
Development and Validation of an HPLC-UV Method for the Dissolution Studies of 3D-Printed Paracetamol Formulations in Milk-Containing Simulated Gastrointestinal Media

 ,
,  ,
, 
 and
and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Method Development
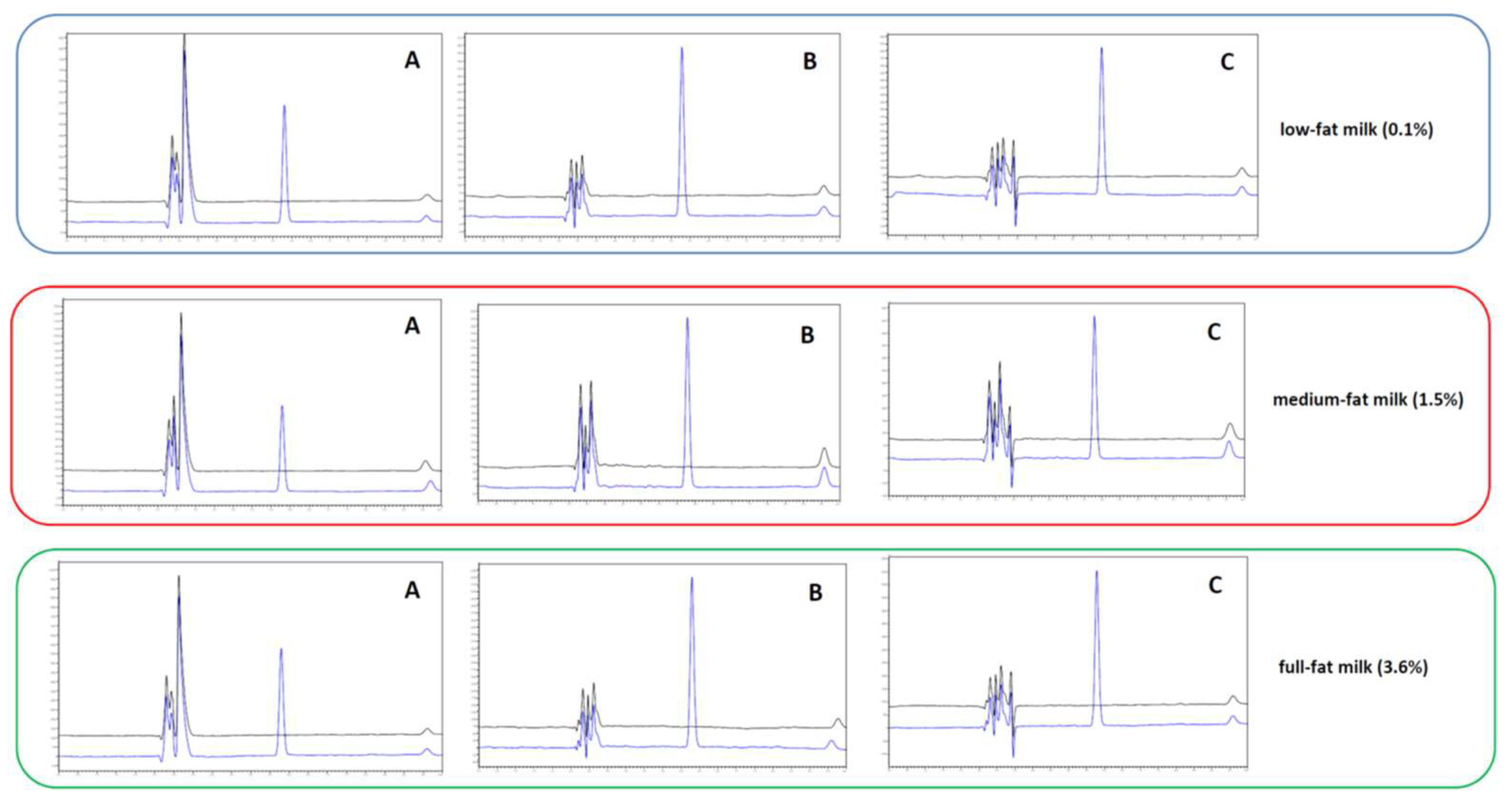
2.2. Optimization of Sample Preparation
2.3. Method Validation
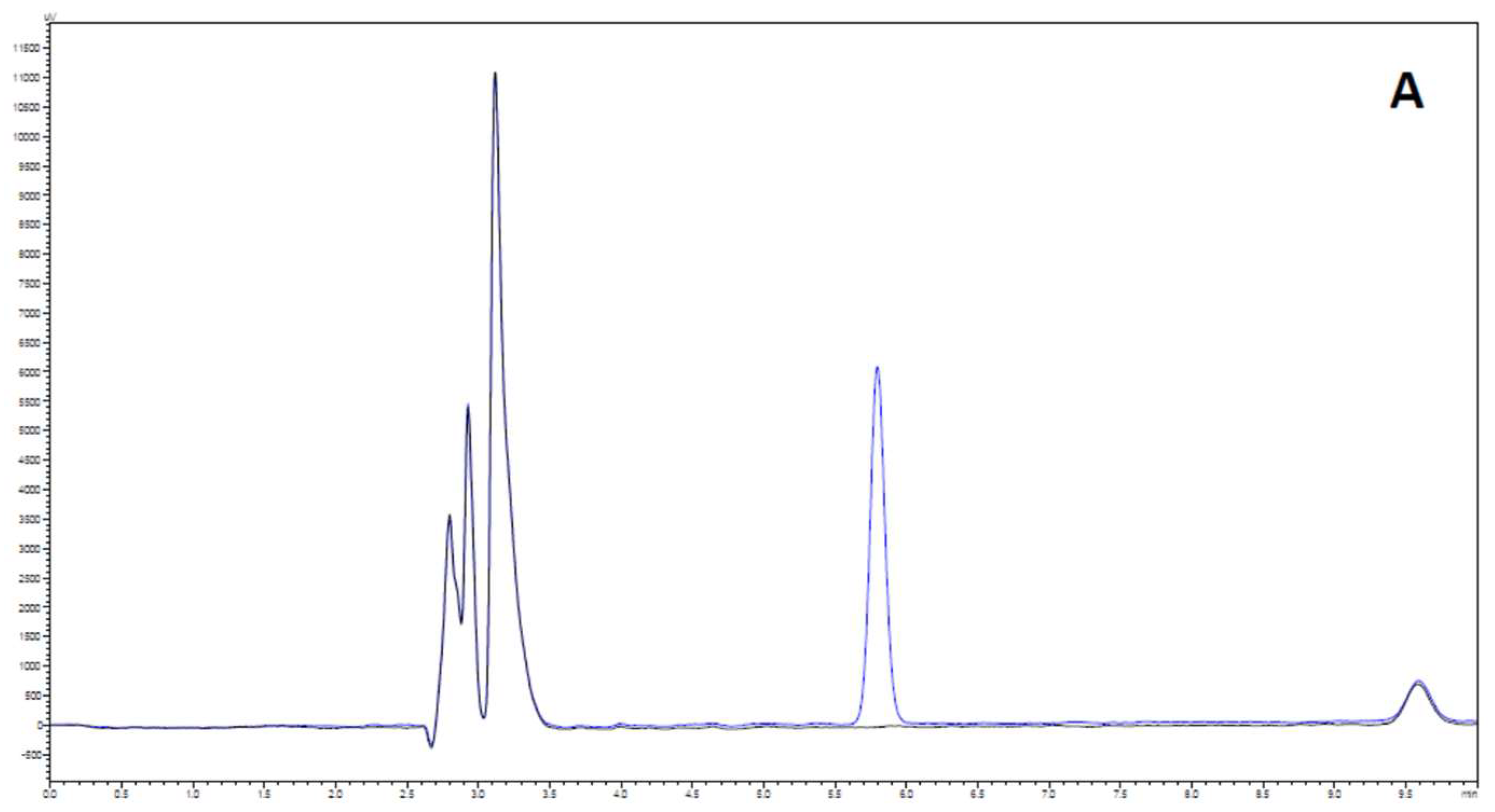
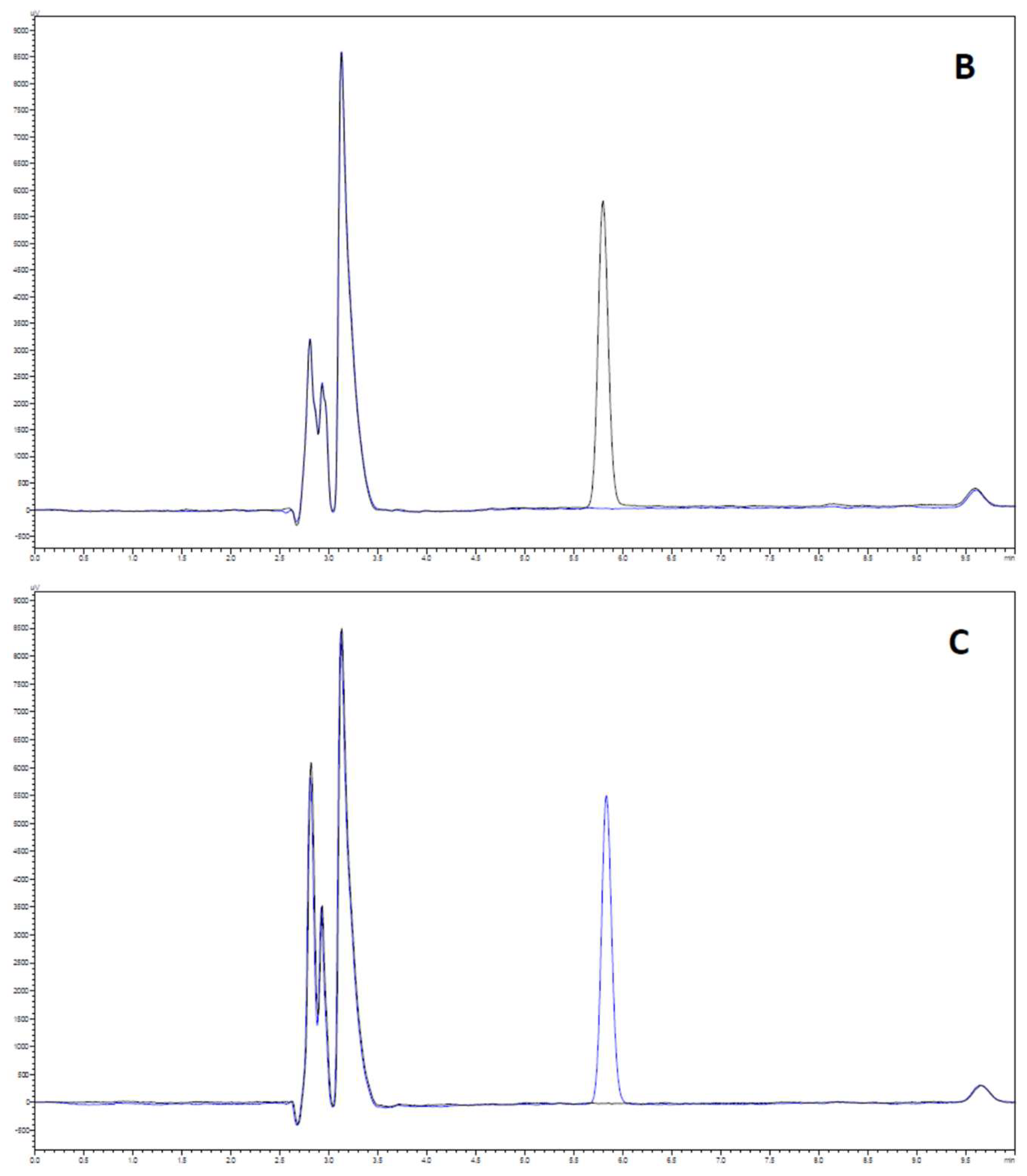
2.3.1. System Suitability
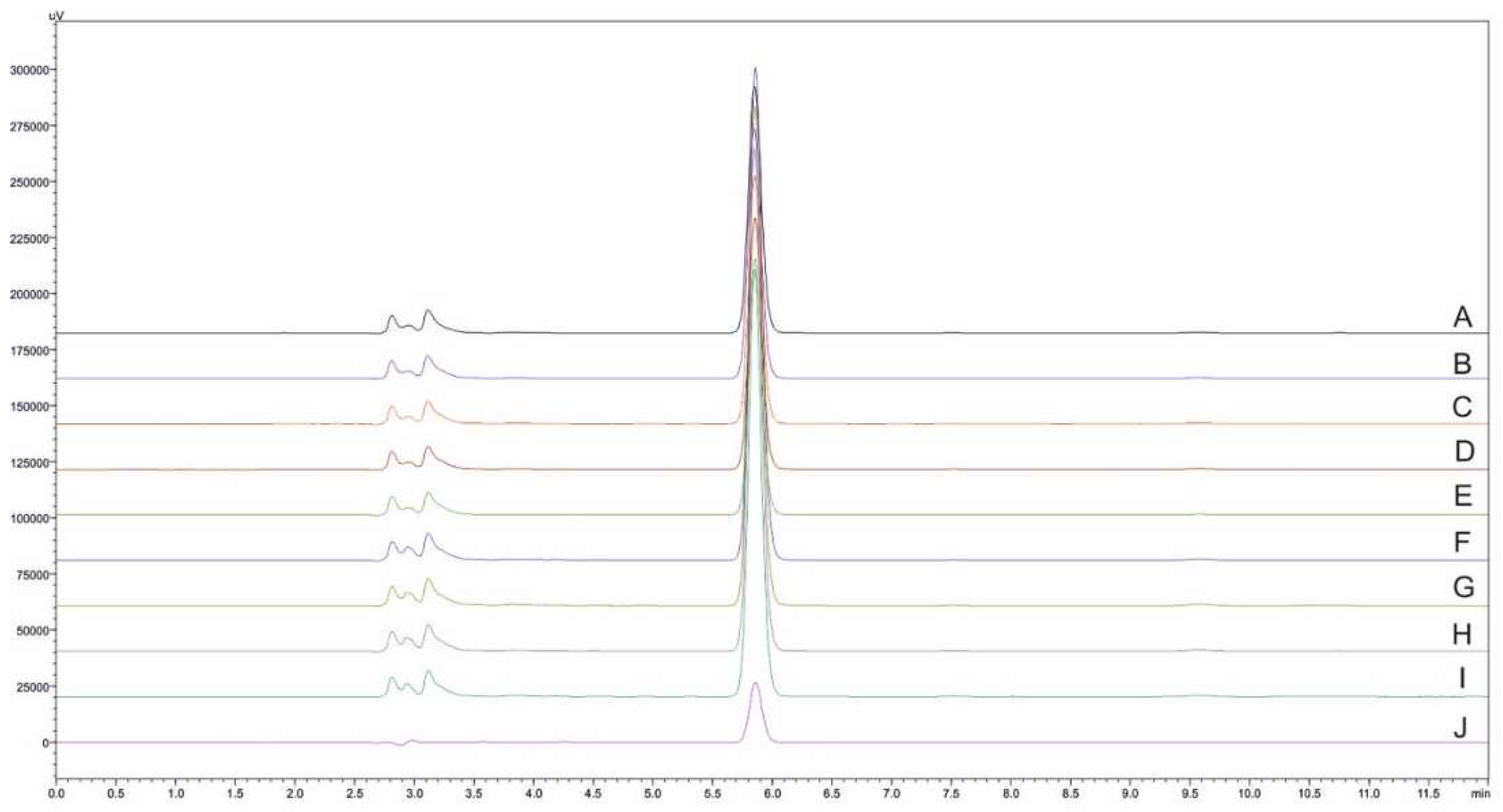
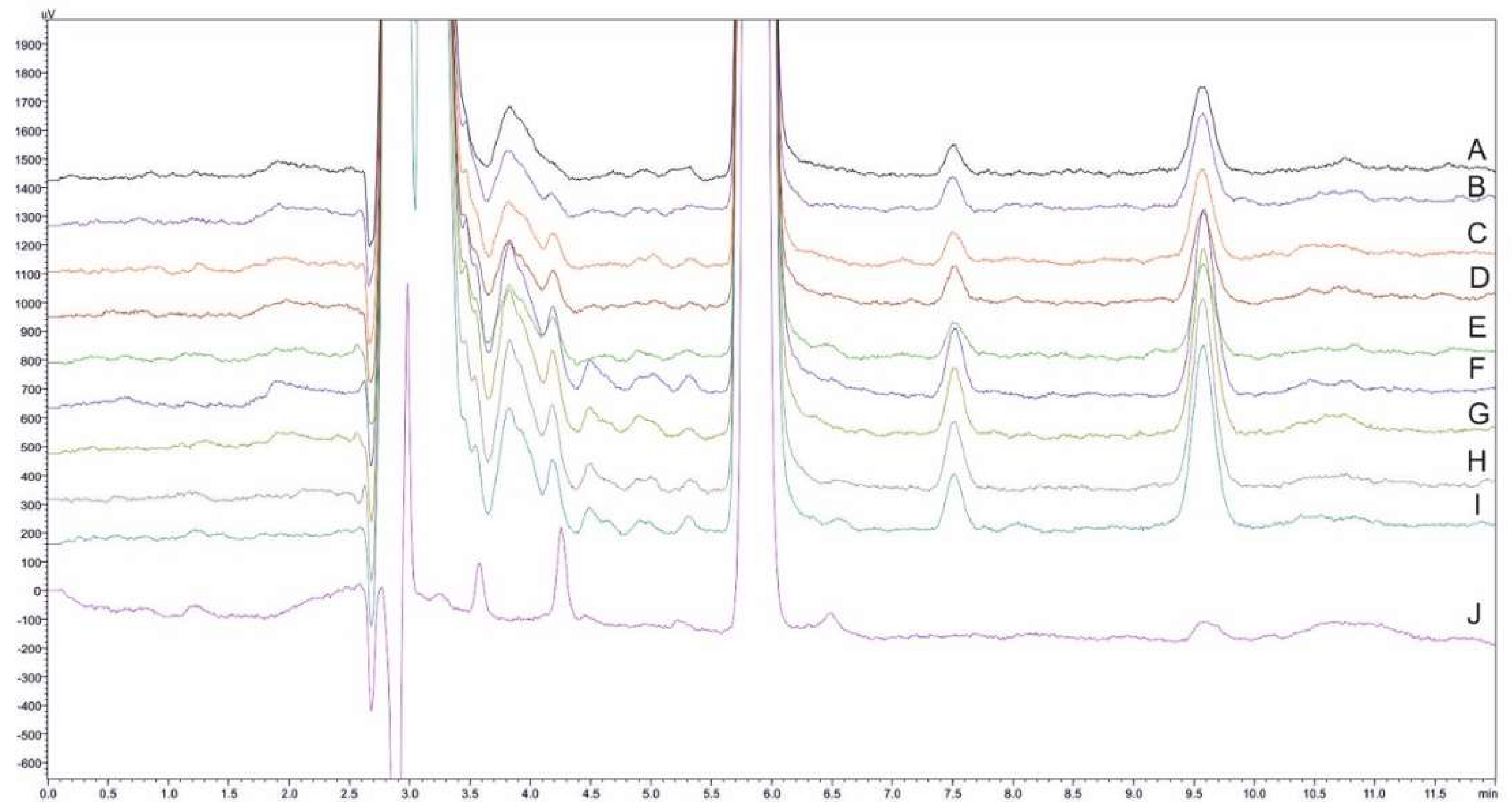
2.3.2. Selectivity and Carry-Over
2.3.3. Selection of the Response Function
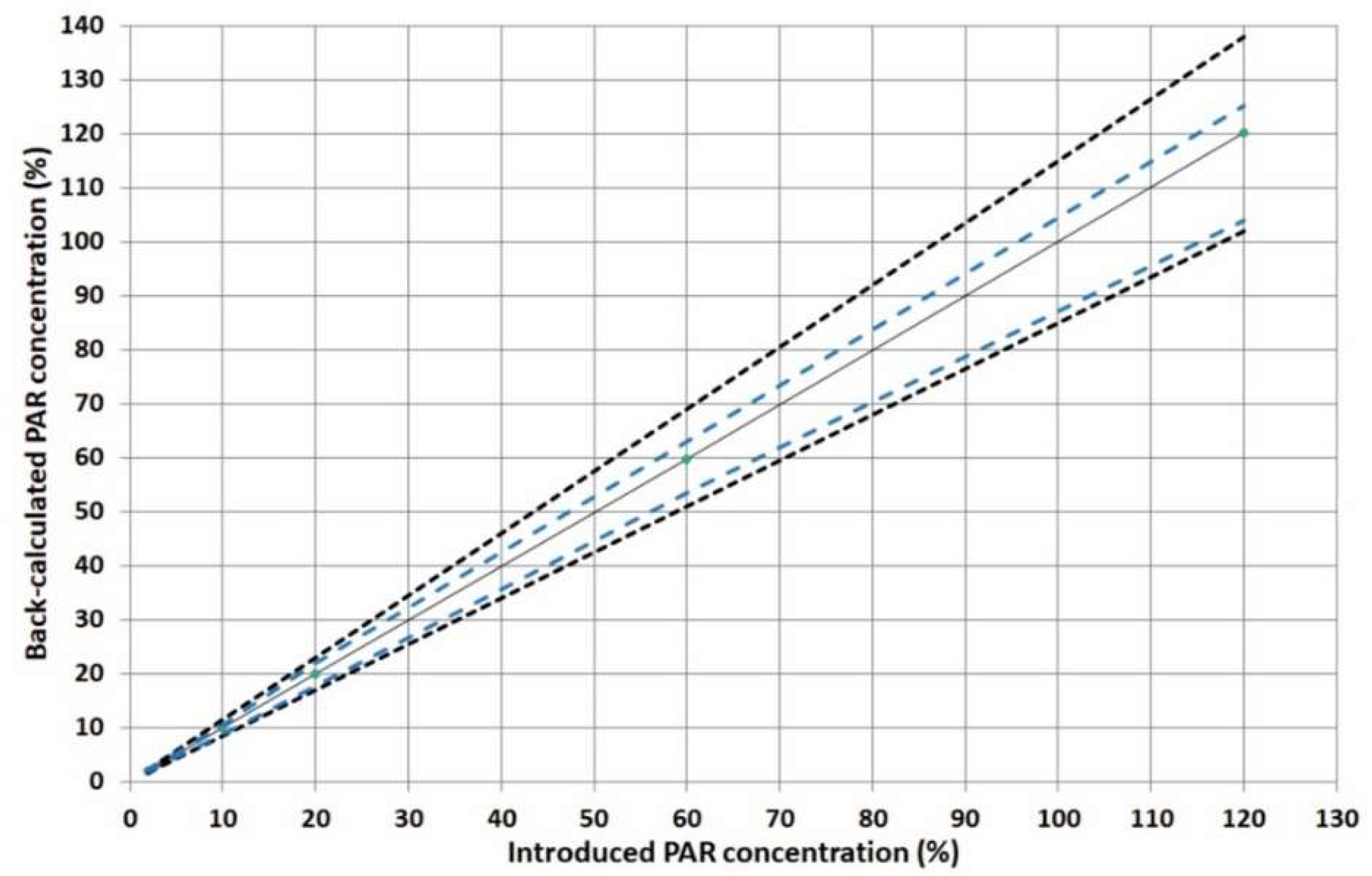
2.3.4. Linearity
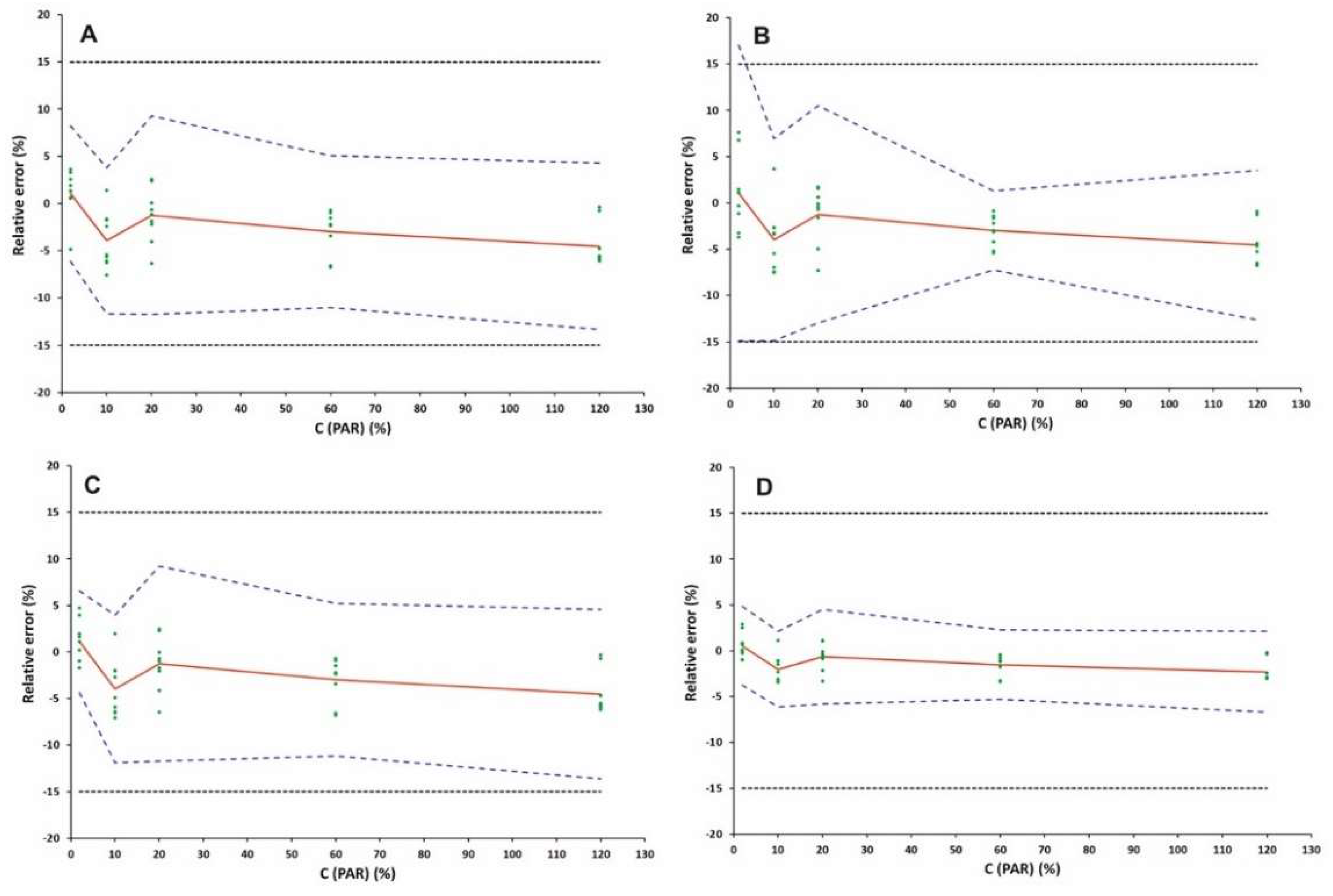
2.3.5. Accuracy, Precision and Trueness
2.3.6. LODs and LOQs
2.3.7. Solution Stability
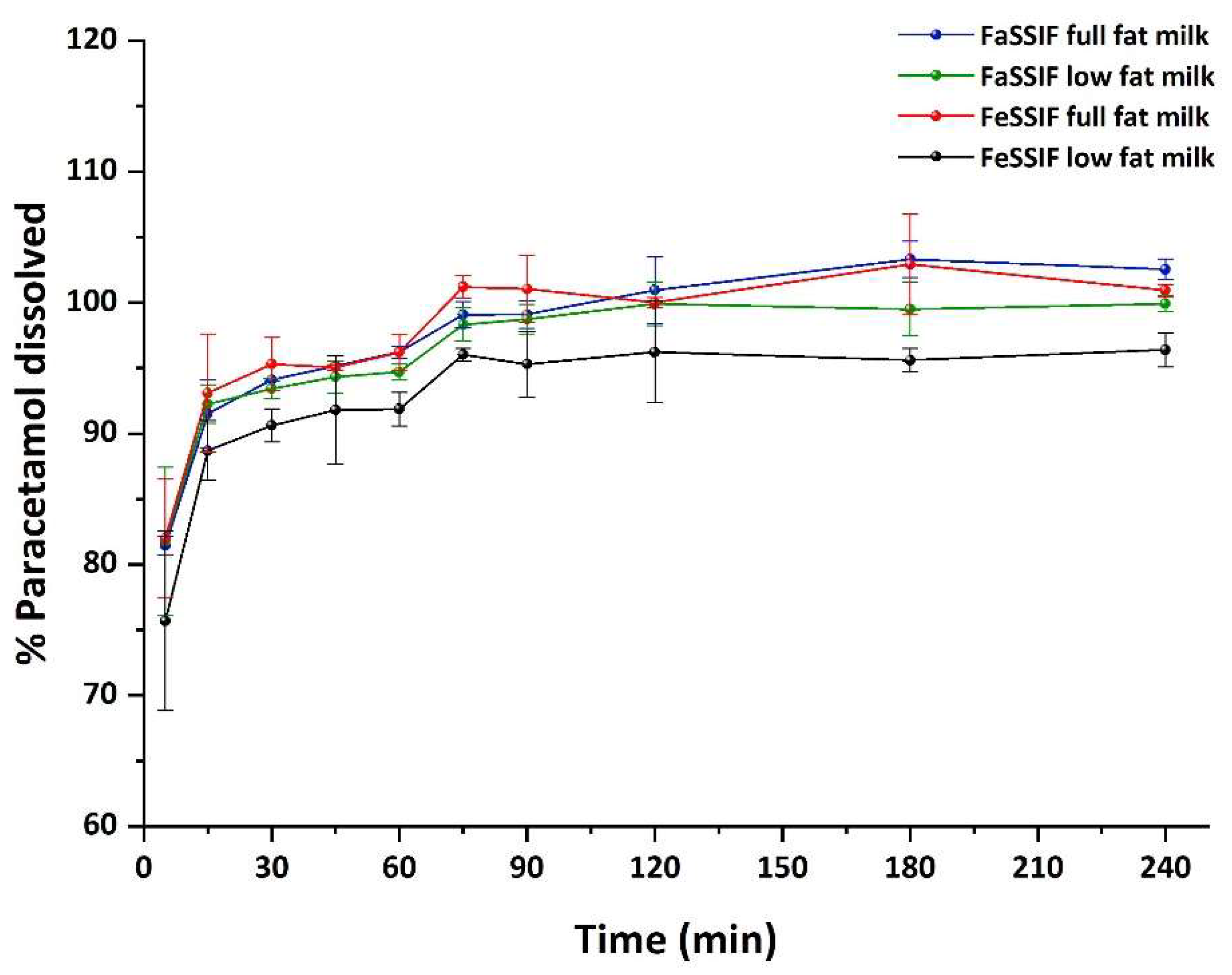
2.4. Method Application—Dissolution Study
3. Materials and Methods
3.1. Chemicals and Solutions
3.2. Instrumentation and Chromatographic Conditions
3.3. Preparation of Calibration (CS) and Validation Standards (VS)
3.4. Sample Preparation
3.5. Method Validation
- -
- is the estimate of the mean results of the jth concentration level;
- -
- is the unknown “true value”;
- -
- p is the number of series;
- -
- n is the number of independent replicates per series;
- -
- Qt (ν; is the β quantile of the t-Student distribution with ν degrees of freedom;
- -
- is the within-series variance;
- -
- is the between-series variance.
3.6. System Suitability Test
3.7. Application of the Proposed Method—Dissolution Test
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef] [Green Version]
- US Food and Drug Administration Guidance for Industry: Extended Release Oral Dosage Forms, Development Evaluation and Application of In Vitro/In Vivo Correlations. Cent. Drug Eval. Res. 1997.
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution Media Simulating Conditions in the Proximal Human Gastrointestinal Tract: An Update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef]
- Wang, Q.; Fotaki, N.; Mao, Y. Biorelevant Dissolution: Methodology and Application in Drug Development. Dissolution Technol. 2009, 16, 6–12. [Google Scholar] [CrossRef]
- Elkasabgy, N.A.; Mahmoud, A.A.; Maged, A. 3D printing: An appealing route for customized drug delivery systems. Int. J. Pharm. 2020, 588, 119732. [Google Scholar] [CrossRef]
- Perrot, S.; Cittée, J.; Louis, P.; Quentin, B.; Robert, C.; Milon, J.; Bismut, H.; Baumelou, A. Self-medication in pain management: The state of the art of pharmacists’ role for optimal Over-The-Counter analgesic use. Eur. J. Pain 2019, 23, 1747–1762. [Google Scholar] [CrossRef]
- Karavasili, C.; Zgouro, P.; Manousi, N.; Lazaridou, A.; Zacharis, C.K.; Bouropoulos, N.; Moschakis, T.; Fatouros, D.G. Cereal-Based 3D Printed Dosage Forms for Drug Administration During Breakfast in Pediatric Patients within a Hospital Setting. J. Pharm. Sci. 2022. [Google Scholar] [CrossRef]
- Baxevanis, F.; Kuiper, J.; Fotaki, N. Strategic drug analysis in fed-state gastric biorelevant media based on drug physicochemical properties. Eur. J. Pharm. Biopharm. 2018, 127, 326–341. [Google Scholar] [CrossRef]
- Koesukwiwat, U.; Jayanta, S.; Leepipatpiboon, N. Solid-phase extraction for multiresidue determination of sulfonamides, tetracyclines, and pyrimethamine in Bovine’s milk. J. Chromatogr. A 2007, 1149, 102–111. [Google Scholar] [CrossRef]
- Nász, S.; Debreczeni, L.; Rikker, T.; Eke, Z. Development and validation of a liquid chromatographic-tandem mass spectrometric method for determination of eleven coccidiostats in milk. Food Chem. 2012, 133, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, M.; Ishida, S.; Hirata, Y.; Hirata, K.; Watanabe, K.; Seno, H.; Ishii, A. Simple analysis of acetaminophen in human plasma by solid-phase microextraction and gas chromatography. Jpn. J. Forensic Toxicol. 2004, 22, 22–26. [Google Scholar]
- Kishida, K. Simplified extraction of tetracycline antibiotics from milk using a centrifugal ultrafiltration device. Food Chem. 2011, 126, 687–690. [Google Scholar] [CrossRef]
- Franek, F.; Holm, P.; Larsen, F.; Steffansen, B. Interaction between fed gastric media (Ensure Plus®) and different hypromellose based caffeine controlled release tablets: Comparison and mechanistic study of caffeine release in fed and fasted media versus water using the USP dissolution apparatus 3. Int. J. Pharm. 2014, 461, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, F.; Kuiper, J.; Fotaki, N. Fed-state gastric media and drug analysis techniques: Current status and points to consider. Eur. J. Pharm. Biopharm. 2016, 107, 234–248. [Google Scholar] [CrossRef] [Green Version]
- ICH Harmonised Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2 (R1). 2005. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 10 June 2022).
- Valko, K.; Nunhuck, S.; Bevan, C.; Abraham, M.H.; Reynolds, D.P. Fast Gradient HPLC Method to Determine Compounds Binding to Human Serum Albumin. Relationships with Octanol/Water and Immobilized Artificial Membrane Lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. [Google Scholar] [CrossRef]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.A.; Dewe, W.; Feinberg, M.; Lallier, M.; et al. Validation of quantitative analytical procedure, harmonization of approaches. STP Pharma Prat. 2003, 13, 101–138. [Google Scholar]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.A.; Dewe, W.; Feinberg, M.; Lallier, M.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal—Part I. J. Pharm. Biomed. Anal. 2004, 36, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Bouabidi, A.; Talbi, M.; Bourichi, H.; Bouklouze, A.; El Karbane, M.; Boulanger, B.; Brik, Y.; Hubert, P.; Rozet, E. Flexibility and applicability of β-expectation tolerance interval approach to assess the fitness of purpose of pharmaceutical analytical methods. Drug Test. Anal. 2012, 4, 1014–1027. [Google Scholar] [CrossRef]
- Hubert, P.; Nguyen-Huu, J.-J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.-A.; Dewé, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal—Part III. J. Pharm. Biomed. Anal. 2007, 45, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Nguyen-Huu, J.-J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.-A.; Dewé, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal—Part IV. J. Pharm. Biomed. Anal. 2008, 48, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M. Validation of analytical methods based on accuracy profiles. J. Chromatogr. A 2007, 1158, 174–183. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calibration Curve 1 | Slope ± SD 2 (×103) | |||||
|---|---|---|---|---|---|---|
| Aqueous | 21.1 ± 0.10 | |||||
| Precipitation reagent/fat content | MeOH | ACN | 10% TFA | |||
| Slope ± SD (×103) | ME 3 (%) | Slope ± SD (×103) | ME% | Slope ± SD (×103) | ME% | |
| [FaSSGF:milk, 4:1] | ||||||
| 0.1% | 20.7 ± 0.31 | 98.1 | 21.4 ± 0.26 | 101.4 | 20.8 ± 0.31 | 98.6 |
| 1.5% | 20.7 ± 0.10 | 98.1 | 20.9 ± 0.07 | 99.1 | 20.4 ± 0.07 | 96.7 |
| 3.6% | 21.0 ± 0.08 | 99.5 | 22.0 ± 0.28 | 104.2 | 21.4 ± 0.16 | 101.4 |
| [FaSSIF:milk, 4:1] | ||||||
| 0.1% | 19.4 ± 0.28 | 91.9 | 20.8 ± 0.36 | 98.6 | 20.2 ± 0.17 | 95.7 |
| 1.5% | 20.3 ± 0.06 | 96.2 | 19.0 ± 0.22 | 90.0 | 19.6 ± 0.22 | 92.9 |
| 3.6% | 20.9 ± 0.17 | 99.1 | 21.5 ± 0.12 | 101.9 | 19.7 ± 0.14 | 93.4 |
| [FeSSIF:milk, 4:1] | ||||||
| 0.1% | 19.5 ± 0.31 | 92.4 | 20.8 ± 0.45 | 98.6 | 20.1 ± 0.26 | 95.3 |
| 1.5% | 20.6 ± 0.28 | 97.6 | 19.7 ± 0.17 | 93.3 | 19.2 ± 0.43 | 91.0 |
| 3.6% | 20.5 ± 0.23 | 97.2 | 21.3 ± 0.20 | 101.0 | 20.1 ± 0.35 | 95.3 |
| Validation Criteria | |||
|---|---|---|---|
| Response function (linear regression) | Slope (×103) | Intercept (×103) | r2 |
| (k 1 = 3; m = 5; n = 3) (2–120%) | |||
| Day 1 | 21.07 | −2.483 | 0.9997 |
| Day 2 | 21.13 | −0.238 | 0.9999 |
| Day 3 | 21.18 | −3.135 | 1.0000 |
| Precision (k = 3; n = 3) | |||
| C (μg mL−1) | sr (%) 2 | sR (%) 3 | |
| 10 | 2.1 | 2.6 | |
| 50 | 2.7 | 3.0 | |
| 100 | 1.9 | 3.1 | |
| 300 | 1.5 | 2.4 | |
| 600 | 1.4 | 2.5 | |
| Trueness (k = 3; n = 3) | |||
| C (μg mL−1) | Relative bias (%) | ||
| 10 | +1.1 | ||
| 50 | +3.9 | ||
| 100 | −1.2 | ||
| 300 | −3.0 | ||
| 600 | −4.5 | ||
| Accuracy (k = 3; n = 3) | |||
| C (μg mL−1) | Relative β-ΕΤΙ (%) | ||
| 10 | [−6.13, 8.25] | ||
| 50 | [−11.67, 3.81] | ||
| 100 | [−11.75, 9.28] | ||
| 300 | [−11.00, 5.10] | ||
| 600 | [−13.34, 4.29] | ||
| Linearity (k = 3; n = 3; m = 5) (2–120%) | |||
| Slope | 1.000 | ||
| Intercept | −0.0004 | ||
| r2 | 0.9999 | ||
| LOD (μg mL−1) | 0.02 | ||
| LLOQ/ULOQ (%) | 2/120 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manousi, N.; Karavasili, C.; Fatouros, D.G.; Tzanavaras, P.D.; Zacharis, C.K. Development and Validation of an HPLC-UV Method for the Dissolution Studies of 3D-Printed Paracetamol Formulations in Milk-Containing Simulated Gastrointestinal Media. Pharmaceuticals 2022, 15, 755. https://doi.org/10.3390/ph15060755
Manousi N, Karavasili C, Fatouros DG, Tzanavaras PD, Zacharis CK. Development and Validation of an HPLC-UV Method for the Dissolution Studies of 3D-Printed Paracetamol Formulations in Milk-Containing Simulated Gastrointestinal Media. Pharmaceuticals. 2022; 15(6):755. https://doi.org/10.3390/ph15060755
Chicago/Turabian StyleManousi, Natalia, Christina Karavasili, Dimitrios G. Fatouros, Paraskevas D. Tzanavaras, and Constantinos K. Zacharis. 2022. "Development and Validation of an HPLC-UV Method for the Dissolution Studies of 3D-Printed Paracetamol Formulations in Milk-Containing Simulated Gastrointestinal Media" Pharmaceuticals 15, no. 6: 755. https://doi.org/10.3390/ph15060755
APA StyleManousi, N., Karavasili, C., Fatouros, D. G., Tzanavaras, P. D., & Zacharis, C. K. (2022). Development and Validation of an HPLC-UV Method for the Dissolution Studies of 3D-Printed Paracetamol Formulations in Milk-Containing Simulated Gastrointestinal Media. Pharmaceuticals, 15(6), 755. https://doi.org/10.3390/ph15060755