Diethylpyrocarbonate: Sensitive to the Residue Microenvironment, Providing Improved Analysis of Protein Higher Order Structure by Mass Spectrometry. Anal. Chem. 2019, 91, 8516–8523. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.; Bender, J.; Kundlacz, T.; Schmidt, C. Evaluation of NHS-Acetate and DEPC labelling for determination of solvent accessible amino acid residues in protein complexes. J. Proteom. 2020, 222, 103793. [Google Scholar] [CrossRef] [PubMed]
- Limpikirati, P.; Liu, T.; Vachet, R.W. Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods 2018, 144, 79–93. [Google Scholar] [CrossRef]
- Gau, B.C.; Sharp, J.S.; Rempel, D.L.; Gross, M.L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 2009, 81, 6563–6571. [Google Scholar] [CrossRef]
- Liu, X.R.; Rempel, D.L.; Gross, M.L. Protein higher-order-structure determination by fast photochemical oxidation of proteins and mass spectrometry analysis. Nat. Protoc. 2020, 15, 3942–3970. [Google Scholar] [CrossRef] [PubMed]
- Borotto, N.B.; Zhou, Y.; Hollingsworth, S.R.; Hale, J.E.; Graban, E.M.; Vaughan, R.C.; Vachet, R.W. Investigating Therapeutic Protein Structure with Diethylpyrocarbonate Labeling and Mass Spectrometry. Anal. Chem. 2015, 87, 10627–10634. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, C.Y.; Limpikirati, P.; Vachet, R.W. Complementary Structural Information for Stressed Antibodies from Hydrogen-Deuterium Exchange and Covalent Labeling Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2021, 32, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, V.L.; Vachet, R.W. Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Anal. Chem. 2008, 80, 2895–2904. [Google Scholar] [CrossRef]
- Zhou, Y.; Vachet, R.W. Diethylpyrocarbonate labeling for the structural analysis of proteins: Label scrambling in solution and how to avoid it. J. Am. Soc. Mass Spectrom. 2012, 23, 899–907. [Google Scholar] [CrossRef]
- Mendoza, V.L.; Vachet, R.W. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev. 2009, 28, 785–815. [Google Scholar] [CrossRef]
- Pan, X.; Tran, T.; Kirsch, Z.J.; Thompson, L.K.; Vachet, R.W. Diethylpyrocarbonate-Based Covalent Labeling Mass Spectrometry of Protein Interactions in a Membrane Complex System. J. Am. Soc. Mass Spectrom. 2022, 34, 82–91. [Google Scholar] [CrossRef]
- Guo, C.; Cheng, M.; Li, W.; Gross, M.L. Diethylpyrocarbonate Footprints a Membrane Protein in Micelles. J. Am. Soc. Mass Spectrom. 2021, 32, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Biehn, S.E.; Limpikirati, P.; Vachet, R.W.; Lindert, S. Utilization of Hydrophobic Microenvironment Sensitivity in Diethylpyrocarbonate Labeling for Protein Structure Prediction. Anal. Chem. 2021, 93, 8188–8195. [Google Scholar] [CrossRef]
- Kaur, P.; Tomechko, S.E.; Kiselar, J.; Shi, W.; Deperalta, G.; Wecksler, A.T.; Gokulrangan, G.; Ling, V.; Chance, M.R. Characterizing monoclonal antibody structure by carboxyl group footprinting. mAbs 2015, 7, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, C.Y.; Kirsch, Z.J.; Vachet, R.W. Epitope Mapping with Diethylpyrocarbonate Covalent Labeling-Mass Spectrometry. Anal. Chem. 2021, 94, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Augener, W.; Grey, H.M. Studies on the mechanism of heat aggregation of human gamma-G. J. Immunol. 1970, 105, 1024–1030. [Google Scholar] [CrossRef]
- Van Der Linden, R.H.J.; Frenken, L.G.J.; De Geus, B.; Harmsen, M.M.; Ruuls, R.C.; Stok, W.; de Ron, L.; Wilson, S.; Davis, P.; Verrips, C.T. Comparison of physical chemical properties of llama V(HH) antibody fragments and mouse monoclonal antibodies. Biochim. Et Biophys. Acta Protein Struct. Mol. Enzymol. 1999, 1431, 37–46. [Google Scholar] [CrossRef]
- Ladenson, R.C.; Crimmins, D.L.; Landt, Y.; Ladenson, J.H. Isolation and characterization of a thermally stable recombinant anti-caffeine heavy-chain antibody fragment. Anal. Chem. 2006, 78, 4501–4508. [Google Scholar] [CrossRef]
- Akazawa-Ogawa, Y.; Takashima, M.; Lee, Y.H.; Ikegami, T.; Goto, Y.; Uegaki, K.; Hagihara, Y. Heat-induced irreversible denaturation of the camelid single domain vhh antibody is governed by chemical modifications. J. Biol. Chem. 2014, 289, 15666–15679. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.A.; Keire, D.A.; Ye, H. The impact of standard accelerated stability conditions on antibody higher order structure as assessed by mass spectrometry. mAbs 2019, 11, 930–941. [Google Scholar] [CrossRef]
- Thiagarajan, G.; Semple, A.; James, J.K.; Cheung, J.K.; Shameem, M. A comparison of biophysical characterization techniques in predicting monoclonal antibody stability. mAbs 2016, 8, 1088–1097. [Google Scholar] [CrossRef]
- Halley, J.; Chou, Y.R.; Cicchino, C.; Huang, M.; Sharma, V.; Tan, N.C.; Thakkar, S.; Zhou, L.L.; Al-Azzam, W.; Cornen, S.; et al. An Industry Perspective on Forced Degradation Studies of Biopharmaceuticals: Survey Outcome and Recommendations. J. Pharm. Sci. 2019, 109, 6–21. [Google Scholar] [CrossRef]
- Limpikirati, P.K.; Zhao, B.; Pan, X.; Eyles, S.J.; Vachet, R.W. Covalent Labeling/Mass Spectrometry of Monoclonal Antibodies with Diethylpyrocarbonate: Reaction Kinetics for Ensuring Protein Structural Integrity. J. Am. Soc. Mass Spectrom. 2020, 31, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Limpikirati, P.; Chen, H.; Liu, T.; Vachet, R.W. Higher-Order Structure Influences the Kinetics of Diethylpyrocarbonate Covalent Labeling of Proteins. J. Am. Soc. Mass Spectrom. 2020, 31, 658–665. [Google Scholar] [CrossRef]
- Torrente-López, A.; Hermosilla, J.; Salmerón-García, A.; Cabeza, J.; Navas, N. Comprehensive Analysis of Nivolumab, A Therapeutic Anti-Pd-1 Monoclonal Antibody: Impact of Handling and Stress. Pharmaceutics 2022, 14, 692. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Das, T.K.; Singh, S.K.; Kumar, S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. mAbs 2009, 1, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Limpikirati, P.; Hale, J.E.; Hazelbaker, M.; Huang, Y.; Jia, Z.; Yazdani, M.; Graban, E.M.; Vaughan, R.C.; Vachet, R.W. Covalent labeling and mass spectrometry reveal subtle higher order structural changes for antibody therapeutics. mAbs 2019, 11, 463–476. [Google Scholar] [CrossRef]
- Zhang, H.; Dalby, P.A. Stability enhancement in a mAb and Fab coformulation. Sci. Rep. 2020, 10, 21129. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Cheung, J.K.; Dellatore, S.M.; Katiyar, A.; Bhat, R.; Sun, J.; Ponniah, G.; Neill, A.; Mason, B.; Beck, A.; et al. Forced degradation of recombinant monoclonal antibodies: A practical guide. mAbs 2017, 9, 1217–1230. [Google Scholar] [CrossRef]
- Li, X.; Xiao, L.; Kochert, B.; Donnelly, D.P.; Gao, X.; Richardson, D. Extended characterization of unpaired cysteines in an IgG1 monoclonal antibody by LC-MS analysis. Anal. Biochem. 2021, 622, 114172. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, J.; Hewitt, D.; Tran, B.; Gao, X.; Qiu, Z.J.; Tejada, M.; Gazzano-Santoro, H.; Kao, Y.-H. Identification and characterization of buried unpaired cysteines in a recombinant monoclonal igg1 antibody. Anal. Chem. 2012, 84, 7112–7123. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Yates, Z.; Balland, A.; Kleemann, G.R. Human IgG1 hinge fragmentation as the result of H2O2-mediated radical cleavage. J. Biol. Chem. 2009, 284, 35390–35402. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.C.; Chang, C.W.; Huang, S.Y.; Wei, T.Y.; Huang, Y.L.; Lin, Y.H.; Chen, H.-M.; Chen, S.-F. Evaluation of disulfide scrambling during the enzymatic digestion of bevacizumab at various pH values using mass spectrometry. Biochim. Et Biophys. Acta Proteins Proteom. 2016, 1864, 1188–1194. [Google Scholar] [CrossRef]
- Coghlan, J.; Benet, A.; Kumaran, P.; Ford, M.; Veale, L.; Skilton, S.J.; Saveliev, S.; Schwendeman, A.A. Streamlining the Characterization of Disulfide Bond Shuffling and Protein Degradation in IgG1 Biopharmaceuticals Under Native and Stressed Conditions. Front. Bioeng. Biotechnol. 2022, 10, 862456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, Q.; Wu, S.; Karger, B.L.; Hancock, W.S. Characterization and Comparison of Disulfide Linkages and Using LC-MS with Electron Transfer Dissociation. Anal. Chem. 2011, 83, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Vlasak, J.; Ionescu, R. Fragmentation of monoclonal antibodies. mAbs 2011, 3, 253–263. [Google Scholar] [CrossRef]
- Gao, S.X.; Zhang, Y.; Stansberry-Perkins, K.; Buko, A.; Bai, S.; Nguyen, V.; Brader, M.L. Fragmentation of a highly purified monoclonal antibody attributed to residual CHO cell protease activity. Biotechnol. Bioeng. 2011, 108, 977–982. [Google Scholar] [CrossRef]
- Pikal-Cleland, K.A.; Carpenter, J.F. Lyophilization-induced protein denaturation in phosphate buffer systems: Monomeric and tetrameric β-galactosidase. J. Pharm. Sci. 2001, 90, 1255–1268. [Google Scholar] [CrossRef]
- Kirsch, Z.J.; Arden, B.G.; Vachet, R.W.; Limpikirati, P. Covalent Labeling with Diethylpyrocarbonate for Studying Protein Higher-Order Structure by Mass Spectrometry. J. Vis. Exp. 2021, 172, e61983. [Google Scholar]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaikwad, M.; Richter, F.; Götz, R.; Dörrbaum, A.; Schumacher, L.; Tonillo, J.; Frech, C.; Kellner, R.; Hopf, C. Site-Specific Structural Changes in Long-Term-Stressed Monoclonal Antibody Revealed with DEPC Covalent-Labeling and Quantitative Mass Spectrometry. Pharmaceuticals 2023, 16, 1418. https://doi.org/10.3390/ph16101418
Gaikwad M, Richter F, Götz R, Dörrbaum A, Schumacher L, Tonillo J, Frech C, Kellner R, Hopf C. Site-Specific Structural Changes in Long-Term-Stressed Monoclonal Antibody Revealed with DEPC Covalent-Labeling and Quantitative Mass Spectrometry. Pharmaceuticals. 2023; 16(10):1418. https://doi.org/10.3390/ph16101418
Chicago/Turabian StyleGaikwad, Manasi, Florian Richter, Rabea Götz, Aline Dörrbaum, Lena Schumacher, Jason Tonillo, Christian Frech, Roland Kellner, and Carsten Hopf. 2023. "Site-Specific Structural Changes in Long-Term-Stressed Monoclonal Antibody Revealed with DEPC Covalent-Labeling and Quantitative Mass Spectrometry" Pharmaceuticals 16, no. 10: 1418. https://doi.org/10.3390/ph16101418
APA StyleGaikwad, M., Richter, F., Götz, R., Dörrbaum, A., Schumacher, L., Tonillo, J., Frech, C., Kellner, R., & Hopf, C. (2023). Site-Specific Structural Changes in Long-Term-Stressed Monoclonal Antibody Revealed with DEPC Covalent-Labeling and Quantitative Mass Spectrometry. Pharmaceuticals, 16(10), 1418. https://doi.org/10.3390/ph16101418