
Carboxyl Group-Modified Myoglobin Induces TNF-α-Mediated Apoptosis in Leukemia Cells

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Death Receptor-Mediated Apoptotic Pathway Is Involved in SEM-Mb-Mediated Cytotoxicity
2.2. SEM-Mb Induces Ca2+-Elicited ROS Generation, Which in Turn Reduces U937 Cell Viability
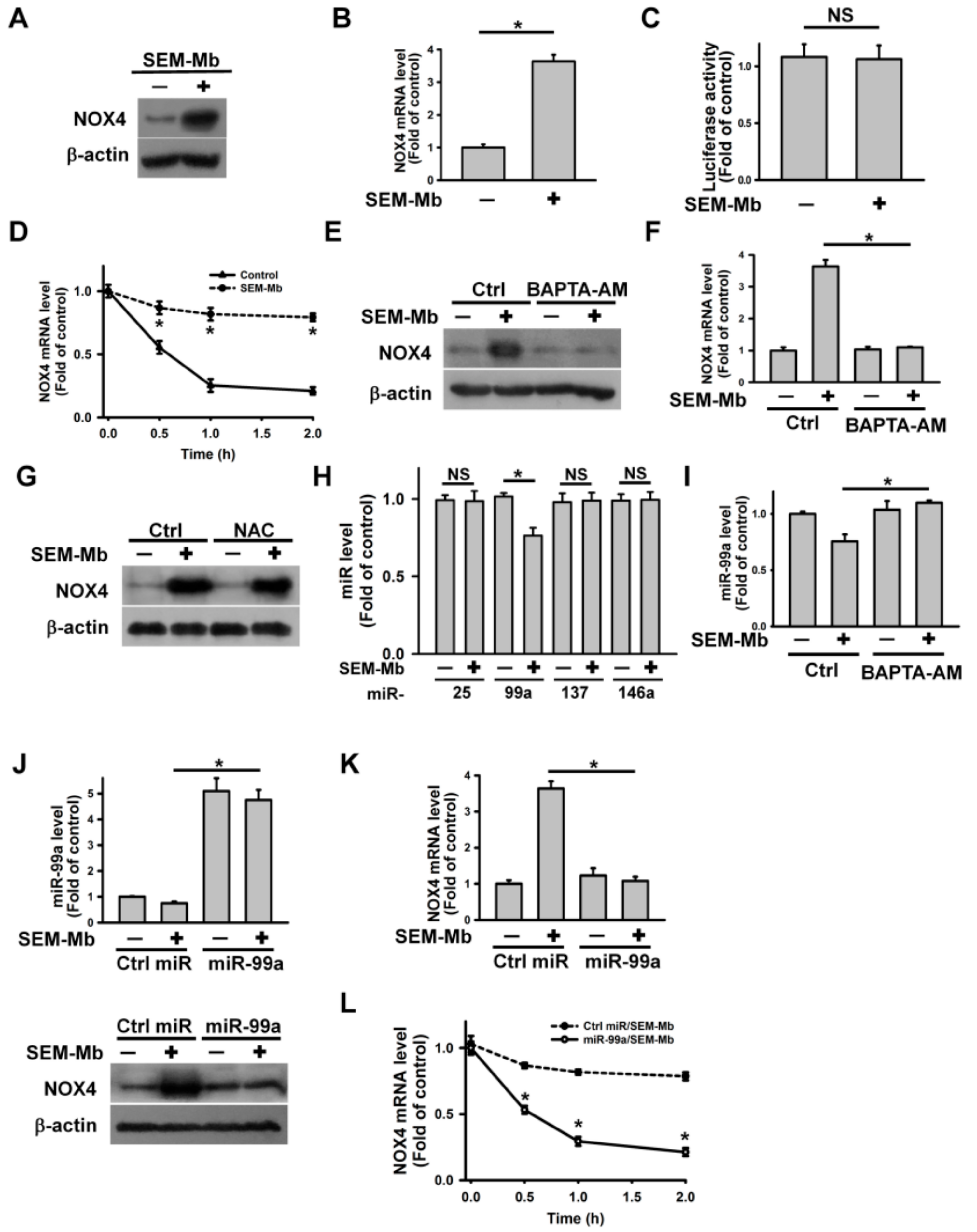
2.3. SEM-Mb Induces Ca2+-Mediated Downregulation of miR-99a, Leading to Upregulation of NOX4 Expression
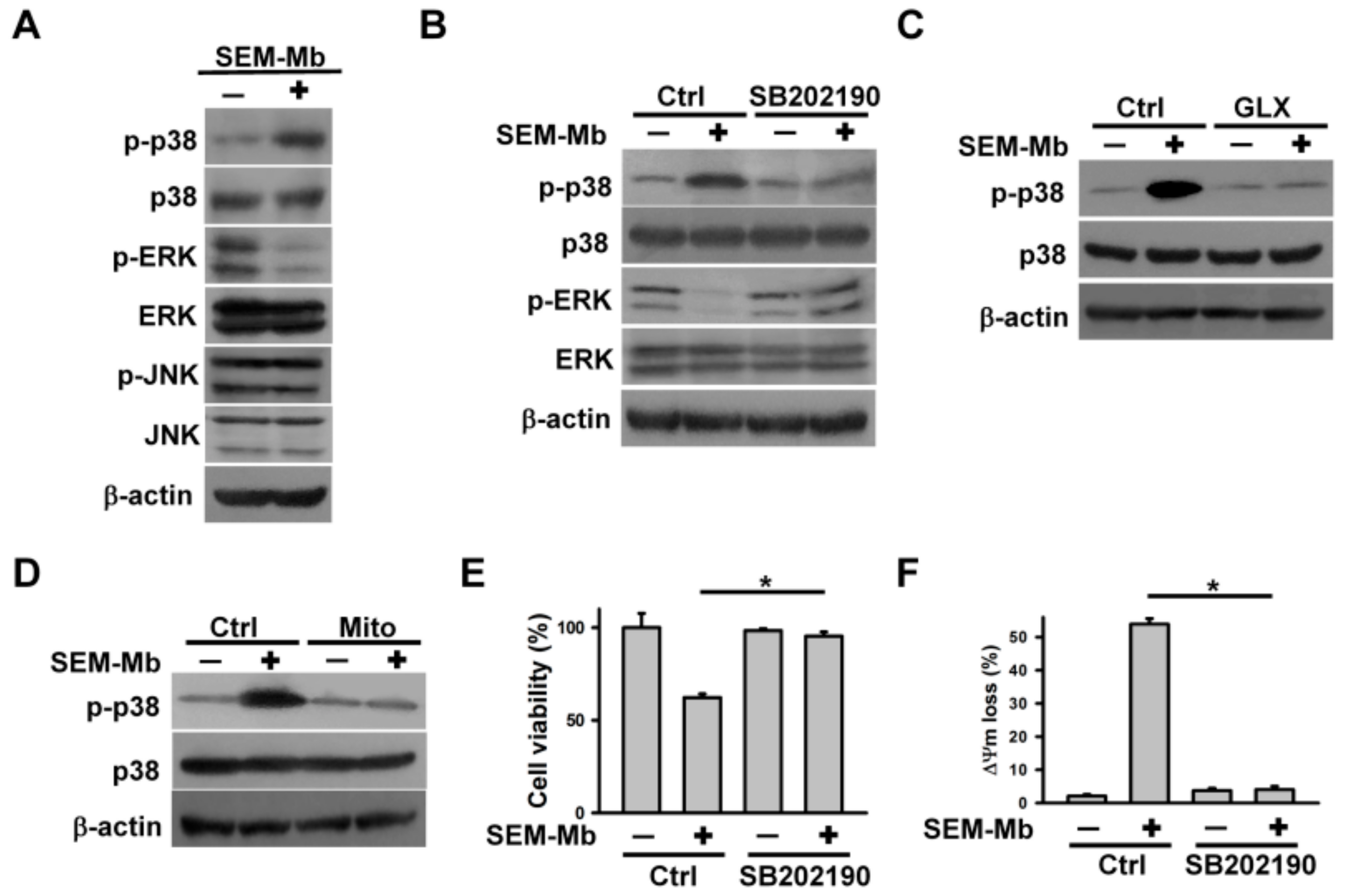
2.4. The Cytotoxicity of SEM-Mb Is Related to ROS-Mediated p38 MAPK Activation
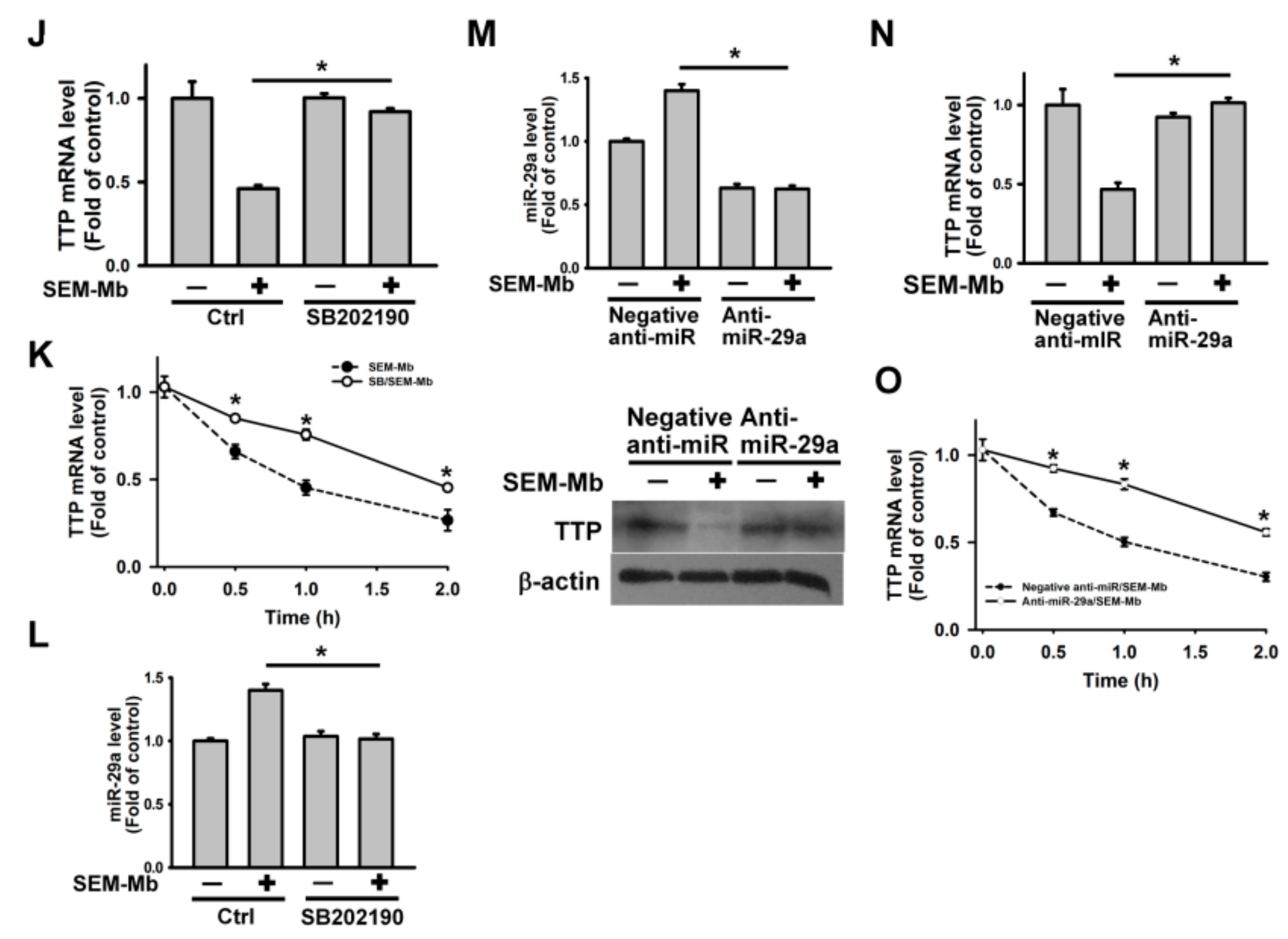
2.5. SEM-Mb Promotes TNF-α Expression via A p38 MAPK-Dependent Post-Transcriptional Pathway
2.6. SEM-Mb-Induced p38 MAPK Activation Increases miR-29a Expression, Leading to the Inhibition of TTP Expression
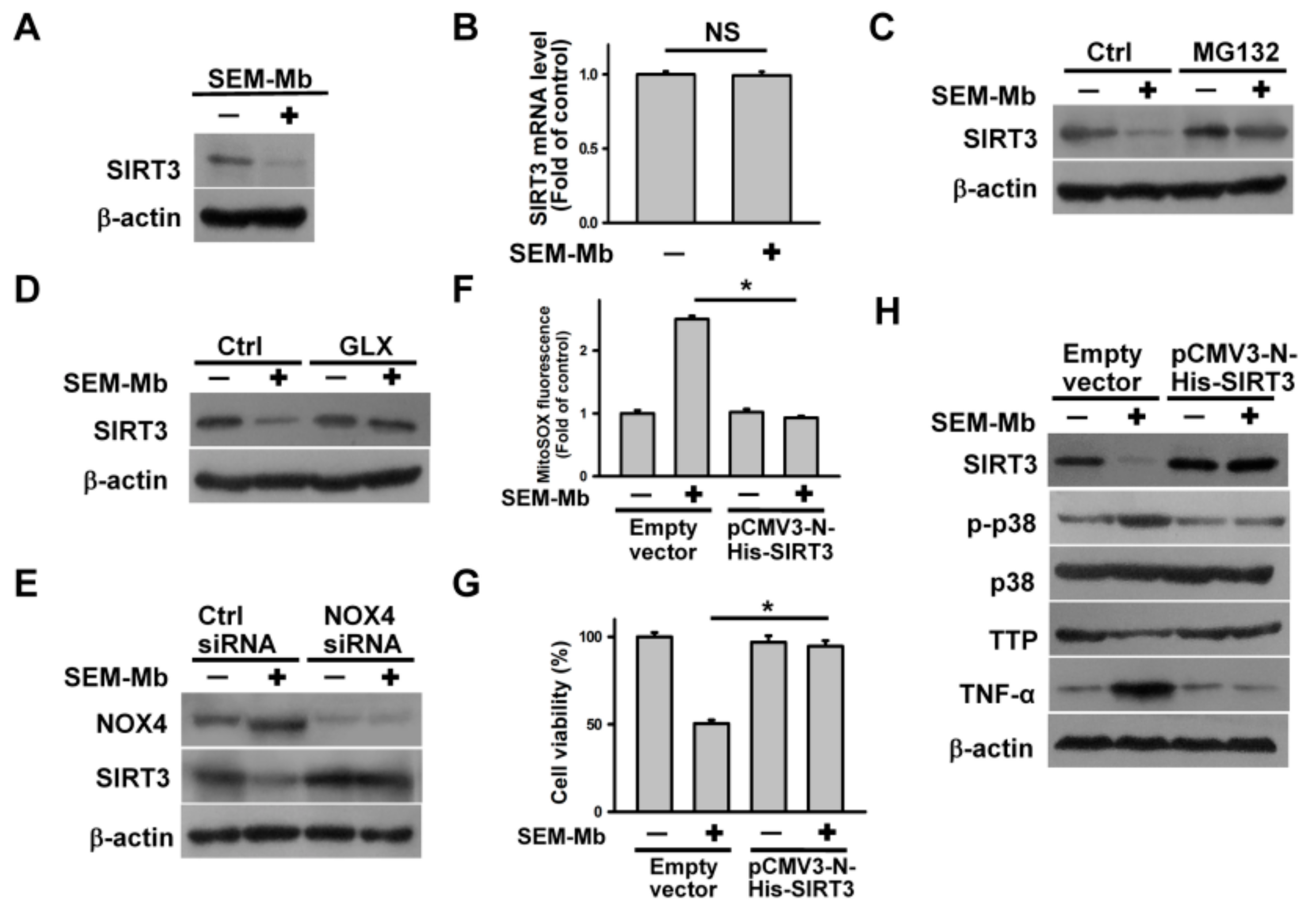
2.7. SEM-Mb Triggers p38 MAPK/TTP Axis-Dependent TNF-α Expression by Promoting SIRT3 Degradation
2.8. SEM-Mb Induces Apoptosis in HL-60 Cells via NOX4/SIRT3/p38 MAPK/TTP Axis-Dependent TNF-α Expression
2.9. SEM-Mb Induces Death of THP-1, K562, Jurkat, and ABT-199-Resistant U937 Cells via p38 MAPK-Mediated TNF-α Expression
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Modification of Mb with Semicarbazide
4.3. Cell Culture
4.4. Detection of ROS, [Ca2+]i, and Mitochondrial Depolarization
4.5. Immunoblotting
4.6. qRT-PCR
4.7. Transfection with Plasmid and siRNA
4.8. Stem-Loop qRT-PCR for miRNAs
4.9. Transient Transfection of miRNA Mimic and Inhibitor
4.10. Luciferase Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baslé, E.; Joubert, N.; Pucheault, M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat. Commun. 2014, 5, 4740. [Google Scholar] [CrossRef] [PubMed]
- Naowarojna, N.; Cheng, R.; Lopez, J.; Wong, C.; Qiao, L.; Liu, P. Chemical modifications of proteins and their applications in metalloenzyme studies. Synth. Syst. Biotechnol. 2021, 6, 32–49. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Modulation of immobilized lipase enatioselectivity via chemical amination. Adv. Synth. Catal. 2007, 349, 1119–1127. [Google Scholar] [CrossRef]
- Li, L.; He, L.; Tan, S.; Guo, X.; Lu, H.; Qi, Z.; Pan, C.; An, X.; Jiang, S.; Liu, S. 3-Hydroxyphthalic anhydride-modified chicken ovalbumin exhibits potent and broad anti-HIV-1 activity: A potential microbicide for preventing sexual transmission of HIV-1. Antimicrob. Agents Chemother. 2010, 54, 1700–1711. [Google Scholar] [CrossRef]
- Li, L.; Qiao, P.; Yang, J.; Lu, L.; Tan, S.; Lu, H.; Zhang, X.; Chen, X.; Wu, S.; Jiang, S.; et al. Maleic anhydride-modified chicken ovalbumin as an effective and inexpensive anti-HIV microbicide candidate for prevention of HIV sexual transmission. Retrovirology 2010, 7, 37. [Google Scholar] [CrossRef]
- Yang, S.Y.; Chen, Y.J.; Kao, P.H.; Chang, L.S. Bovine serum albumin with glycated carboxyl groups shows membrane-perturbing activities. Arch. Biochem. Biophys. 2014, 564, 43–51. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Chen, Y.J.; Fu, Y.S.; Chang, L.S. Antibacterial and membrane-damaging activities of mannosylated bovine serum albumin. Arch. Biochem. Biophys. 2015, 573, 14–22. [Google Scholar] [CrossRef]
- Tang, C.C.; Shi, Y.J.; Chen, Y.J.; Chang, L.S. Ovalbumin with glycated carboxyl groups shows membrane-damaging activity. Int. J. Mol. Sci. 2017, 18, 520. [Google Scholar] [CrossRef]
- Shi, Y.J.; Wang, R.T.; Chu, Y.H.; Chen, Y.J.; Tang, C.C.; Fu, Y.S.; Lee, Y.C.; Wang, L.J.; Huang, C.H.; Chang, L.S. Membrane-damaging activities of mannosylated ovalbumin are involved in its antibacterial action. Arch. Biochem. Biophys. 2018, 639, 1–8. [Google Scholar] [CrossRef]
- Shi, Y.J.; Chiou, J.T.; Huang, C.H.; Lee, Y.C.; Wang, L.J.; Chang, L.S. Modification of carboxyl groups converts α-lactalbumin into an active molten globule state with membrane-perturbing activity and cytotoxicity. Int. J. Biol. Macromol. 2020, 163, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Chiou, J.T.; Shi, Y.J.; Lee, Y.C.; Wang, L.J.; Chen, Y.J.; Chang, L.S. Carboxyl group-modified α-lactalbumin induces TNF-α-mediated apoptosis in leukemia and breast cancer cells through the NOX4/p38 MAPK/PP2A axis. Int. J. Biol. Macromol. 2021, 187, 513–527. [Google Scholar] [CrossRef]
- Brinkmann, C.R.; Thiel, S.; Otzen, D.E. Protein-fatty acid complexes: Biochemistry, biophysics and function. FEBS J. 2013, 280, 1733–1749. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.C.S.; Nadeem, A.; Svanborg, C. HAMLET-A protein-lipid complex with broad tumoricidal activity. Biochem. Biophys. Res. Commun. 2017, 482, 454–458. [Google Scholar] [CrossRef]
- Postnikova, G.B.; Shekhovtsova, E.A. Myoglobin: Oxygen depot or oxygen transporter to mitochondria? A novel mechanism of myoglobin deoxygenation in cells (review). Biochemistry (Moscow) 2018, 83, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Narita, M.; Itsuno, Y.; Itsuno, S. Autonomous sequences in myoglobin emerging from x-ray structure of holomyoglobin. ACS Omega 2019, 4, 992–999. [Google Scholar] [CrossRef]
- Mak, P.; Szewczyk, A.; Mickowska, B.; Kicinska, A.; Dubin, A. Effect of antimicrobial apomyoglobin 56-131 peptide on liposomes and planar lipid bilayer membrane. Int. J. Antimicrob. Agents 2001, 17, 137–142. [Google Scholar] [CrossRef]
- Flonta, S.E.; Arena, S.; Pisacane, A.; Michieli, P.; Bardelli, A. Expression and functional regulation of myoglobin in epithelial cancers. Am. J. Pathol. 2009, 175, 201–206. [Google Scholar] [CrossRef]
- Oleksiewicz, U.; Daskoulidou, N.; Liloglou, T.; Tasopoulou, K.; Bryan, J.; Gosney, J.R.; Field, J.K.; Xinarianos, G. Neuroglobin and myoglobin in non-small cell lung cancer: Expression, regulation and prognosis. Lung Cancer 2011, 74, 411–418. [Google Scholar] [CrossRef]
- Behnes, C.L.; Bedke, J.; Schneider, S.; Küffer, S.; Strauss, A.; Bremmer, F.; Ströbel, P.; Radzun, H.J. Myoglobin expression in renal cell carcinoma is regulated by hypoxia. Exp. Mol. Pathol. 2013, 95, 307–312. [Google Scholar] [CrossRef]
- Meller, S.; Bicker, A.; Montani, M.; Ikenberg, K.; Rostamzadeh, B.; Sailer, V.; Wild, P.; Dietrich, D.; Uhl, B.; Sulser, T.; et al. Myoglobin expression in prostate cancer is correlated to androgen receptor expression and markers of tumor hypoxia. Virchows Arch. 2014, 465, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Meller, S.; Van Ellen, A.; Gevensleben, H.; Bicker, A.; Hankeln, T.; Gorr, T.A.; Sailer, V.; Dröge, F.; Schröck, F.; Bootz, F.; et al. Ectopic myoglobin expression is associated with a favourable outcome in head and neck squamous cell carcinoma patients. Anticancer. Res. 2016, 36, 6235–6241. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, G.; Rose, M.; Geisler, C.; Fritzsche, F.R.; Gerhardt, J.; Lüke, C.; Ladhoff, A.M.; Knüchel, R.; Dietel, M.; Moch, H.; et al. Endogenous myoglobin in human breast cancer is a hallmark of luminal cancer phenotype. Br. J. Cancer 2010, 102, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Burkhart, K. Myoglobin toxicity in proximal human kidney cells: Roles of Fe, Ca2+, H2O2, and terminal mitochondrial electron transport. Kidney Int. 1997, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.J.; Lee, Y.C.; Wang, L.J.; Chiou, J.T.; Tseng, B.Y. Chang, L.S. Carboxyl group-modified myoglobin shows membrane-permeabilizing activity. Arch. Biochim. Biophys. 2022, 728, 109371. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledanom, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Y.; Wang, Z.; Wang, L.; Wei, X.; Zhang, B.; Wen, Z.; Fang, H.; Pang, Q.; Yi, F. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am. J. Nephrol. 2010, 32, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Sun, Q.; Jiang, Y.; Zhang, X.; Chen, L.; Xie, C.; Qin, F.; Chen, Y.; Lv, H.; Chen, W.; et al. MiR-146a affects the alteration in myeloid differentiation induced by hydroquinone in human CD34+ hematopoietic progenitor cells and HL-60 cells. Toxicol. Res. 2016, 5, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.O.; Reiisi, S.; Shareef, S. miRNAs, oxidative stress, and cancer: A comprehensive and updated review. J. Cell. Physiol. 2020, 35, 8812–8825. [Google Scholar] [CrossRef]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- White, C.D.; Sacks, D.B. Regulation of MAP kinase signaling by calcium. Methods Mol. Biol. 2010, 661, 151–165. [Google Scholar]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How can ROS activate MAPK pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Chen, Y.J.; Cheng, T.L.; Lin, S.R.; Chang, L.S. Cross talk between p38MAPK and ERK is mediated through MAPK-mediated protein phosphatase 2A catalytic subunit α and MAPK phosphatase-1 expression in human leukemia U937 cells. Cell. Signal. 2013, 25, 1845–1851. [Google Scholar] [CrossRef]
- Liu, W.H.; Chou, W.M.; Chang, L.S. p38 MAPK/PP2Acα/TTP pathway on the connection of TNF-α and caspases activation on hydroquinone-induced apoptosis. Carcinogenesis 2013, 34, 818–827. [Google Scholar] [CrossRef]
- Gebeshuber, C.A.; Zatloukal, K.; Martinez, J. miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep. 2009, 10, 400–405. [Google Scholar] [CrossRef]
- Wang, L.J.; Lee, Y.C.; Huang, C.H.; Shi, Y.J.; Chen, Y.J.; Pei, S.N.; Chou, Y.W.; Chang, L.S. Non-mitotic effect of albendazole triggers apoptosis of human leukemia cells via SIRT3/ROS/p38 MAPK/TTP axis-mediated TNF-α upregulation. Biochem. Pharmacol. 2019, 162, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Magina, K.N.; Pregartner, G.; Zebisch, A.; Wölfler, A.; Neumeister, P.; Greinix, H.T.; Berghold, A.; Sill, H. Cytarabine dose in the consolidation treatment of AML: A systematic review and meta-analysis. Blood 2017, 130, 946–948. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Reszka, K.J.; Wagner, B.A.; Teesch, L.M.; Britigan, B.E.; Spitz, D.R.; Burns, C.P. Inactivation of anthracyclines by cellular peroxidase. Cancer Res. 2005, 65, 6346–6353. [Google Scholar] [CrossRef]
- Hosseini, M.; Rezvani, H.R.; Aroua, N.; Bosc, C.; Farge, T.; Saland, E.; Guyonnet-Dupérat, V.; Zaghdoudi, S.; Jarrou, L.; Larrue, C.; et al. Targeting myeloperoxidase disrupts mitochondrial redox balance and overcomes cytarabine resistance in human acute myeloid leukemia. Cancer Res. 2019, 79, 5191–5203. [Google Scholar] [CrossRef]
- Cerella, C.; Dicato, M.; Diederich, M. BH3 Mimetics in AML Therapy: Death and Beyond? Trends Pharmacol. Sci. 2020, 41, 793–814. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Chiou, J.T.; Lee, Y.C.; Chang, L.S. Docetaxel-triggered SIDT2/NOX4/JNK/HuR signaling axis is associated with TNF-α-mediated apoptosis of cancer cells. Biochem. Pharmacol. 2022, 195, 114865. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Iwahara, T.; Bonasio, R.; Narendra, V.; Reinberg, D. SIRT3 functions in the nucleus in the control of stress-related gene expression. Mol. Cell. Biol. 2012, 32, 5022–5034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florean, C.L.; Song, S.; Dicato, M.; Diederich, M. Redox biology of regulated cell death in cancer: A focus on necroptosis and ferroptosis. Free Radic. Biol. Med. 2019, 134, 177–189. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer. 2019, 18, 100. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J.; Svistunenko, D.A.; Cooper, C.E.; Wilson, M.T. The radical and redox chemistry of myoglobin and hemoglobin: From in vitro studies to human pathology. Antioxid. Redox. Signal. 2004, 6, 954–966. [Google Scholar]
- Richards, M.P. Redox reactions of myoglobin. Antioxid. Redox. Signal. 2013, 18, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Braganza, A.; Quesnelle, K.; Bickta, J.; Reyes, C.; Wang, Y.; Jessup, M.; St Croix, C.; Arlotti, J.; Singh, S.V.; Shiva, S. Myoglobin induces mitochondrial fusion, thereby inhibiting breast cancer cell proliferation. J. Biol. Chem. 2019, 294, 7269–7282. [Google Scholar] [CrossRef] [PubMed]
- Gburek, J.; Birn, H.; Verroust, P.J.; Goj, B.; Jacobsen, C.; Moestrup, S.K.; Willnow, T.E.; Christensen, E.I. Renal uptake of myoglobin is mediated by the endocytic receptors megalin and cubilin. Am. J. Physiol. Renal. Physiol. 2003, 285, F451–F458. [Google Scholar] [CrossRef]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef]
- Svanborg, C.; Agerstam, H.; Aronson, A.; Bjerkvig, R.; Düringer, C.; Fischer, W.; Gustafsson, L.; Hallgren, O.; Leijonhuvud, I.; Linse, S.; et al. HAMLET kills tumor cells by an apoptosis-like mechanism--cellular, molecular, and therapeutic aspects. Adv. Cancer Res. 2003, 88, 1–29. [Google Scholar]
- Lin, L.; Pinker, R.J.; Forde, K.; Rose, G.D.; Kallenbach, N.R. Molten globular characteristics of the native state of apomyoglobin. Nat. Struct. Biol. 1994, 1, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Aizawa, T.; Kariya, R.; Okada, S.; Demura, M.; Kawano, K.; Makabe, K.; Kuwajima, K. Molecular mechanisms of the cytotoxicity of human α-lactalbumin made lethal to tumor cells (HAMLET) and other protein-oleic acid complexes. J. Biol. Chem. 2013, 288, 14408–14416. [Google Scholar] [CrossRef] [PubMed]
- Van Beers, M.M.; Bardor, M. Minimizing immunogenicity of biopharmaceuticals by controlling critical quality attributes of proteins. Biotechnol. J. 2012, 7, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Dingman, R.; Balu-Iyer, S.V. Immunogenicity of protein pharmaceuticals. J. Pharm. Sci. 2019, 108, 1637–1654. [Google Scholar] [CrossRef]
- El-Maksoud, A.A.A.; Makhlouf, A.I.A.; Altemimi, A.B.; El-Ghany, I.H.A.; Nassrallah, A.; Cacciola, F.; Abedelmaksoud, T.G. Nano milk protein-mucilage complexes: Characterization and anticancer effect. Molecules 2021, 26, 6372. [Google Scholar] [CrossRef]
- Van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]
- Chiou, J.T.; Lee, Y.C.; Wang, L.J.; Chang, L.S. BCL2 inhibitor ABT-199 and BCL2L1 inhibitor WEHI-539 coordinately promote NOXA-mediated degradation of MCL1 in human leukemia cells. Chem. Biol. Interact. 2022, 361, 109978. [Google Scholar] [CrossRef]
- Lee, Y.C.; Wang, L.J.; Huang, C.H.; Shi, Y.J.; Chang, L.S. ABT-263-induced MCL1 upregulation depends on autophagy-mediated 4EBP1 downregulation in human leukemia cells. Cancer Lett. 2018, 432, 191–204. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chang, L.S. Arecoline-induced death of human leukemia K562 cells is associated with surface up-modulation of TNFR2. J. Cell. Physiol. 2012, 227, 2240–2251. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-C.; Chiou, J.-T.; Wang, L.-J.; Shi, Y.-J.; Chen, Y.-J.; Chang, L.-S. Carboxyl Group-Modified Myoglobin Induces TNF-α-Mediated Apoptosis in Leukemia Cells. Pharmaceuticals 2022, 15, 1066. https://doi.org/10.3390/ph15091066
Lee Y-C, Chiou J-T, Wang L-J, Shi Y-J, Chen Y-J, Chang L-S. Carboxyl Group-Modified Myoglobin Induces TNF-α-Mediated Apoptosis in Leukemia Cells. Pharmaceuticals. 2022; 15(9):1066. https://doi.org/10.3390/ph15091066
Chicago/Turabian StyleLee, Yuan-Chin, Jing-Ting Chiou, Liang-Jun Wang, Yi-Jun Shi, Ying-Jung Chen, and Long-Sen Chang. 2022. "Carboxyl Group-Modified Myoglobin Induces TNF-α-Mediated Apoptosis in Leukemia Cells" Pharmaceuticals 15, no. 9: 1066. https://doi.org/10.3390/ph15091066
APA StyleLee, Y.-C., Chiou, J.-T., Wang, L.-J., Shi, Y.-J., Chen, Y.-J., & Chang, L.-S. (2022). Carboxyl Group-Modified Myoglobin Induces TNF-α-Mediated Apoptosis in Leukemia Cells. Pharmaceuticals, 15(9), 1066. https://doi.org/10.3390/ph15091066