
Total Synthesis of the Antimycobacterial Natural Product Chlorflavonin and Analogs via a Late-Stage Ruthenium(II)-Catalyzed ortho-C(sp2)-H-Hydroxylation

 ,
,  , , ,
, , ,
Abstract

1. Introduction
2. Results and Discussion
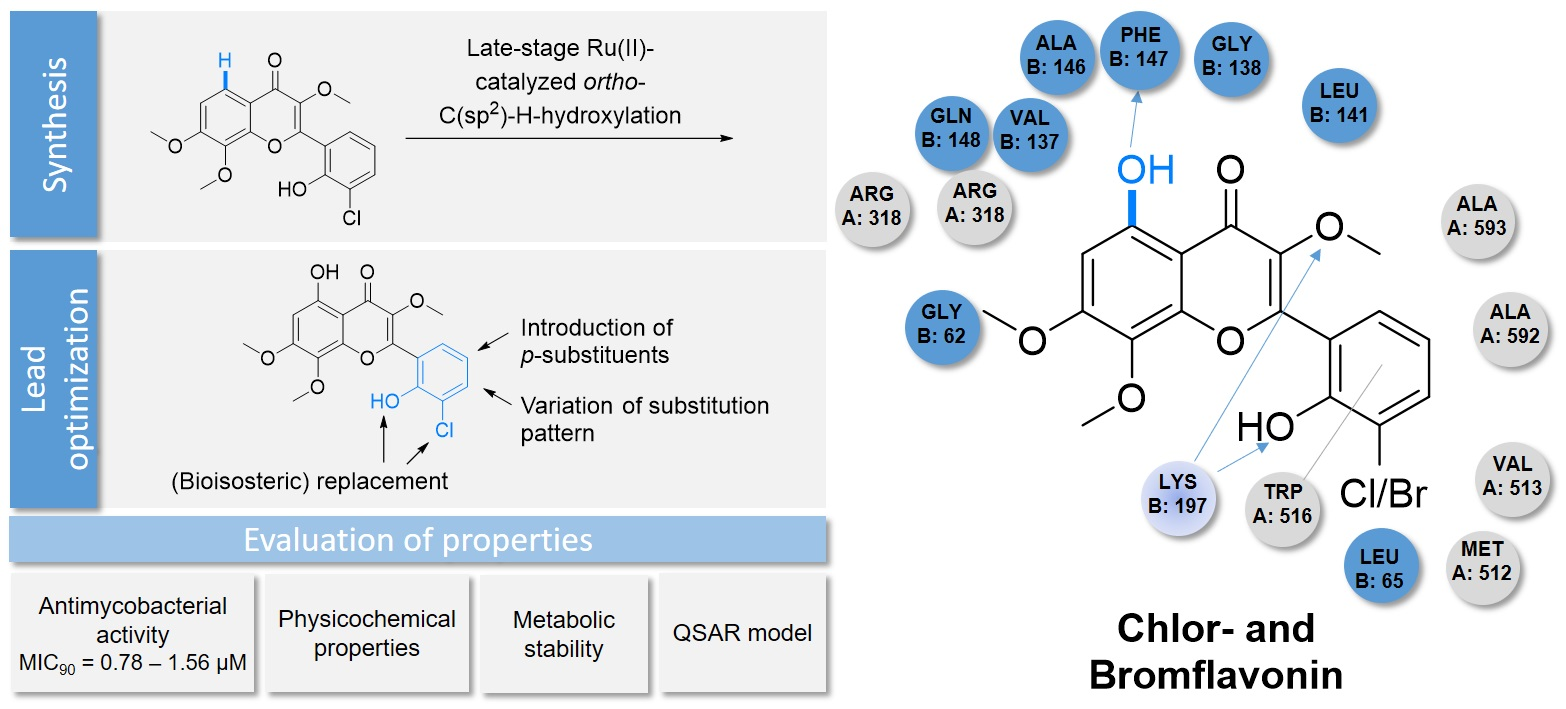
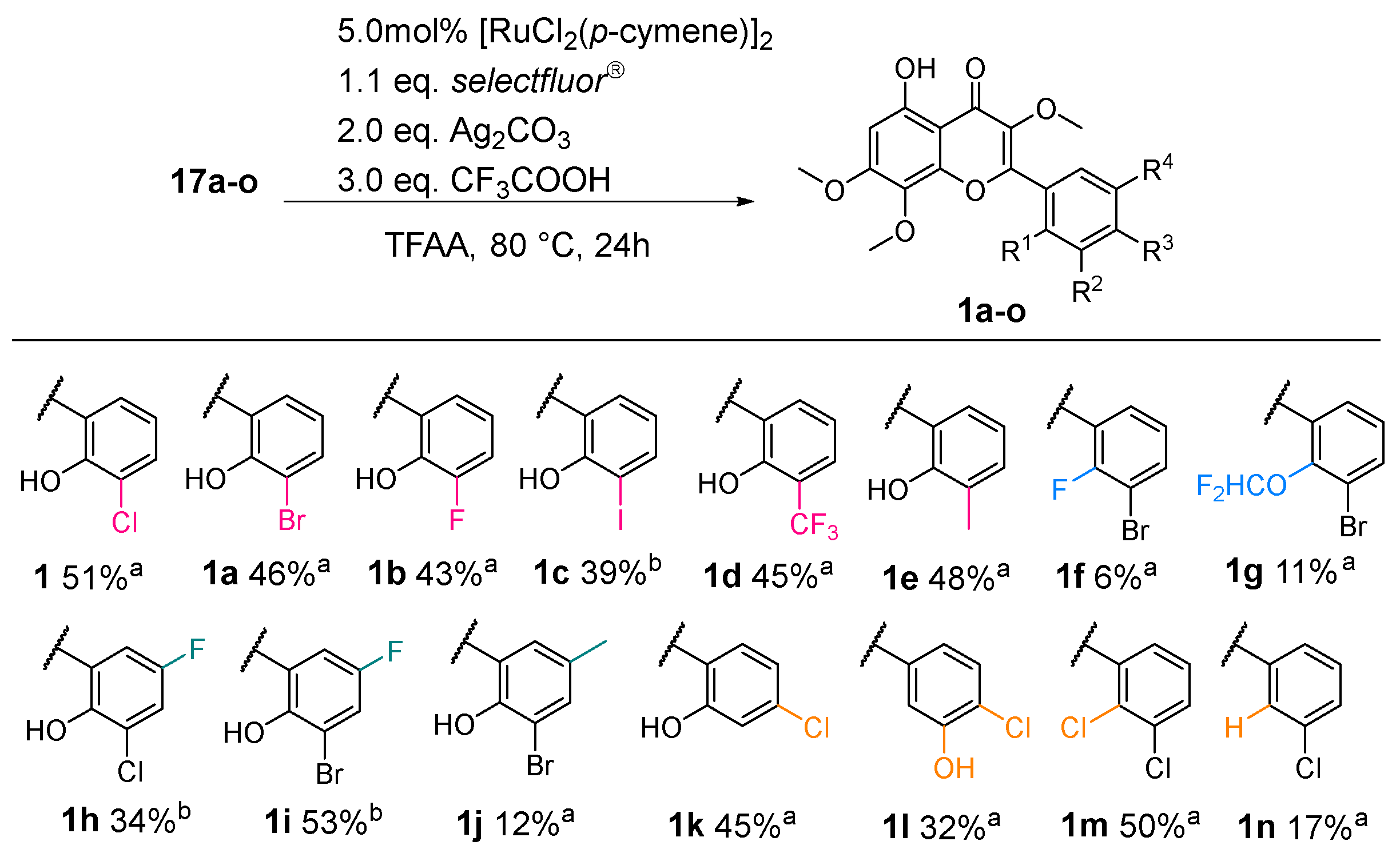
2.1. Synthesis of CF
2.2. Lead Optimization of CF
2.3. Evaluation of Selected Physicochemical Properties, Microsomal Stability, and Computations
3. Materials and Methods
3.1. Chemistry
3.2. Determination of Minimal Inhibitory Concentration (MIC)
3.3. Determination of Cytotoxcity
3.4. Evaluation of Solubility
3.5. Evaluation of Compound Chemical Stability
3.6. Evaluation of Compound Metabolic Stability
3.7. Determination of Compound pKa Values
3.8. Analysis of a Quantitative Structure-Activity Relationship Model
3.9. Solubility and Partition Coefficient Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gandhi, N.R.; Brust, J.C.M.; Shah, N.S. A new era for treatment of drug-resistant tuberculosis. Eur. Respir. J. 2018, 52, 1801350. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, E.H.; Dooley, K.E. New Drugs for the Treatment of Tuberculosis. Clin. Chest Med. 2019, 40, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, F.; Zhang, W. Bedaquiline and delamanid in the treatment of multidrug-resistant tuberculosis: Promising but challenging. Drug Dev. Res. 2018, 80, 98–105. [Google Scholar] [CrossRef]
- Richards, M.; Bird, A.E.; Munden, J.E. Chlorflavonin, A New Antifungal Antibiotic. J. Antibiot. 1969, 22, 388. [Google Scholar] [CrossRef]
- Bird, A.E.; Marshall, A.C. Structure of chlorflavonin. J. Chem. Soc. Perkin 1 1969, 18, 2418–2420. [Google Scholar] [CrossRef] [PubMed]
- Sampson, S.L.; Mansfield, K.G.; Carville, A.; Magee, D.M.; Quitugua, T.; Howerth, E.W.; Bloom, B.R.; Hondalus, M.K. Extended safety and efficacy studies of a live attenuated double leucine and pantothenate auxotroph of Mycobacterium tuberculosis as a vaccine candidate. Vaccine 2011, 29, 4839–4847. [Google Scholar] [CrossRef] [PubMed]
- Sampson, S.L.; Dascher, C.C.; Sambandamurthy, V.K.; Russell, R.G.; Jacobs, W.R.; Bloom, B.R.; Hondalus, M.K. Protection Elicited by a Double Leucine and Pantothenate Auxotroph of Mycobacterium tuberculosis in Guinea Pigs. Infect. Immun. 2004, 72, 3031–3037. [Google Scholar] [CrossRef]
- Hondalus, M.K.; Bardarov, S.; Russell, R.; Chan, J.; Jacobs, W.R.; Bloom, B.R. Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect. Immun. 2000, 68, 2888–2898. [Google Scholar] [CrossRef]
- Radhakrishnan, A.N.; Wagner, R.P.; Snell, E.E. Biosynthesis of Valine and Isoleucine: III. α-Keto-β-hydroxy acid reductase and α-hydroxy-β-keto acid reductoisomerase. J. Biol. Chem. 1960, 235, 2322–2331. [Google Scholar] [CrossRef]
- Leaviti, R.I. Isoleucine and Valine Metabolism in Escherichia coli XII. J. Bacteriol. 1964, 88, 172–178. [Google Scholar] [CrossRef]
- Steinmetz, A.; Vyazmensky, M.; Meyer, D.; Barak, Z.E.; Golbik, R.; Chipman, D.M.; Tittmann, K. Valine 375 and Phenylalanine 109 Confer Affinity and Specificity for Pyruvate as Donor Substrate in Acetohydroxy Acid Synthase Isozyme II from Escherichia coli. Biochemistry 2010, 49, 5188–5199. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-J.; Yu, Y.G.; Hahn, H.G.; Choi, J.-D.; Yoon, M.-Y. Characterization of acetohydroxyacid synthase from Mycobacterium tuberculosis and the identification of its new inhibitor from the screening of a chemical library. FEBS Lett. 2005, 579, 4903–4910. [Google Scholar] [CrossRef] [PubMed]
- Umbarger, H.E.; Brown, B. Isoleucine and Valine Metabolism in Escherichia coli: VIII. The Formation of Acetolactate. J. Biol. Chem. 1958, 233, 1156–1160. [Google Scholar] [CrossRef]
- Rehberg, N.; Akone, H.S.; Ioerger, T.R.; Erlenkamp, G.; Daletos, G.; Gohlke, H.; Proksch, P.; Kalscheuer, R. Chlorflavonin Targets Acetohydroxyacid Synthase Catalytic Subunit IlvB1 for Synergistic Killing of Mycobacterium tuberculosis. ACS Infect. Dis. 2018, 4, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Grandoni, J.A.; Marta, P.T.; Schloss, J.V. Inhibitors of branched-chain amino acid biosynthesis as potential antituberculosis agents. J. Antimicrob. Chemother. 1998, 42, 475–482. [Google Scholar] [CrossRef]
- Awasthy, D.; Gaonkar, S.; Shandil, R.K.; Yadav, R.; Bharath, S.; Marcel, N.; Subbulakshmi, V.; Sharma, U. Inactivation of the ilvB1 gene in Mycobacterium tuberculosis leads to branched-chain amino acid auxotrophy and attenuation of virulence in mice. Microbiology 2009, 155, 2978–2987. [Google Scholar] [CrossRef]
- Gokhale, K.; Tilak, B. Mechanisms of bacterial acetohydroxyacid synthase (AHAS) and specific inhibitors of Mycobacterium tuberculosis AHAS as potential drug candidates against tuberculosis. Curr. Drug Targets 2015, 16, 689–699. [Google Scholar] [CrossRef]
- Martins, B.T.; Correia da Silva, M.; Pinto, M.; Cidade, H.; Kijjoa, A. Marine natural flavonoids: Chemistry and biological activities. Nat. Prod. Res. 2018, 33, 3260–3272. [Google Scholar] [CrossRef]
- Veitch, N.C.; Grayer, R.J. Flavonoids and their glycosides, including anthocyanins. Nat. Prod. Rep. 2011, 28, 1626–1695. [Google Scholar] [CrossRef]
- Toekes, A.L.; Bognar, R. Synthesis of chlorflavonin. Acta Chim. Acade. Sci. Hung. 1981, 107, 365–368. [Google Scholar]
- Zhao, X.; Liu, J.; Xie, Z.; Li, Y. A One-Pot Synthesis of Aurones from Substituted Acetophenones and Benz aldehydes: A Concise Synthesis of Aureusidin. Synthesis 2012, 44, 2217–2224. [Google Scholar] [CrossRef][Green Version]
- Ferreira, D.; Brandt, E.V.; du Volsteedt, F.R.; Roux, D.G. Parameters regulating the α- and β-cyclization of chalcones. J. Chem. Soc. Perkin Trans. 1 1975, 1437–1446. [Google Scholar] [CrossRef]
- Serdiuk, I.E.; Roshal, A.D.; Błażejowski, J. Quantum-Chemical Analysis of the Algar–Flynn–Oyamada Reaction Mechanism. Chem. Heterocycl. Compd. 2014, 50, 396–403. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Dröge, T.; Liu, F.; Glorius, F. Towards mild metal-catalyzed C–H bond activation. Chem. Soc. Rev. 2011, 40, 4740–4761. [Google Scholar] [CrossRef]
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C–H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef]
- Chen, S.; Ranjan, P.; Voskressensky, L.G.; Van der Eycken, E.V.; Sharma, U.K. Recent Developments in Transition-Metal Catalyzed Direct C-H Alkenylation, Alkylation, and Alkynylation of Azoles. Molecules 2020, 25, 4970. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Chang, S. Ruthenium-catalyzed direct C-H amidation of arenes including weakly coordinating aromatic ketones. Chemistry 2013, 19, 7328–7333. [Google Scholar] [CrossRef]
- Shin, Y.; Han, S.; De, U.; Park, J.; Sharma, S.; Mishra, N.K.; Lee, E.-K.; Lee, Y.; Kim, H.S.; Kim, I.S. Ru(II)-Catalyzed Selective C–H Amination of Xanthones and Chromones with Sulfonyl Azides: Synthesis and Anticancer Evaluation. J. Org. Chem. 2014, 79, 9262–9271. [Google Scholar] [CrossRef]
- Kim, K.; Choe, H.; Jeong, Y.; Lee, J.H.; Hong, S. Ru(II)-Catalyzed Site-Selective Hydroxylation of Flavone and Chromone Derivatives: The Importance of the 5-Hydroxyl Motif for the Inhibition of Aurora Kinases. Org. Lett. 2015, 17, 2550–2553. [Google Scholar] [CrossRef]
- Da Silva Júnior, E.N.; Jardim, G.A.M.; Gomes, R.S.; Liang, Y.-F.; Ackermann, L. Weakly-coordinating N-oxide and carbonyl groups for metal-catalyzed C–H activation: The case of A-ring functionalization. Chem. Commun. 2018, 54, 7398–7411. [Google Scholar] [CrossRef]
- Das, R.; Kumar, G.S.; Kapur, M. Amides as Weak Coordinating Groups in Proximal C–H Bond Activation. Eur. J. Org. Chem. 2017, 2017, 5439–5459. [Google Scholar] [CrossRef]
- Leitch, J.A.; Wilson, P.B.; McMullin, C.L.; Mahon, M.F.; Bhonoah, Y.; Williams, I.H.; Frost, C.G. Ruthenium(II)-Catalyzed C–H Functionalization Using the Oxazolidinone Heterocycle as a Weakly Coordinating Directing Group: Experimental and Computational Insights. ACS Catal. 2016, 6, 5520–5529. [Google Scholar] [CrossRef]
- De Sarkar, S.; Liu, W.; Kozhushkov, S.I.; Ackermann, L. Weakly Coordinating Directing Groups for Ruthenium(II)-Catalyzed C-H Activation. Adv. Synth. Catal. 2014, 356, 1461–1479. [Google Scholar] [CrossRef]
- Shan, G.; Yang, X.; Ma, L.; Rao, Y. Pd-Catalyzed C-H Oxygenation with TFA/TFAA: Expedient Access to Oxygen-Containing Heterocycles and Late-Stage Drug Modification. Angew. Chem. Int. Ed. 2012, 51, 13070–13074. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; et al. CF2H, a Functional Group-Dependent Hydrogen-Bond Donor: Is It a More or Less Lipophilic Bioisostere of OH, SH, and CH3? J. Med. Chem. 2019, 62, 5628–5637. [Google Scholar] [CrossRef]
- Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. J. Med. Chem. 2017, 60, 797–804. [Google Scholar] [CrossRef]
- Kobayashi, S.; Arai, K.; Shimizu, H.; Ihori, Y.; Ishitani, H.; Yamashita, Y. A Novel Dinuclear Chiral Niobium Complex for Lewis Acid Catalyzed Enantioselective Reactions: Design of a Tridentate Ligand and Elucidation of the Catalyst Structure. Angew. Chem. Int. Ed. 2005, 44, 761–764. [Google Scholar] [CrossRef]
- Daly, A.M.; Gilheany, D.G. The synthesis and use in asymmetric epoxidation of metal salen complexes derived from enantiopure trans-cyclopentane- and cyclobutane-1,2-diamine. Tetrahedron Asymmetry 2003, 14, 127–137. [Google Scholar] [CrossRef]
- Christensen, H. Preparation of Salicylaldehydes via the Ortho-Lithio Derivatives of Methoxymethyl-Protected Phenols. Synth. Commun. 1975, 5, 65–78. [Google Scholar] [CrossRef]
- Takeuchi, D.; Chiba, Y.; Takano, S.; Osakada, K. Double-Decker-Type Dinuclear Nickel Catalyst for Olefin Polymerization: Efficient Incorporation of Functional Co-monomers. Angew. Chem. Int. Ed. 2013, 52, 12536–12540. [Google Scholar] [CrossRef] [PubMed]
- Bischof, D.; Tripp, M.W.; Hofmann, P.E.; Ip, C.-H.; Ivlev, S.I.; Gerhard, M.; Koert, U.; Witte, G. Regioselective Fluorination of Acenes: Tailoring of Molecular Electronic Levels and Solid-State Properties. Chemistry 2022, 28, e202103653. [Google Scholar] [CrossRef] [PubMed]
- Badetti, E.; Lloveras, V.; Amadio, E.; Di Lorenzo, R.; Olivares-Marín, M.; Tesio, A.Y.; Zhang, S.; Pan, F.; Rissanen, K.; Veciana, J.; et al. Organic Polyradicals as Redox Mediators: Effect of Intramolecular Radical Interactions on Their Efficiency. ACS Appl. Mater. Interfaces 2020, 12, 45968–45975. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Qiu, J.; Miyazaki, R.; Aoyama, H.; Murai, K.; Hasegawa, J.-Y.; Arisawa, M. Ni-Catalyzed Cycloisomerization between 3-Phenoxy Acrylic Acid Derivatives and Alkynes via Intramolecular Cleavage and Formation of the C–O Bond To Give 2,3-Disubstituted Benzofurans. Org. Lett. 2019, 21, 8400–8403. [Google Scholar] [CrossRef]
- Li, L.; Wang, F.; Ni, C.; Hu, J. Synthesis of gem-Difluorocyclopropa(e)nes and O-, S-, N-, and P-Difluoromethylated Compounds with TMSCF2Br. Angew. Chem. Int. Ed. 2013, 52, 12390–12394. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Gao, N.; Li, S.Q.; Huang, Y.; Bourassa, J.L.; Huryn, D.M. Experimental Design on Single-Time-Point High-Throughput Microsomal Stability Assay. J. Pharm. Sci. 2004, 93, 1537–1544. [Google Scholar] [CrossRef]
- Obach, R.S. Prediction of Human Clearance of Twenty-Nine Drugs from Hepatic Microsomal Intrinsic Clearance Data: An Examination of In Vitro Half-Life Approach and Nonspecific Binding to Microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar]
- Kaus, J.W.; Pierce, L.; Walker, R.C.; McCammont, J.A. Improving the Efficiency of Free Energy Calculations in the Amber Molecular Dynamics Package. J. Chem. Theory Comput. 2013, 9, 4131–4139. [Google Scholar] [CrossRef]
- Homeyer, N.; Stoll, F.; Hillisch, A.; Gohlke, H. Binding Free Energy Calculations for Lead Optimization: Assessment of Their Accuracy in an Industrial Drug Design Context. J. Chem. Theory Comput. 2014, 10, 3331–3344. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. FEW: A workflow tool for free energy calculations of ligand binding. J. Comput. Chem. 2013, 34, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Alelyunas, Y.W.; Liu, R.; Pelosi-Kilby, L.; Shen, C. Application of a Dried-DMSO rapid throughput 24-h equilibrium solubility in advancing discovery candidates. Eur. J. Pharm. Sci. 2009, 37, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, L.; Tilton, S.; Wang, J. Development of a high throughput equilibrium solubility assay using miniaturized shake-flask method in early drug discovery. J. Pharm. Sci. 2007, 96, 3052–3071. [Google Scholar] [CrossRef] [PubMed]
- Gift, A.D.; Stewart, S.M.; Kwete Bokashanga, P. Experimental Determination of pKa Values by Use of NMR Chemical Shifts, Revisited. J. Chem. Educ. 2012, 89, 1458–1460. [Google Scholar] [CrossRef]
- ROCS, 3.4.2.1; OpenEye Scientific Software: Santa Fe, NM, USA, 2020.
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Kruskal, J.B. On the shortest spanning subtree of a graph and the traveling salesman problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2006, 50, 74–82. [Google Scholar] [CrossRef]
- QikProp, Release 2021-4; Schrödinger: New York, NY, USA, 2021.
- Ioannidis, H.; Drakopoulos, A.; Tzitzoglaki, C.; Homeyer, N.; Kolarov, F.; Gkeka, P.; Freudenberger, K.; Liolios, C.; Gauglitz, G.; Cournia, Z.; et al. Alchemical Free Energy Calculations and Isothermal Titration Calorimetry Measurements of Aminoadamantanes Bound to the Closed State of Influenza A/M2TM. J. Chem. Inf. Modeling 2016, 56, 862–876. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry a | Catalyst | Oxidant (Equiv.) | Additive (Equiv.) | Solvent (0.07 M) | T (°C) | Time (h) | Conversion (%) |
| 1 | Pd(TFA)2 | PhI(TFA)2 (1.2) | - | DCE | 80 | 8 | trace |
| 2 | Pd(TFA)2 | PhI(TFA)2 (2.0) | - | DCE | 80 | 16 | decomp. |
| 3 | Pd(TFA)2 | PhI(TFA)2 (1.2) | - | 9:1 TFA/TFAA | 80 | 8 | 48 |
| 4 | Pd(TFA)2 | PhI(TFA)2 (1.2) | - | 1:66 TFA/TFAA b | 80 | 8 | 52 |
| 5 | [RuCl2(p-cymene)]2 | PhI(TFA)2 (1.5) | Ag2CO3 (1.5) | 1:66 TFA/TFAA b | 80 | 24 | 43 |
| 6 | [RuCl2(p-cymene)]2 | selectfluor (1.5) | Ag2CO3 (1.5) | 1:66 TFA/TFAA b | 80 | 16 | 53 |
| 7 | [RuCl2(p-cymene)]2 | selectfluor (2.0) | Ag2CO3 (2.0) | 1:66 TFA/TFAA b | 80 | 16 | 61 |
| 8 | [RuCl2(p-cymene)]2 | selectfluor (1.1) | Ag2CO3 (2.0) | 1:66 TFA/TFAA b | 80 | 24 | 72 |
| 9 | [RuCl2(p-cymene)]2 | selectfluor (1.5) | Ag2CO3 (1.5) | 1:66 TFA/TFAA b | 60 | 16 | trace |
| 10 | [RuCl2(p-cymene)]2 | selectfluor (1.2) | Ag2CO3 (1.2) | 1:66 TFA/TFAA b | 120 | 2 | 74 |
| CF | BF | |
|---|---|---|
| Water solubility at pH 7.4 a | <5 μM | <5 μM |
| pKa1 (2′-OH) b pKa2 (5-OH) b | 6.80 ± 0.07 10.40 ± 0.03 | 6.74 ± 0.04 10.30 ± 0.04 |
| remaining after 30 min c | 48% | 60% |
| CLint, app d | 76.3 mL/min/mg | 53.4 mL/min/mg |
| EH d | 0.78 | 0.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berger, A.; Knak, T.; Kiffe-Delf, A.-L.; Mudrovcic, K.; Singh, V.; Njoroge, M.; Burckhardt, B.B.; Gopalswamy, M.; Lungerich, B.; Ackermann, L.; et al. Total Synthesis of the Antimycobacterial Natural Product Chlorflavonin and Analogs via a Late-Stage Ruthenium(II)-Catalyzed ortho-C(sp2)-H-Hydroxylation. Pharmaceuticals 2022, 15, 984. https://doi.org/10.3390/ph15080984
Berger A, Knak T, Kiffe-Delf A-L, Mudrovcic K, Singh V, Njoroge M, Burckhardt BB, Gopalswamy M, Lungerich B, Ackermann L, et al. Total Synthesis of the Antimycobacterial Natural Product Chlorflavonin and Analogs via a Late-Stage Ruthenium(II)-Catalyzed ortho-C(sp2)-H-Hydroxylation. Pharmaceuticals. 2022; 15(8):984. https://doi.org/10.3390/ph15080984
Chicago/Turabian StyleBerger, Alexander, Talea Knak, Anna-Lene Kiffe-Delf, Korana Mudrovcic, Vinayak Singh, Mathew Njoroge, Bjoern B. Burckhardt, Mohanraj Gopalswamy, Beate Lungerich, Lutz Ackermann, and et al. 2022. "Total Synthesis of the Antimycobacterial Natural Product Chlorflavonin and Analogs via a Late-Stage Ruthenium(II)-Catalyzed ortho-C(sp2)-H-Hydroxylation" Pharmaceuticals 15, no. 8: 984. https://doi.org/10.3390/ph15080984
APA StyleBerger, A., Knak, T., Kiffe-Delf, A.-L., Mudrovcic, K., Singh, V., Njoroge, M., Burckhardt, B. B., Gopalswamy, M., Lungerich, B., Ackermann, L., Gohlke, H., Chibale, K., Kalscheuer, R., & Kurz, T. (2022). Total Synthesis of the Antimycobacterial Natural Product Chlorflavonin and Analogs via a Late-Stage Ruthenium(II)-Catalyzed ortho-C(sp2)-H-Hydroxylation. Pharmaceuticals, 15(8), 984. https://doi.org/10.3390/ph15080984