
Computational Methods in Cooperation with Experimental Approaches to Design Protein Tyrosine Phosphatase 1B Inhibitors in Type 2 Diabetes Drug Design: A Review of the Achievements of This Century

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
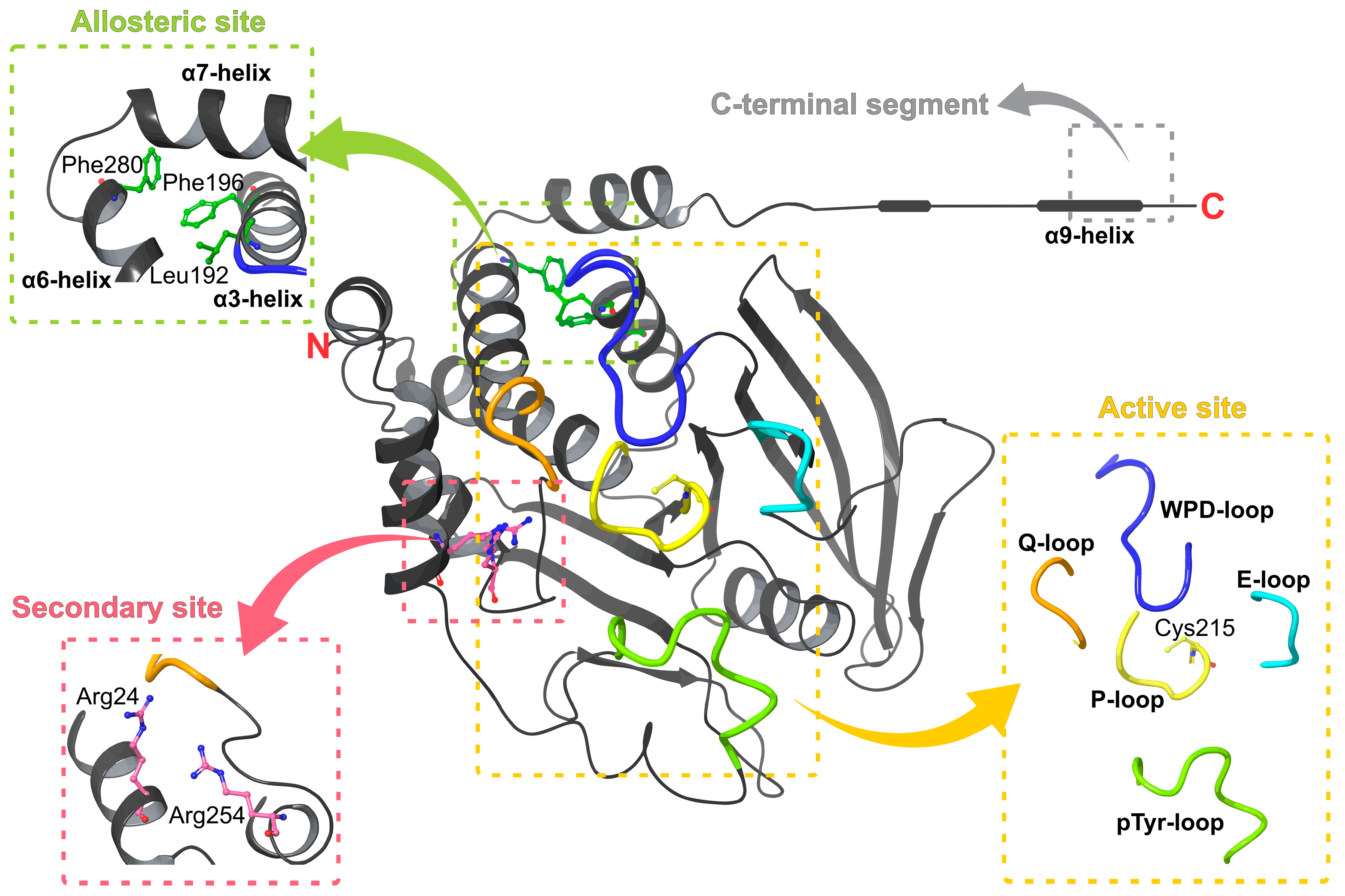
2. PTP1B Structure and Target Sites
2.1. Active and Secondary Sites
2.2. Allosteric Sites
3. Computational Strategies Applied to Discover PTP1B Inhibitors
3.1. Virtual Screening
3.2. Molecular Docking
3.3. Pharmacophore Modeling
3.4. QSAR
4. Development of PTP1B Inhibitors through Computational Approaches
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. 2020. Available online: https://www.who.int/ (accessed on 11 December 2021).
- Garg, S.; Maurer, H.; Reed, K.; Selagamsetty, R. Diabetes and cancer: Two diseases with obesity as a common risk factor. Diabetes Obes. Metab. 2014, 16, 97–110. [Google Scholar] [CrossRef]
- Stavrovskaya, A. Cellular mechanisms of multidrug resistance of tumor cells. Biochem. C/C Biokhimiia 2000, 65, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A.; Bristol, M.L.; Yalowich, J.C. Toxicity issues in cancer drug development. Curr. Opin. Investig. Drugs 2010, 11, 612–614. [Google Scholar]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycemia in type 2 diabetes: A patient-centered approach: Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012, 35, 1364–1379. [Google Scholar] [CrossRef] [Green Version]
- Kalra, S.; Ghosh, S.; Aamir, A.; Ahmed, M.T.; Amin, M.F.; Bajaj, S.; Baruah, M.P.; Bulugahapitiya, U.; Das, A.; Giri, M. Safe and pragmatic use of sodium-glucose co-transporter 2 inhibitors in type 2 diabetes mellitus: South Asian Federation of Endocrine Societies consensus statement. Indian J. Endocrinol. Metab. 2017, 21, 210–230. [Google Scholar] [CrossRef]
- Kang, J.G.; Park, C.Y. Anti-obesity drugs: A review about their effects and safety. Diabetes Metab. J. 2012, 36, 13–25. [Google Scholar] [CrossRef] [Green Version]
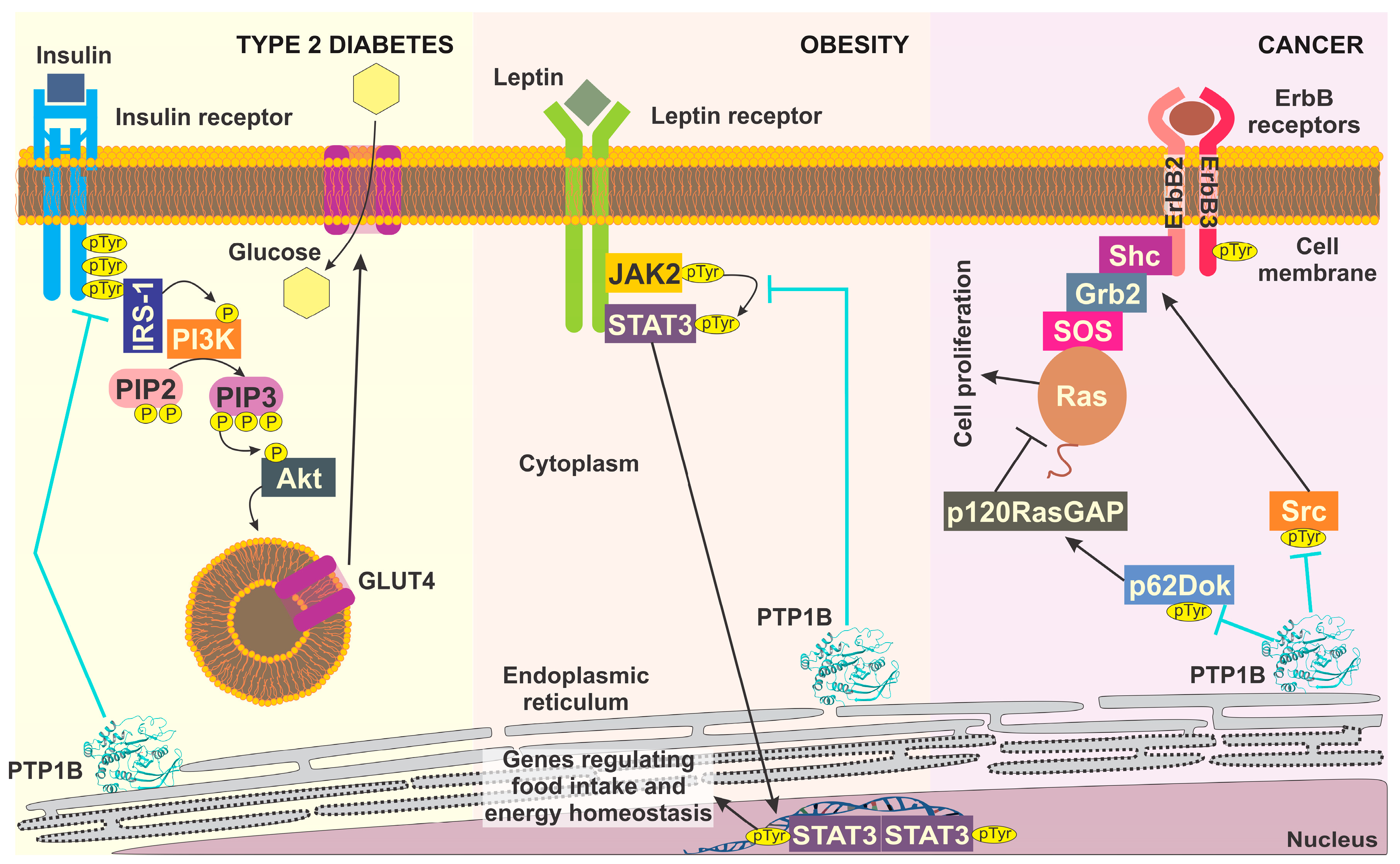
- Asante-Appiah, E.; Kennedy, B.P. Protein tyrosine phosphatases: The quest for negative regulators of insulin action. Am. J. Physiol.-Endocrinol. Metab. 2003, 284, E663–E670. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Kim, Y.-B.; Lee, A.; Toschi, E.; Bonner-Weir, S.; Kahn, C.R.; Neel, B.G.; Kahn, B.B. Protein-tyrosine phosphatase 1B deficiency reduces insulin resistance and the diabetic phenotype in mice with polygenic insulin resistance. J. Biol. Chem. 2007, 282, 23829–23840. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Uetani, N.; Simoncic, P.D.; Chaubey, V.P.; Lee-Loy, A.; McGlade, C.J.; Kennedy, B.P.; Tremblay, M.L. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev. Cell 2002, 2, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.-B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B. PTP1B regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wu, Y.; Zhu, S.; Liang, W.; Wang, Z.; Wang, Y.; Lv, T.; Yao, Y.; Yuan, D.; Song, Y. PTP1B promotes cell proliferation and metastasis through activating src and ERK1/2 in non-small cell lung cancer. Cancer Lett. 2015, 359, 218–225. [Google Scholar] [CrossRef]
- Bjorge, J.D.; Pang, A.; Fujita, D.J. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 2000, 275, 41439–41446. [Google Scholar] [CrossRef] [Green Version]
- Lessard, L.; Stuible, M.; Tremblay, M.L. The two faces of PTP1B in cancer. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 613–619. [Google Scholar] [CrossRef]
- Stuible, M.; Doody, K.M.; Tremblay, M.L. PTP1B and TC-PTP: Regulators of transformation and tumorigenesis. Cancer Metastasis Rev. 2008, 27, 215–230. [Google Scholar] [CrossRef]
- Bakke, J.; Haj, F.G. Protein-tyrosine phosphatase 1B substrates and metabolic regulation. Semin. Cell Dev. Biol. 2015, 37, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef]
- Barr, A.J. Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Med. Chem. 2010, 2, 1563–1576. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; VanEtten, R. Pre-steady-state and steady-state kinetic analysis of the low molecular weight phosphotyrosyl protein phosphatase from bovine heart. J. Biol. Chem. 1991, 266, 1516–1525. [Google Scholar] [CrossRef]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Møller, N.P.H. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [Green Version]
- Ibarra-Sánchez, M.J.; Simoncic, P.D.; Nestel, F.R.; Duplay, P.; Lapp, W.S.; Tremblay, M.L. The T-cell protein tyrosine phosphatase. Semin. Immunol. 2000, 12, 379–386. [Google Scholar] [CrossRef]
- Iversen, L.F.; Møller, K.B.; Pedersen, A.K.; Peters, G.H.; Petersen, A.S.; Andersen, H.S.; Branner, S.; Mortensen, S.B.; Møller, N.P.H. Structure determination of T cell protein-tyrosine phosphatase. J. Biol. Chem. 2002, 277, 19982–19990. [Google Scholar] [CrossRef] [Green Version]
- You-Ten, K.E.; Muise, E.S.; Itié, A.; Michaliszyn, E.; Wagner, J.; Jothy, S.; Lapp, W.S.; Tremblay, M.L. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase–deficient mice. J. Exp. Med. 1997, 186, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef]
- Chernoff, J.; Schievella, A.R.; Jost, C.A.; Erikson, R.; Neel, B.G. Cloning of a cDNA for a major human protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. USA 1990, 87, 2735–2739. [Google Scholar] [CrossRef] [Green Version]
- Frangioni, J.V.; Beahm, P.H.; Shifrin, V.; Jost, C.A.; Neel, B.G. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell 1992, 68, 545–560. [Google Scholar] [CrossRef]
- Tonks, N.K.; Diltz, C.; Fischer, E. Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 1988, 263, 6722–6730. [Google Scholar] [CrossRef]
- Lorenzen, J.A.; Dadabay, C.Y.; Fischer, E.H. COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus. J. Cell Biol. 1995, 131, 631–643. [Google Scholar] [CrossRef]
- Gjörloff-Wingren, A.; Saxena, M.; Han, S.; Wang, X.; Alonso, A.; Renedo, M.; Oh, P.; Williams, S.; Schnitzer, J.; Mustelin, T. Subcellular localization of intracellular protein tyrosine phosphatases in T cells. Eur. J. Immunol. 2000, 30, 2412–2421. [Google Scholar] [CrossRef]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Barford, D.; Flint, A.J.; Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 1995, 268, 1754–1758. [Google Scholar] [CrossRef]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Müller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Tautz, L.; Critton, D.A.; Grotegut, S. Protein tyrosine phosphatases: Structure, function, and implication in human disease. In Phosphatase Modulators; Springer: Berlin/Heidelberg, Germany, 2013; pp. 179–221. [Google Scholar] [CrossRef]
- Critton, D.A.; Tautz, L.; Page, R. Visualizing active-site dynamics in single crystals of HePTP: Opening of the WPD loop involves coordinated movement of the E loop. J. Mol. Biol. 2011, 405, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Chen, X.; Luechapanichkul, R.; Selner, N.G.; Meyer, T.M.; Wavreille, A.S.; Chan, R.; Iorio, C.; Zhou, X.; Neel, B.G. Substrate specificity of protein tyrosine phosphatases 1B, RPTPα, SHP-1, and SHP-2. Biochemistry 2011, 50, 2339–2356. [Google Scholar] [CrossRef] [Green Version]
- Peti, W.; Page, R. Strategies to make protein serine/threonine (PP1, calcineurin) and tyrosine phosphatases (PTP1B) druggable: Achieving specificity by targeting substrate and regulatory protein interaction sites. Bioorg. Med. Chem. 2015, 23, 2781–2785. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Niu, T.; Zhang, A.; Mishra, A.K.; Zhao, Z.J.; Zhou, G.W. Relation between the flexibility of the WPD loop and the activity of the catalytic domain of protein tyrosine phosphatase SHP-1. J. Cell. Biochem. 2002, 84, 47–55. [Google Scholar] [CrossRef]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [Green Version]
- Low, J.L.; Chai, C.L.; Yao, S.Q. Bidentate inhibitors of protein tyrosine phosphatases. Antioxid. Redox Signal. 2014, 20, 2225–2250. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Jin, Y.Y.; Wang, Y.L.; Wang, R.L.; Lu, X.H.; Kong, D.X.; Xu, W.R. The Discovery of a Novel and Selective Inhibitor of PTP 1B Over TCPTP: 3D QSAR Pharmacophore Modeling, Virtual Screening, Synthesis, and Biological Evaluation. Chem. Biol. Drug. Des. 2014, 83, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Shinde, R.N.; Kumar, G.S.; Eqbal, S.; Sobhia, M.E. Screening and identification of potential PTP1B allosteric inhibitors using in silico and in vitro approaches. PLoS ONE 2018, 13, e0199020. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Zhang, S.E.; Nie, F.; Yang, Y.; Tang, Y.B.; Yin, W.; Tian, J.Y.; Ye, F.; Xiao, Z. Discovery of novel PTP1B inhibitors via pharmacophore-oriented scaffold hopping from Ertiprotafib. Bioorg. Med. Chem. Lett. 2013, 23, 6217–6222. [Google Scholar] [CrossRef]
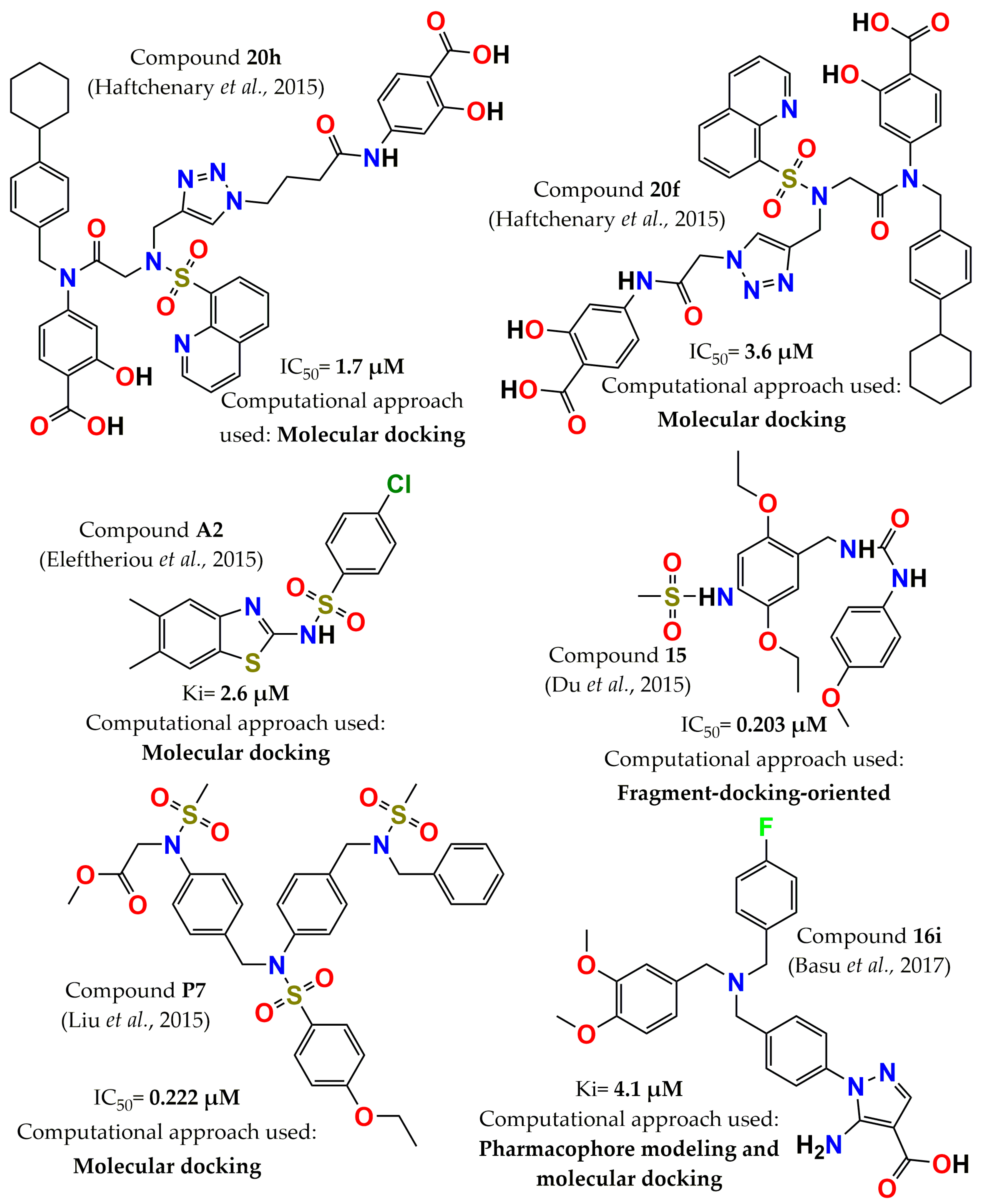
- Du, Y.; Ling, H.; Shen, J.; Li, Q. Discovery of novel, potent, selective and cellular active ADC type PTP1B inhibitors via fragment-docking-oriented de novel design. Bioorg. Med. Chem. 2015, 23, 4891–4898. [Google Scholar] [CrossRef]
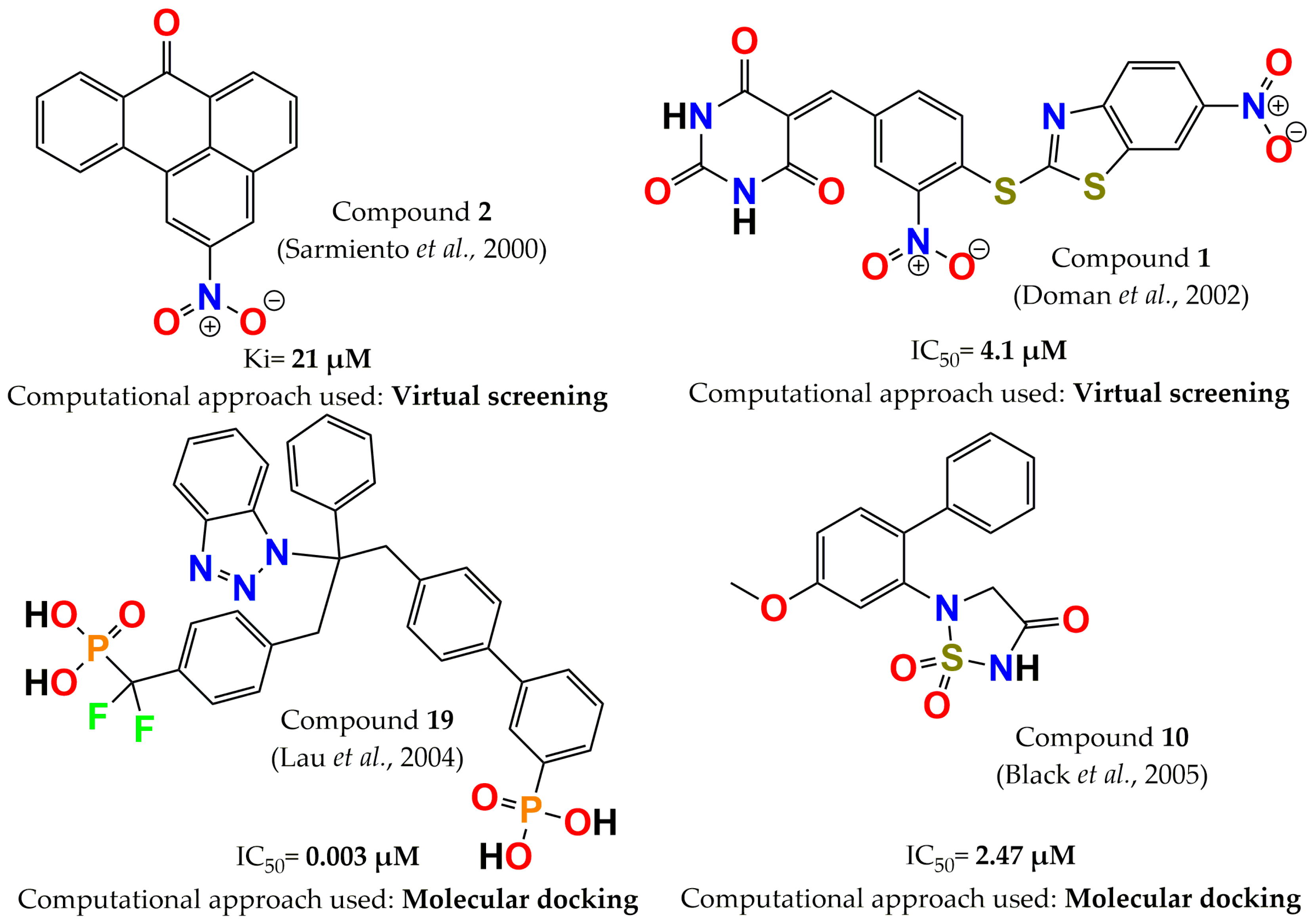
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef]
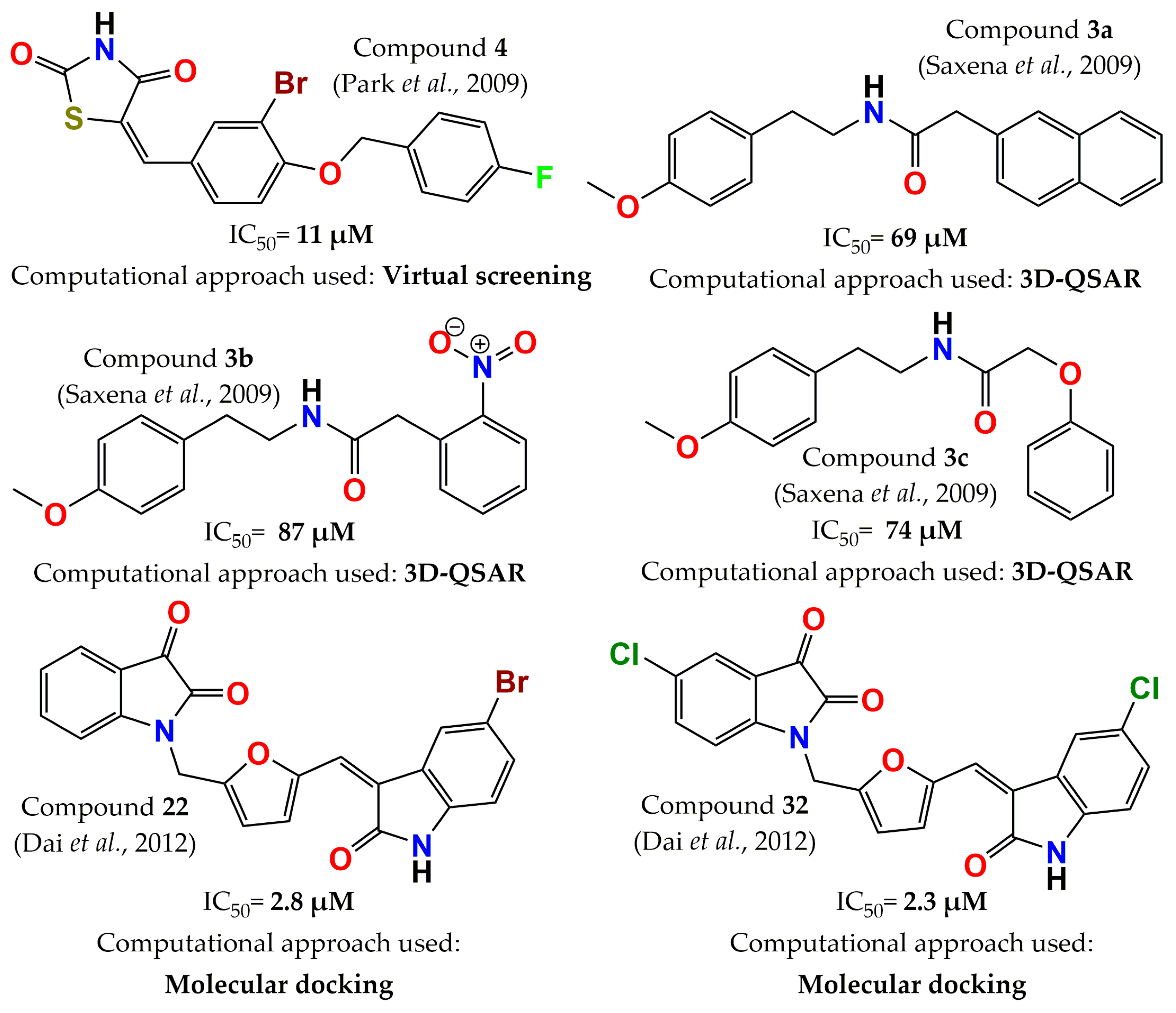
- Park, H.; Bhattarai, B.R.; Ham, S.W.; Cho, H. Structure-based virtual screening approach to identify novel classes of PTP1B inhibitors. Eur. J. Med. Chem. 2009, 44, 3280–3284. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Kumar, K.K.; Kumar, Y.N.; Bhaskar, M. Modeling, molecular dynamics, and docking assessment of transcription factor rho: A potential drug target in Brucella melitensis 16M. Drug Des. Devel. Ther. 2015, 9, 1897–1912. [Google Scholar] [CrossRef] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Stumpfe, D.; Bajorath, J. Current trends, overlooked issues, and unmet challenges in virtual screening. J. Chem. Inf. Model. 2020, 60, 4112–4115. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Gesto, D.; Oliveira, E.F.; Santos-Martins, D.; Brás, N.F.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys. 2015, 582, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Slater, O.; Kontoyianni, M. The compromise of virtual screening and its impact on drug discovery. Expert Opin. Drug Discov. 2019, 14, 619–637. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput.-Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor–ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 2002, 47, 409–443. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. In Molecular Modeling of Proteins. Methods Molecular Biology™; Kukol, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; Volume 443, pp. 365–382. [Google Scholar]
- Zhou, Z.; Felts, A.K.; Friesner, R.A.; Levy, R.M. Comparative performance of several flexible docking programs and scoring functions: Enrichment studies for a diverse set of pharmaceutically relevant targets. J. Chem. Inf. Model. 2007, 47, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.D.; Strizhev, A.; Leonard, J.M.; Blake, J.F.; Matthew, J.B. Consensus scoring for ligand/protein interactions. J. Mol. Graph. Model. 2002, 20, 281–295. [Google Scholar] [CrossRef]
- Chen, Y.; Pohlhaus, D.T. In silico docking and scoring of fragments. Drug Discov. Today Technol. 2010, 7, e149–e156. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.H.; Sodero, A.C.; Jofily, P.; Silva-Jr, F.P. Key topics in molecular docking for drug design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [Green Version]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.Y.; Coumar, M.S.; Shiao, H.Y.; Wang, W.C.; Chen, C.W.; Song, J.S.; Chen, C.H.; Lin, W.H.; Wu, S.H.; Hsu, J.T. Ligand efficiency based approach for efficient virtual screening of compound libraries. Eur. J. Med. Chem. 2014, 83, 226–235. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Structure based design and molecular docking studies for phosphorylated tau inhibitors in Alzheimer’s disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [Green Version]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1468–e1488. [Google Scholar] [CrossRef] [Green Version]
- Selassie, C.; Verma, R.P. History of quantitative structure-activity relationships. In Burger’s Medicinal Chemistry and Drug Discovery; Abraham, D.J., Ed.; Wiley: Hoboken, NJ, USA, 2003; Volume 1, pp. 1–48. [Google Scholar]
- Yang, G.F.; Huang, X. Development of quantitative structure-activity relationships and its application in rational drug design. Curr. Pharm. Des. 2006, 12, 4601–4611. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Wood, D. Modern 2D QSAR for drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 505–522. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [Green Version]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef]
- Achary, P.G. Applications of quantitative structure-Activity relationships (QSAR) based virtual screening in drug design: A review. Mini Rev. Med. Chem. 2020, 20, 1375–1388. [Google Scholar] [CrossRef]
- Sarmiento, M.; Wu, L.; Keng, Y.-F.; Song, L.; Luo, Z.; Huang, Z.; Wu, G.Z.; Yuan, A.K.; Zhang, Z.Y. Structure-based discovery of small molecule inhibitors targeted to protein tyrosine phosphatase 1B. J. Med. Chem. 2000, 43, 146–155. [Google Scholar] [CrossRef]
- Lau, C.K.; Bayly, C.I.; Gauthier, J.Y.; Li, C.S.; Therien, M.; Asante-Appiah, E.; Cromlish, W.; Boie, Y.; Forghani, F.; Desmarais, S. Structure based design of a series of potent and selective non peptidic PTP-1B inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 1043–1048. [Google Scholar] [CrossRef]
- Black, E.; Breed, J.; Breeze, A.L.; Embrey, K.; Garcia, R.; Gero, T.W.; Godfrey, L.; Kenny, P.W.; Morley, A.D.; Minshull, C.A. Structure-based design of protein tyrosine phosphatase-1B inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2503–2507. [Google Scholar] [CrossRef]
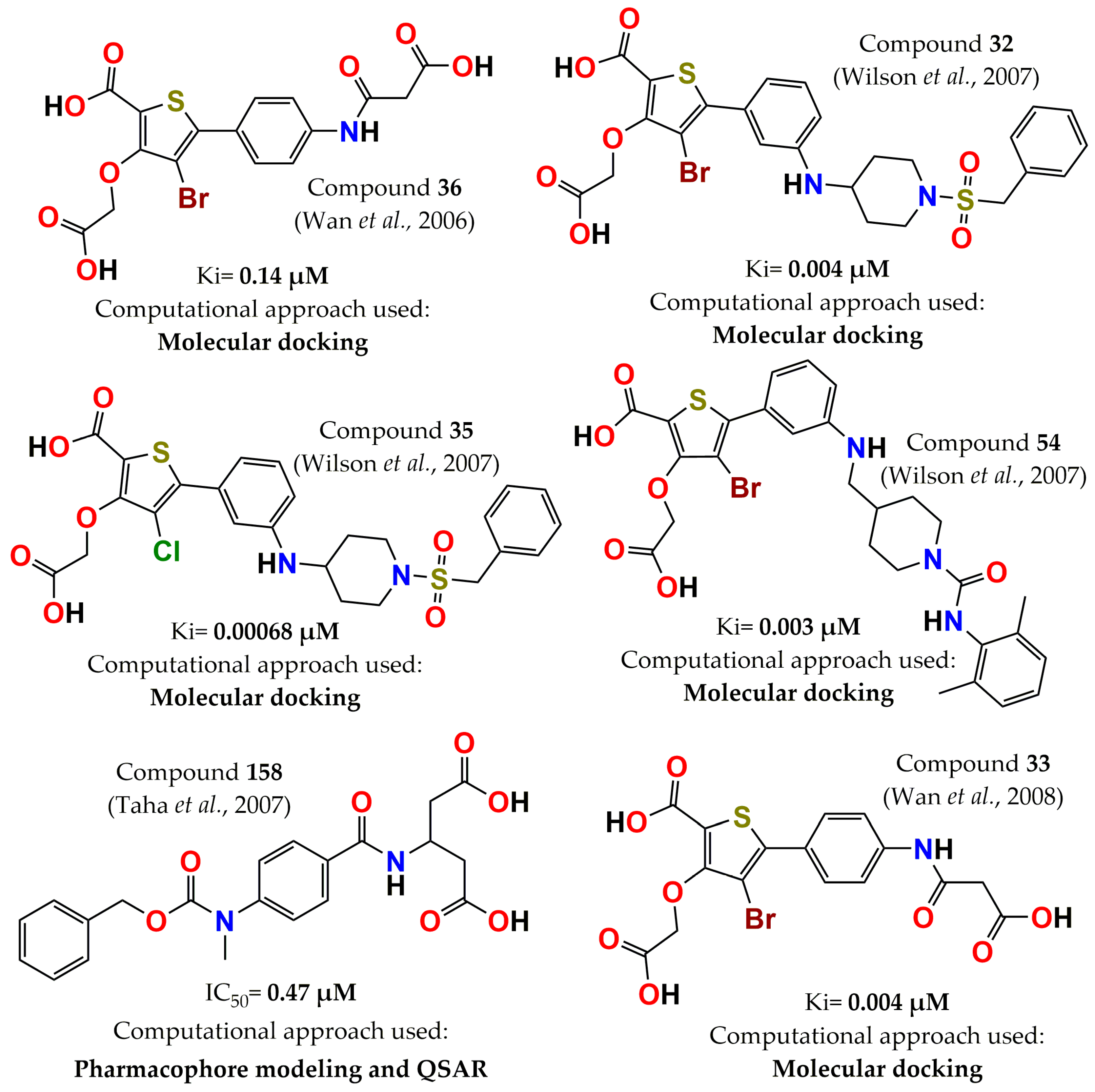
- Wan, Z.K.; Lee, J.; Xu, W.; Erbe, D.V.; Joseph-McCarthy, D.; Follows, B.C.; Zhang, Y.L. Monocyclic thiophenes as protein tyrosine phosphatase 1B inhibitors: Capturing interactions with Asp48. Bioorg. Med. Chem. Lett. 2006, 16, 4941–4945. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.K.; Lee, J.; Hotchandani, R.; Moretto, A.; Binnun, E.; Wilson, D.P.; Kirincich, S.J.; Follows, B.C.; Ipek, M.; Xu, W. Structure-based optimization of protein tyrosine phosphatase-1B inhibitors: Capturing interactions with arginine 24. ChemMedChem 2008, 3, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.P.; Wan, Z.K.; Xu, W.X.; Kirincich, S.J.; Follows, B.C.; Joseph-McCarthy, D.; Foreman, K.; Moretto, A.; Wu, J.; Zhu, M. Structure-based optimization of protein tyrosine phosphatase 1B inhibitors: From the active site to the second phosphotyrosine binding site. J. Med. Chem. 2007, 50, 4681–4698. [Google Scholar] [CrossRef]
- Taha, M.O.; Bustanji, Y.; Al-Bakri, A.G.; Yousef, A.M.; Zalloum, W.A.; Al-Masri, I.M.; Atallah, N. Discovery of new potent human protein tyrosine phosphatase inhibitors via pharmacophore and QSAR analysis followed by in silico screening. J. Mol. Graph. Model. 2007, 25, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.K.; Pandey, G.; Gupta, S.; Singh, A.B.; Srivastava, A.K. Synthesis of protein tyrosine phosphatase 1B inhibitors: Model validation and docking studies. Bioorg. Med. Chem. Lett. 2009, 19, 2320–2323. [Google Scholar] [CrossRef]
- Dai, H.-l.; Gao, L.-X.; Yang, Y.; Li, J.-Y.; Cheng, J.-G.; Li, J.; Wen, R.; Peng, Y.-Q.; Zheng, J.-B. Discovery of di-indolinone as a novel scaffold for protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7440–7443. [Google Scholar] [CrossRef]
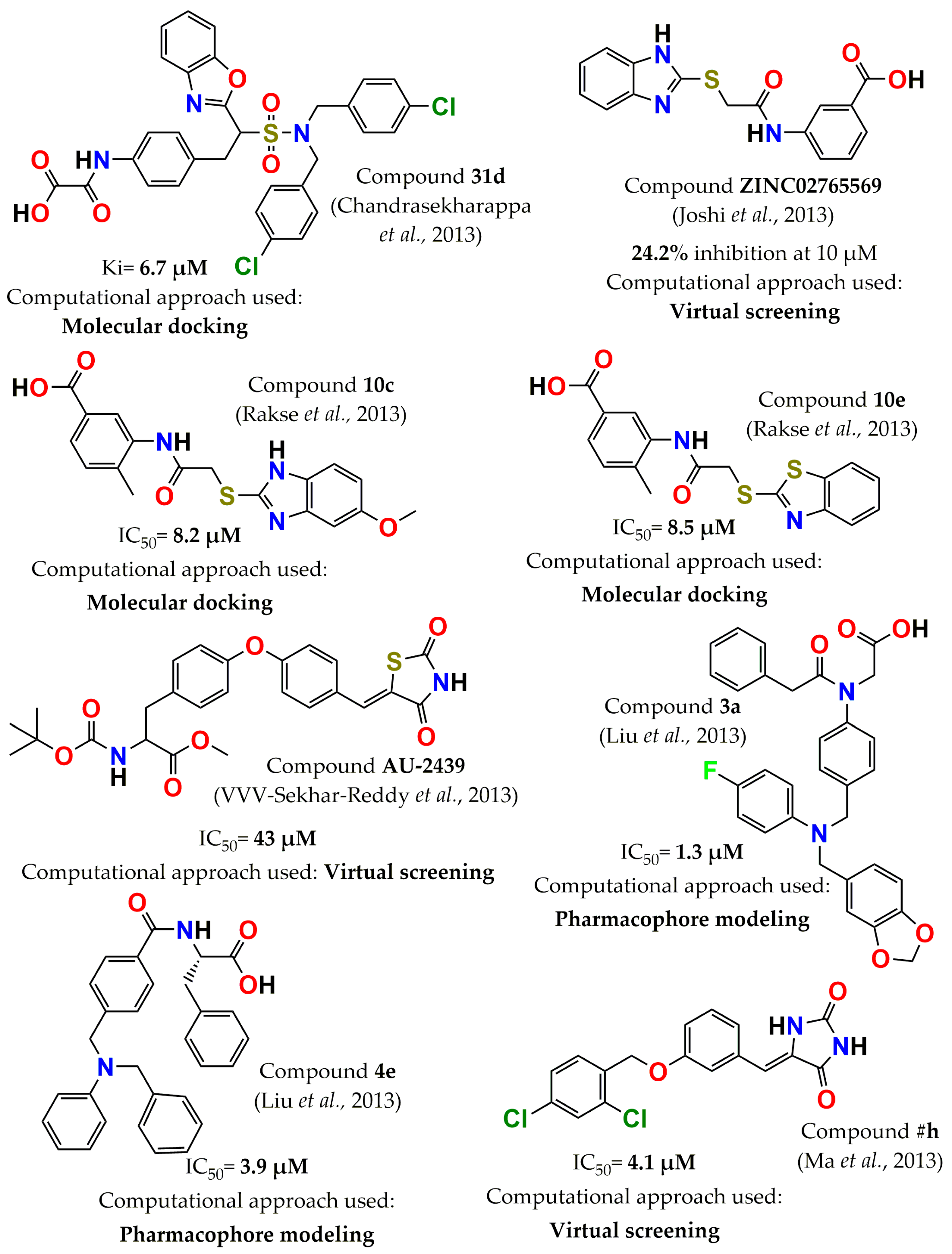
- Chandrasekharappa, A.P.; Badiger, S.E.; Dubey, P.K.; Panigrahi, S.K.; Manukonda, S.R.V. Design and synthesis of 2-substituted benzoxazoles as novel PTP1B inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2579–2584. [Google Scholar] [CrossRef]
- Joshi, P.; Deora, G.S.; Rathore, V.; Tanwar, O.; Rawat, A.K.; Srivastava, A.; Jain, D. Identification of ZINC02765569: A potent inhibitor of PTP1B by vHTS. Med. Chem. Res. 2013, 22, 28–34. [Google Scholar] [CrossRef]
- Rakse, M.; Karthikeyan, C.; Deora, G.S.; Moorthy, N.; Rathore, V.; Rawat, A.K.; Srivastava, A.; Trivedi, P. Design, synthesis and molecular modelling studies of novel 3-acetamido-4-methyl benzoic acid derivatives as inhibitors of protein tyrosine phosphatase 1B. Eur. J. Med. Chem. 2013, 70, 469–476. [Google Scholar] [CrossRef]
- VVV-Sekhar-Reddy, M.; Ghadiyaram, C.; Kumar-Panigrahi, S.; Hosahalli, S.; Narasu-Mangamoori, L. Diphenylether derivative as selective inhibitor of protein tyrosine phosphatase 1B (PTP1B) over T-cell protein tyrosine phosphatase (TCPTP) identified through virtual screening. Mini Rev. Med. Chem. 2013, 13, 1602–1606. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, S.X.; Cheng, X.C.; Wang, S.Q.; Dong, W.L.; Wang, R.L.; Xu, W.R. Design and Synthesis of Imidazolidine-2, 4-Dione Derivatives as Selective Inhibitors by Targeting Protein Tyrosine Phosphatase-1 B Over T-Cell Protein Tyrosine Phosphatase. Chem. Biol. Drug. Des. 2013, 82, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Balaramnavar, V.M.; Srivastava, R.; Rahuja, N.; Gupta, S.; Rawat, A.K.; Varshney, S.; Chandasana, H.; Chhonker, Y.S.; Doharey, P.K.; Kumar, S. Identification of novel PTP1B inhibitors by pharmacophore based virtual screening, scaffold hopping and docking. Eur. J. Med. Chem. 2014, 87, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Haftchenary, S.; Jouk, A.O.; Aubry, I.; Lewis, A.M.; Landry, M.; Ball, D.P.; Shouksmith, A.E.; Collins, C.V.; Tremblay, M.L.; Gunning, P.T. Identification of bidentate salicylic acid inhibitors of PTP1B. ACS Med. Chem. Lett. 2015, 6, 982–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriou, P.; Petrou, A.; Geronikaki, A.; Liaras, K.; Dirnali, S.; Anna, M. Prediction of enzyme inhibition and mode of inhibitory action based on calculation of distances between hydrogen bond donor/acceptor groups of the molecule and docking analysis: An application on the discovery of novel effective PTP1B inhibitors. SAR QSAR Environ. Res. 2015, 26, 557–576. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Du, Y.; Song, L.; Shen, J.; Li, Q. Novel, potent, selective and cellular active ABC type PTP1B inhibitors containing (methanesulfonyl-phenyl-amino)-acetic acid methyl ester phosphotyrosine mimetic. Bioorg. Med. Chem. 2015, 23, 7079–7088. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Prathipati, P.; Thorat, S.; Ansari, S.; Patel, M.; Jain, V.; Jinugu, R.R.; Niranjan, S.; De, S.; Reddy, S. Rational design, synthesis, and structure-activity relationships of 5-amino-1H-pyrazole-4-carboxylic acid derivatives as protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. 2017, 25, 67–74. [Google Scholar] [CrossRef]
- Ganou, C.; Eleftheriou, P.T.; Theodosis-Nobelos, P.; Fesatidou, M.; Geronikaki, A.; Lialiaris, T.; Rekka, E. Docking analysis targeted to the whole enzyme: An application to the prediction of inhibition of PTP1B by thiomorpholine and thiazolyl derivatives. SAR QSAR Environ. Res. 2018, 29, 133–149. [Google Scholar] [CrossRef]
- Chen, X.; Gan, Q.; Feng, C.; Liu, X.; Zhang, Q. Virtual screening of novel and selective inhibitors of protein tyrosine phosphatase 1B over T-cell protein tyrosine phosphatase using a bidentate inhibition strategy. J. Chem. Inf. Model. 2018, 58, 837–847. [Google Scholar] [CrossRef]
- Gimeno, A.; Ardid-Ruiz, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Valls, C.; Aragonès, G.; Suárez, M. Combined Ligand-and Receptor-Based Virtual Screening Methodology to Identify Structurally Diverse Protein Tyrosine Phosphatase 1B Inhibitors. ChemMedChem 2018, 13, 1939–1948. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, Y.; Ling, H.; Li, Q.; Shen, J. Discovery of novel high potent and cellular active ADC type PTP1B inhibitors with selectivity over TC-PTP via modification interacting with C site. Eur. J. Med. Chem. 2018, 144, 692–700. [Google Scholar] [CrossRef]
- Xue, W.; Tian, J.; Wang, X.S.; Xia, J.; Wu, S. Discovery of potent PTP1B inhibitors via structure-based drug design, synthesis and in vitro bioassay of Norathyriol derivatives. Bioorg. Chem. 2019, 86, 224–234. [Google Scholar] [CrossRef] [PubMed]
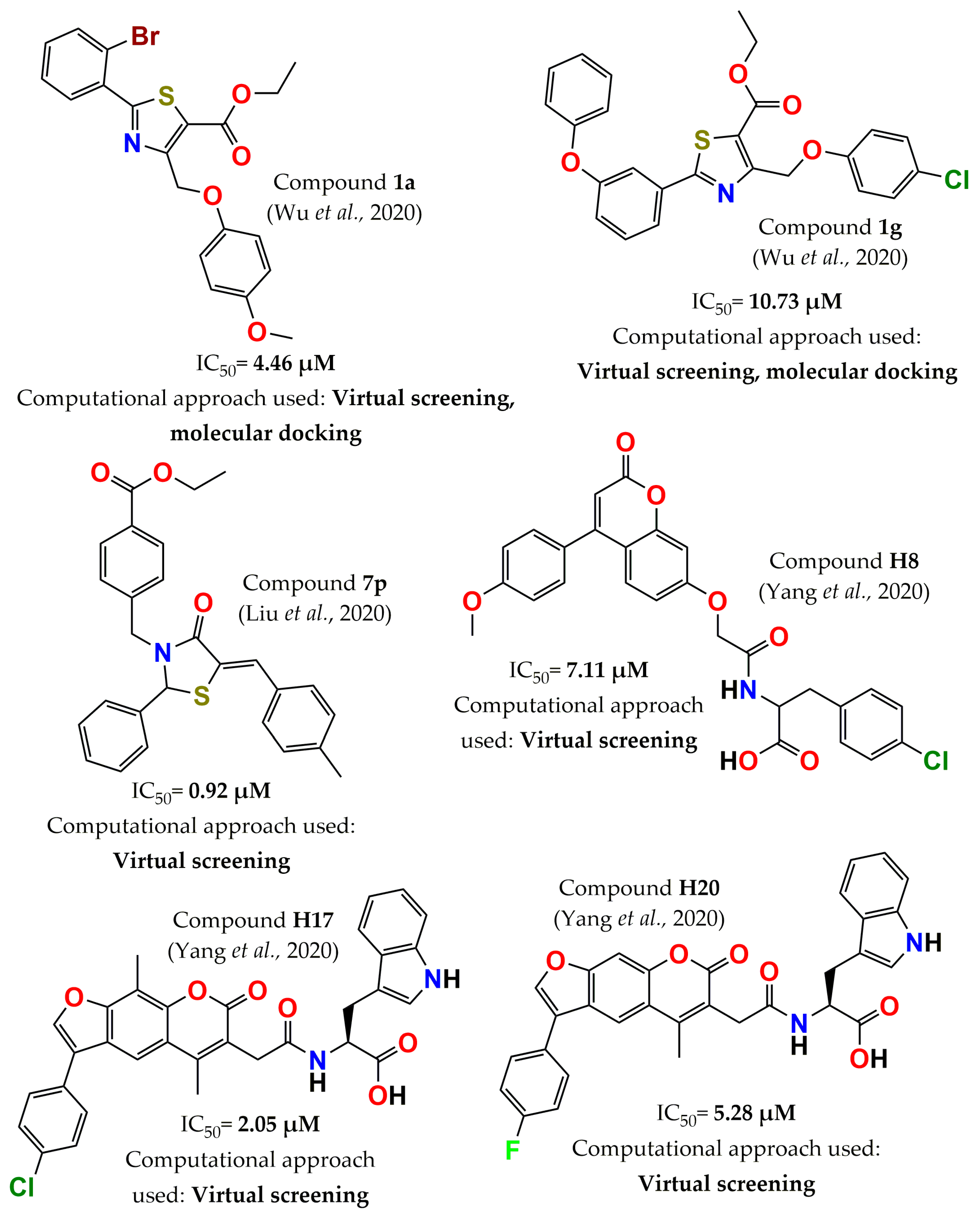
- Wu, J.; Ma, Y.; Zhou, H.; Zhou, L.; Du, S.; Sun, Y.; Li, W.; Dong, W.; Wang, R. Identification of protein tyrosine phosphatase 1B (PTP1B) inhibitors through De Novo Evoluton, synthesis, biological evaluation and molecular dynamics simulation. Biochem. Biophys. Res. Commun. 2020, 526, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.S.; Wang, R.R.; Yue, H.; Zheng, Z.H.; Lu, X.H.; Wang, S.Q.; Dong, W.L.; Wang, R.L. Design, synthesis, biological evaluation and molecular dynamics studies of 4-thiazolinone derivatives as protein tyrosine phosphatase 1B (PTP1B) inhibitors. J. Biomol. Struct. Dyn. 2020, 38, 3814–3824. [Google Scholar] [CrossRef]
- Yang, Y.; Tian, J.Y.; Ye, F.; Xiao, Z. Identification of natural products as selective PTP1B inhibitors via virtual screening. Bioorg. Chem. 2020, 98, 103706. [Google Scholar] [CrossRef] [PubMed]
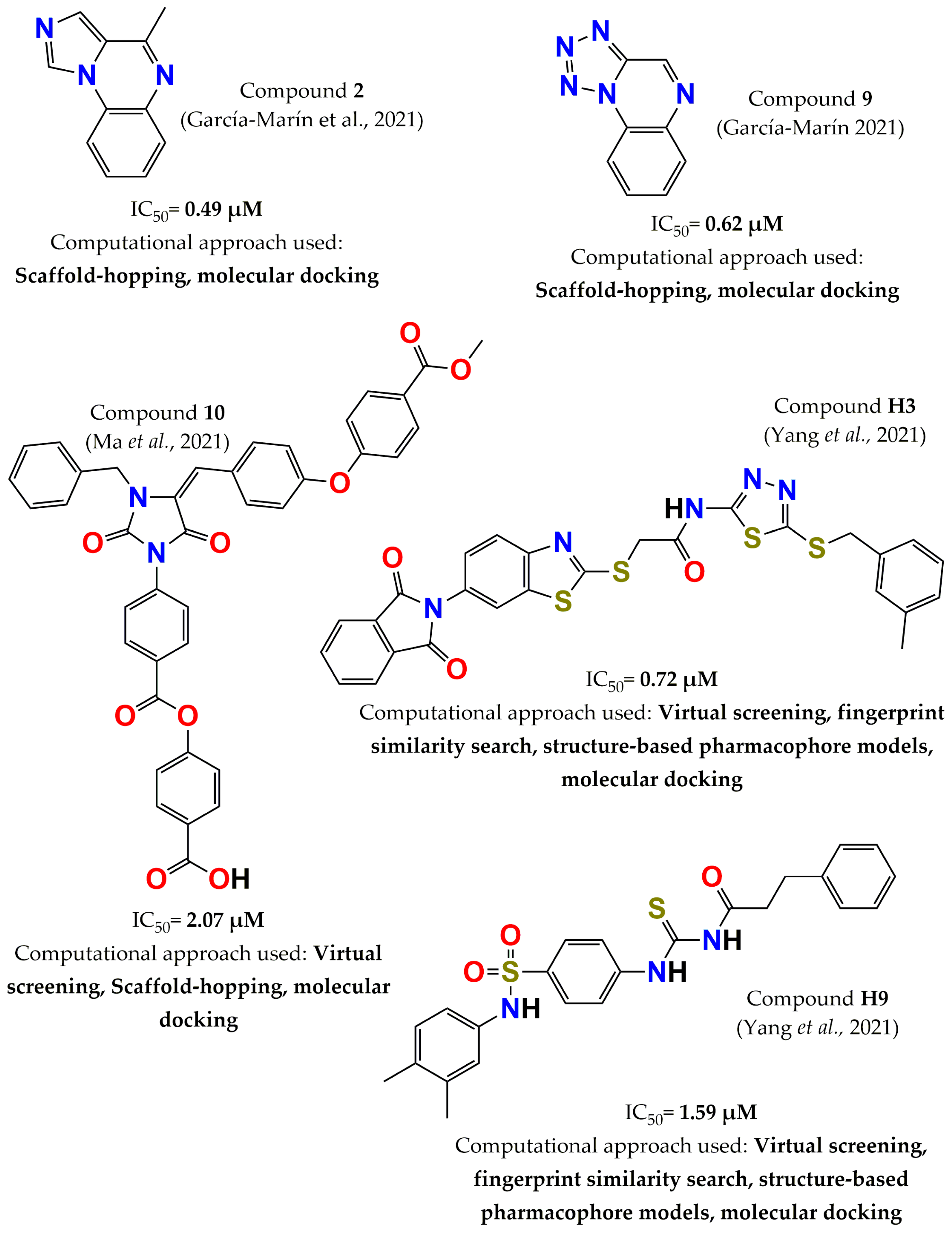
- García-Marín, J.; Griera, M.; Alajarín, R.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; Vaquero, J.J. A Computer-Driven Scaffold-Hopping Approach Generating New PTP1B Inhibitors from the Pyrrolo [1, 2-a] quinoxaline Core. ChemMedChem 2021, 16, 2895–2906. [Google Scholar] [CrossRef]
- Ma, Y.; Ding, T.T.; Liu, Y.Y.; Zheng, Z.H.; Sun, S.X.; Zhang, L.S.; Zhang, H.; Lu, X.-H.; Cheng, X.C.; Wang, R.L. Design, synthesis, biological evaluation and molecular dynamics simulation studies of imidazolidine-2, 4-dione derivatives as novel PTP1B inhibitors. Biochem. Biophys. Res. Commun. 2021, 579, 40–46. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, L.; Tian, J.; Ye, F.; Xiao, Z. Integrated approach to identify selective PTP1B inhibitors targeting the allosteric site. J. Chem. Inf. Model. 2021, 61, 4720–4732. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Almazán, M.I.; Hernández-Campos, A.; Castillo, R.; Sierra-Campos, E.; Valdez-Solana, M.; Avitia-Domínguez, C.; Téllez-Valencia, A. Computational Methods in Cooperation with Experimental Approaches to Design Protein Tyrosine Phosphatase 1B Inhibitors in Type 2 Diabetes Drug Design: A Review of the Achievements of This Century. Pharmaceuticals 2022, 15, 866. https://doi.org/10.3390/ph15070866
Campos-Almazán MI, Hernández-Campos A, Castillo R, Sierra-Campos E, Valdez-Solana M, Avitia-Domínguez C, Téllez-Valencia A. Computational Methods in Cooperation with Experimental Approaches to Design Protein Tyrosine Phosphatase 1B Inhibitors in Type 2 Diabetes Drug Design: A Review of the Achievements of This Century. Pharmaceuticals. 2022; 15(7):866. https://doi.org/10.3390/ph15070866
Chicago/Turabian StyleCampos-Almazán, Mara Ibeth, Alicia Hernández-Campos, Rafael Castillo, Erick Sierra-Campos, Mónica Valdez-Solana, Claudia Avitia-Domínguez, and Alfredo Téllez-Valencia. 2022. "Computational Methods in Cooperation with Experimental Approaches to Design Protein Tyrosine Phosphatase 1B Inhibitors in Type 2 Diabetes Drug Design: A Review of the Achievements of This Century" Pharmaceuticals 15, no. 7: 866. https://doi.org/10.3390/ph15070866
APA StyleCampos-Almazán, M. I., Hernández-Campos, A., Castillo, R., Sierra-Campos, E., Valdez-Solana, M., Avitia-Domínguez, C., & Téllez-Valencia, A. (2022). Computational Methods in Cooperation with Experimental Approaches to Design Protein Tyrosine Phosphatase 1B Inhibitors in Type 2 Diabetes Drug Design: A Review of the Achievements of This Century. Pharmaceuticals, 15(7), 866. https://doi.org/10.3390/ph15070866