
Design and Development of Levodopa Loaded Polymeric Nanoparticles for Intranasal Delivery

 ,
,  ,
,
Abstract
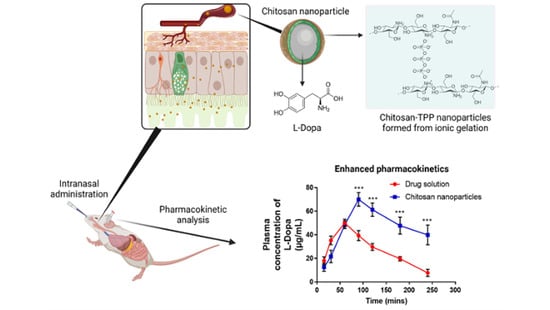
:
1. Introduction
2. Materials and Methods
2.1. Materials
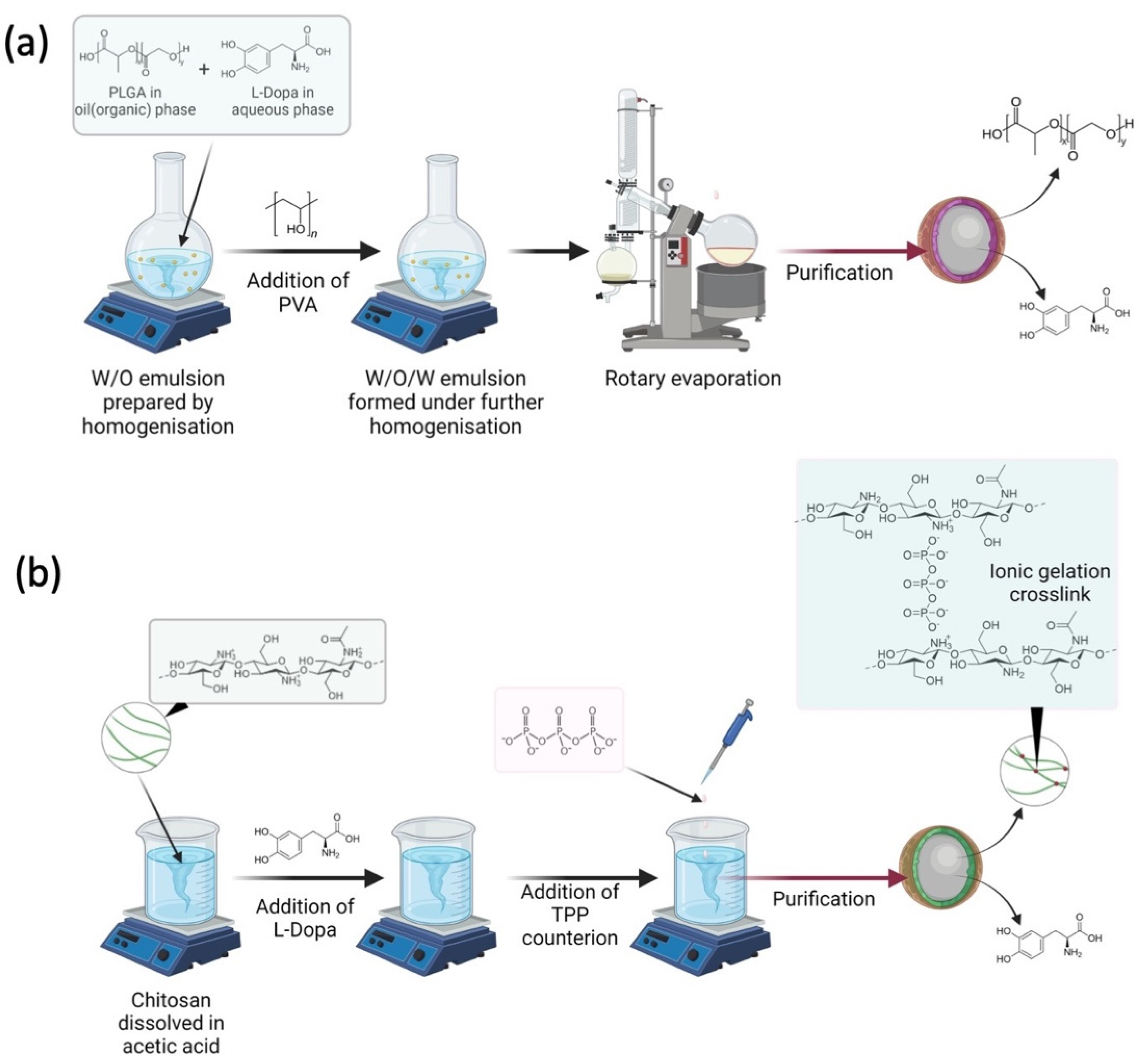
2.2. Preparation of L-Dopa Loaded PLGA Nanoparticles
2.3. Preparation of L-Dopa Loaded Chitosan Nanoparticles
2.4. Characterisation of Nanoparticles
2.4.1. Particle Size Analysis
2.4.2. Drug Entrapment Efficiency
2.4.3. Zeta Potential
2.4.4. Morphology Study
2.4.5. X-ray Powder Diffraction Analysis
2.4.6. Fourier Transform Infrared (FTIR) Analysis
2.4.7. Instrumentation and Chromatographic Condition for the Analytical Method
2.5. In Vivo Nasal Absorption Study
2.6. Statistical Analysis
3. Results and Discussion
3.1. Characterisation of Nanoparticles
3.1.1. Nanoparticle Size and Dispersity
3.1.2. Zeta Potential of Nanoparticles
3.1.3. Drug Entrapment Efficiency
3.1.4. Morphology of L-Dopa Loaded Chitosan Nanoparticle Characterisation
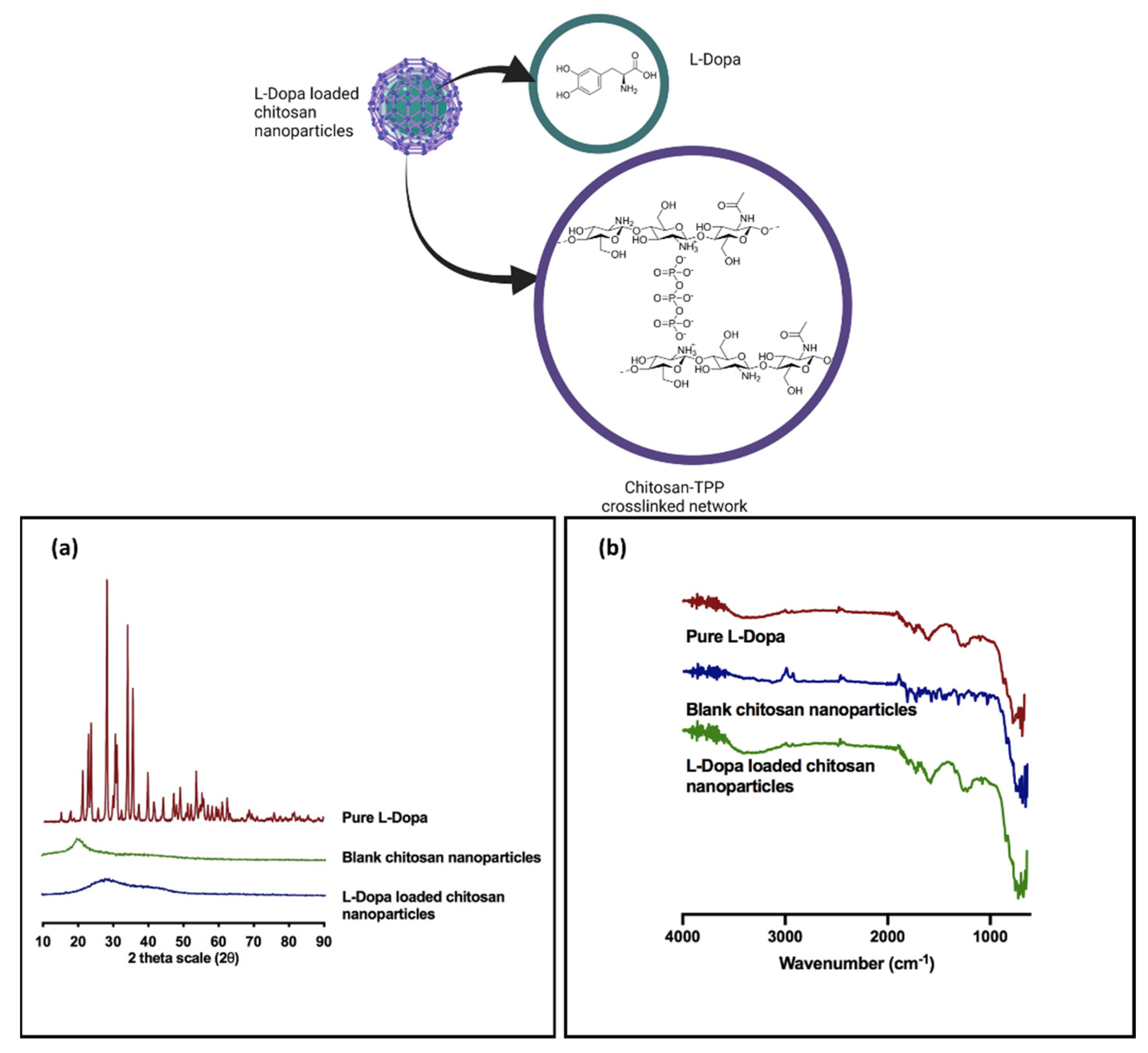
3.1.5. Powder X-ray Diffraction Analysis
3.1.6. Fourier Transform Infrared Analysis
3.2. In Vivo Intranasal Delivery Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pagliaro, L.A.; Pagliaro, A.M.; Pagliaro, L.A.; Pagliaro, A.M. Levodopa⋆ [L-Dopa]. In PNDR: Psychologists’ Neuropsychotropic Drug Reference; Routledge: Oxfordshire, UK, 2020; pp. 185–189. [Google Scholar]
- Lerner, R.P.; Francardo, V.; Fujita, K.; Bimpisidis, Z.; Jourdain, V.A.; Tang, C.C.; Dewey, S.L.; Chaly, T.; Cenci, M.A.; Eidelberg, D. Levodopa-induced abnormal involuntary movements correlate with altered permeability of the blood-brain-barrier in the basal ganglia. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr. Neuropharmacol. 2017, 16, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Suttrup, I.; Warnecke, T. Dysphagia in Parkinson’s Disease. Dysphagia 2016, 31, 24–32. [Google Scholar] [CrossRef]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of Intranasal Delivery Route of Drug Administration for Brain Targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.S.; Illum, L. Absorption Enhancers for Nasal Drug Delivery. Clin. Pharmacokinet. 2003, 42, 1107–1128. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, K.H.; Yoon, I.K.; Lee, K.E.; Chun, I.K.; Rhie, J.Y.; Gwak, H.S. Pharmacokinetic evaluation of formulated levodopa methyl ester nasal delivery systems. Eur. J. Drug Metab. Pharmacokinet. 2014, 39, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, A.; Yan, X.; Chu, L.; Yang, X.; Song, Y.; Sun, K.; Yu, X.; Liu, R.; Wu, Z.; et al. Brain-targeted intranasal delivery of dopamine with borneol and lactoferrin co-modified nanoparticles for treating Parkinson’s disease. Drug Deliv. 2019, 26, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241. [Google Scholar] [CrossRef]
- Zainuddin, S.Z.; Hamid, K.A. Chitosan-Based Oral Drug Delivery System for Peptide, Protein and Vaccine Delivery. In Chitin and Chitosan—Physicochemical Properties and Industrial Applications [Working Title]; InTech: London, UK, 2021. [Google Scholar]
- Yu, S.; Xu, X.; Feng, J.; Liu, M.; Hu, K. Chitosan and chitosan coating nanoparticles for the treatment of brain disease. Int. J. Pharm. 2019, 560, 282–293. [Google Scholar] [CrossRef]
- Comoglu, T.; Arisoy, S.; Atalay, O.; Onal, D.; Pehlivanoglu, B. Development and in vivo application of levodopa loaded polylactic co- gylicolic acid (PLGA) nanoparticles for nazal brain drug delivery. Park. Relat. Disord. 2020, 79, e87. [Google Scholar] [CrossRef]
- Arisoy, S.; Sayiner, O.; Comoglu, T.; Önal, D.; Atalay, O.; Pehlivanoglu, B. In vitro and in vivo evaluation of levodopa-loaded nanoparticles for nose to brain delivery. Pharm. Dev. Technol. 2020, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Lohan, S.; Murthy, R.S.R. Formulation and characterization of intranasal mucoadhesive nanoparticulates and thermo-reversible gel of levodopa for brain delivery. Drug Dev. Ind. Pharm. 2013, 40, 869–878. [Google Scholar] [CrossRef]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Haloperidol-loaded PLGA nanoparticles: Systematic study of particle size and drug content. Int. J. Pharm. 2007, 336, 367–375. [Google Scholar] [CrossRef]
- Rathi Mahesh, C.; Girish Sailor, A.K.; Seth, S.P.C. Development and characterization of quetiapine loaded chitosan nanoparticle. Pharma Sci. Monit. 2013, 4, 150–169. [Google Scholar]
- Mohd, Z.A.; Khuriah, A.H. A Validated High-Performance Liquid Chromatographic Method for The Determination of Levodopa in Rat Plasma & Its Application in Pharmacokinetic Studies. In Proceedings of the 2012 IEEE Symposium on Business, Engineering and Industrial Applications, Bandung, Indonesia, 23–26 September 2012; pp. 136–141. [Google Scholar]
- Mohan, L.J.; McDonald, L.; Daly, J.S.; Ramtoola, Z. Optimising PLGA-PEG Nanoparticle Size and Distribution for Enhanced Drug Targeting to the Inflamed Intestinal Barrier. Pharmaceutics 2020, 12, 1114. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, C. Tuning the Size of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles Fabricated by Nanoprecipitation. Biotechnol. J. 2018, 13, 13. [Google Scholar] [CrossRef]
- Sawtarie, N.; Cai, Y.; Lapitsky, Y. Preparation of chitosan/tripolyphosphate nanoparticles with highly tunable size and low polydispersity. Colloids Surfaces B Biointerfaces 2017, 157, 110–117. [Google Scholar] [CrossRef]
- Rampino, A.; Borgogna, M.; Blasi, P.; Bellich, B.; Cesàro, A. Chitosan nanoparticles: Preparation, size evolution and stability. Int. J. Pharm. 2013, 455, 219–228. [Google Scholar] [CrossRef]
- Cai, Y.; Lapitsky, Y. Analysis of chitosan/tripolyphosphate micro- and nanogel yields is key to understanding their protein uptake performance. J. Colloid Interface Sci. 2017, 494, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Lazaridou, M.; Christodoulou, E.; Nerantzaki, M.; Kostoglou, M.; Lambropoulou, D.A.; Katsarou, A.; Pantopoulos, K.; Bikiaris, D.N. Formulation and In-Vitro Characterization of Chitosan-Nanoparticles Loaded with the Iron Chelator Deferoxamine Mesylate (DFO). Pharmaceutics 2020, 12, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piani, L.; Papo, A. Sodium Tripolyphosphate and Polyphosphate as Dispersing Agents for Alumina Suspensions: Rheological Characterization. J. Eng. 2013, 2013, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Al-Nemrawi, N.K.; Alsharif, S.S.M.; Dave, R.H. Preparation of chitosan-tpp nanoparticles: The influence of chitosan polymeric properties and formulation variables. Int. J. Appl. Pharm. 2018, 10, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Ng, W.K.; Shen, S.; Kim, S.; Tan, R.B. Scalable ionic gelation synthesis of chitosan nanoparticles for drug delivery in static mixers. Carbohydr. Polym. 2013, 94, 940–945. [Google Scholar] [CrossRef]
- Qun, G.; Ajun, W. Effects of molecular weight, degree of acetylation and ionic strength on surface tension of chitosan in dilute solution. Carbohydr. Polym. 2006, 64, 29–36. [Google Scholar] [CrossRef]
- Berger, J.; Reist, M.; Mayer, J.; Felt, O.; Peppas, N.; Gurny, R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Biopharm. 2004, 57, 19–34. [Google Scholar] [CrossRef]
- Fan, W.; Yan, W.; Xu, Z.; Ni, H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surfaces B Biointerfaces 2012, 90, 21–27. [Google Scholar] [CrossRef]
- Lowry, G.V.; Hill, R.J.; Harper, S.; Rawle, A.F.; Hendren, C.O.; Klaessig, F.; Nobbmann, U.; Sayre, P.; Rumble, J. Guidance to improve the scientific value of zeta-potential measurements in nanoEHS. Environ. Sci. Nano 2016, 3, 953–965. [Google Scholar] [CrossRef]
- Al Meslmani, B.M.; Mahmoud, G.F.; Bakowsky, U. Development of expanded polytetrafluoroethylene cardiovascular graft platform based on immobilization of poly lactic- co -glycolic acid nanoparticles using a wet chemical modification technique. Int. J. Pharm. 2017, 529, 238–244. [Google Scholar] [CrossRef]
- Nicolete, R.; dos Santos, D.F.; Faccioli, L.H. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int. Immunopharmacol. 2011, 11, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.I.; Samad, N.A.; Fang, L.; Lim, V. Cytotoxicity of targeted PLGA nanoparticles: A systematic review. RSC Adv. 2021, 11, 9433–9449. [Google Scholar] [CrossRef]
- Parveen, S.; Sahoo, S.K. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur. J. Pharmacol. 2011, 670, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.; Singhvi, G. Multifunctional Nanocrystals for Cancer Therapy: A Potential Nanocarrier; Elsevier BV: Amsterdam, The Netherlands, 2019; ISBN 9780128165058. [Google Scholar]
- Patel, N.; Zariwala, M.G.; Al-Obaidi, H. Fabrication of Biopolymer Based Nanoparticles for the Entrapment of Chromium and Iron Supplements. Processes 2020, 8, 707. [Google Scholar] [CrossRef]
- Karimi, M.; Avci, P.; Ahi, M.; Gazori, T.; Hamblin, M.R.; Naderi-Manesh, H. Evaluation of Chitosan-Tripolyphosphate Nanoparticles as a p-ShRNA Delivery Vector: Formulation, Optimization and Cellular Uptake Study. J. Nanopharm. Drug Deliv. 2014, 1, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliore, M.M.; Vyas, T.K.; Campbell, R.B.; Amiji, M.M.; Waszczak, B.L. Brain delivery of proteins by the intranasal route of administration: A comparison of cationic liposomes versus aqueous solution formulations. J. Pharm. Sci. 2010, 99, 1745–1761. [Google Scholar] [CrossRef] [PubMed]
- Ing, L.Y.; Zin, N.M.; Sarwar, A.; Katas, H. Antifungal Activity of Chitosan Nanoparticles and Correlation with Their Physical Properties. Int. J. Biomater. 2012, 2012, 632698. [Google Scholar] [CrossRef]
- Pawar, V.K.; Asthana, S.; Mishra, N.; Chaurasia, M.; Chourasia, M.K. Chitosan coated hydroxypropyl methylcellulose-ethylcellulose shell based gastroretentive dual working system to improve the bioavailability of norfloxacin. RSC Adv. 2013, 3, 19144–19153. [Google Scholar] [CrossRef]
- Zaki, S.S.O.; Ibrahim, M.N.; Katas, H. Particle Size Affects Concentration-Dependent Cytotoxicity of Chitosan Nanoparticles towards Mouse Hematopoietic Stem Cells. J. Nanotechnol. 2015, 2015, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.-Q.; Xiao, J.-X.; Jia, L.; Yang, J. Complex Coacervation of O-Carboxymethylated Chitosan and Gum Arabic. Int. J. Polym. Mater. Polym. Biomater. 2014, 64, 198–204. [Google Scholar] [CrossRef]
- Swain, S.K.; Dey, R.K.; Islam, M.; Patel, R.; Jha, U.; Patnaik, T.; Airoldi, C. Removal of Fluoride from Aqueous Solution Using Aluminum-Impregnated Chitosan Biopolymer. Sep. Sci. Technol. 2009, 44, 2096–2116. [Google Scholar] [CrossRef]
- Iqbal, M.; Zafar, N.; Fessi, H.; Elaissari, A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int. J. Pharm. 2015, 496, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Ping, Q.; Gao, Y. Effects of formulation factors on encapsulation efficiency and release behaviourin vitroof huperzine A-PLGA microspheres. J. Microencapsul. 2005, 22, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Mishra, A.; Mandal, T.K.; Sa, B.; Chakraborty, J. Optimization of the process parameters for the fabrication of a polymer coated layered double hydroxide-methotrexate nanohybrid for the possible treatment of osteosarcoma. RSC Adv. 2015, 5, 102574–102592. [Google Scholar] [CrossRef]
- Alshamsan, A. Nanoprecipitation Is More Efficient than Emulsion Solvent Evaporation Method to Encapsulate Cucurbitacin I in PLGA Nanoparticles, Elsevier Enhanced Reader. Saudi Pharm. J. 2014, 22, 219–222. [Google Scholar] [CrossRef]
- Al-Maaieh, A.; Flanagan, D.R. Salt and cosolvent effects on ionic drug loading into microspheres using an O/W method. J. Control. Release 2001, 70, 169–181. [Google Scholar] [CrossRef]
- Zhu, H.L.J.K.J. Preparation and characterization of hCG-loaded polylactide or poly(lactide-co-glycolide) microspheres using a modified water-in-oil-in-water (w/o/w) emulsion solvent evaporation technique. J. Microencapsul. 2001, 18, 247–260. [Google Scholar] [CrossRef]
- Pedroso-Santana, S.; Fleitas-Salazar, N. Ionotropic gelation method in the synthesis of nanoparticles/microparticles for biomedical purposes. Polym. Int. 2020, 69, 443–447. [Google Scholar] [CrossRef]
- Xu, Y.; Du, Y. Effect of Molecular Structure of Chitosan on Protein Deli v Ery Properties of Chitosan Nanoparticles. Int. J. Pharm. 2003, 250, 215–226. [Google Scholar] [CrossRef]
- Huang, Y.; Lapitsky, Y. On the kinetics of chitosan/tripolyphosphate micro- and nanogel aggregation and their effects on particle polydispersity. J. Colloid Interface Sci. 2017, 486, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Saini, R.; Srivastava, A.; Gupta, P.; Das, K. pH dependent reversible aggregation of Chitosan and glycol-Chitosan stabilized silver nanoparticles. Chem. Phys. Lett. 2011, 511, 326–330. [Google Scholar] [CrossRef]
- Biró, E.; Németh, Á.S.; Sisak, C.; Feczkó, T.; Gyenis, J. Preparation of chitosan particles suitable for enzyme immobilization. J. Biochem. Biophys. Methods 2008, 70, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Luinstra, M.; Grasmeijer, F.; Hagedoorn, P.; Moes, J.R.; Frijlink, H.W.; de Boer, A.H. A levodopa dry powder inhaler for the treatment of Parkinson’s disease patients in off periods. Eur. J. Pharm. Biopharm. 2015, 97, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledeti, I.; Bolintineanu, S.; Vlase, G.; Circioban, D.; Ledeti, A.; Vlase, T.; Suta, L.-M.; Caunii, A.; Murariu, M. Compatibility study between antiparkinsonian drug Levodopa and excipients by FTIR spectroscopy, X-ray diffraction and thermal analysis. J. Therm. Anal. 2017, 130, 433–441. [Google Scholar] [CrossRef]
- Qi, L.; Xu, Z.; Jiang, X.; Hu, C.; Zou, X. Preparation and antibacterial activity of chitosan nanoparticles. Carbohydr. Res. 2004, 339, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- Kestur, U.S.; Taylor, L.S. Role of polymer chemistry in influencing crystal growth rates from amorphous felodipine. CrystEngComm 2010, 12, 2390–2397. [Google Scholar] [CrossRef]
- Lim, S.-H.; Hudson, S.M. Synthesis and antimicrobial activity of a water-soluble chitosan derivative with a fiber-reactive group. Carbohydr. Res. 2004, 339, 313–319. [Google Scholar] [CrossRef]
- Fernandes Queiroz, M.; Melo, K.R.T.; Sabry, D.A.; Sassaki, G.L.; Rocha, H.A.O. Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation? Mar. Drugs 2015, 13, 141–158. [Google Scholar] [CrossRef]
- Agu, R.U. Challenges in nasal drug absorption: How far have we come? Ther. Deliv. 2016, 7, 495–510. [Google Scholar] [CrossRef]
- Cortés, H.; Alcalá-Alcalá, S.; Caballero-Florán, I.H.; Bernal-Chávez, S.A.; Ávalos-Fuentes, A.; González-Torres, M.; Carmen, M.G.-D.; Figueroa-González, G.; Reyes-Hernández, O.D.; Floran, B.; et al. A Reevaluation of Chitosan-Decorated Nanoparticles to Cross the Blood-Brain Barrier. Membranes 2020, 10, 212. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | L-Dopa (mg/mL) | PLGA (mg/mL) | PVA (% w/v) |
|---|---|---|---|
| P1 | 1 | 1.25 | 0.2 |
| P2 | 1 | 2.50 | 0.2 |
| P3 | 1 | 5.00 | 0.2 |
| Formulation | L-Dopa (% w/v) | Chitosan (% w/v) | Tween® 80 (% w/v) | Sodium TPP (% w/v) |
|---|---|---|---|---|
| C1 | 0.1 | 0.1 | 0.5 | 0.1 |
| C2 | 0.1 | 0.2 | 0.5 | 0.1 |
| C3 | 0.1 | 0.3 | 0.5 | 0.1 |
| C4 | 0.1 | 0.4 | 0.5 | 0.1 |
| Sample | Cmax (µg/mL) | Tmax (min) | Area under the Curve (AUC) | Absolute Bioavailability, F (%) |
|---|---|---|---|---|
| L-dopa | 50.018 ± 3.25 | 60 | 6494.582 ± 272.04 | 26.56 ± 1.11 |
| L-dopa loaded chitosan NP | 70.008 ± 5.77 * | 90 | 11,054.160 ± 1153.50 * | 45.20 ± 4.72 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, M.Z.; Sabri, A.H.B.; Anjani, Q.K.; Domínguez-Robles, J.; Abdul Latip, N.; Hamid, K.A. Design and Development of Levodopa Loaded Polymeric Nanoparticles for Intranasal Delivery. Pharmaceuticals 2022, 15, 370. https://doi.org/10.3390/ph15030370
Ahmad MZ, Sabri AHB, Anjani QK, Domínguez-Robles J, Abdul Latip N, Hamid KA. Design and Development of Levodopa Loaded Polymeric Nanoparticles for Intranasal Delivery. Pharmaceuticals. 2022; 15(3):370. https://doi.org/10.3390/ph15030370
Chicago/Turabian StyleAhmad, Mohd Zulhelmy, Akmal Hidyat Bin Sabri, Qonita Kurnia Anjani, Juan Domínguez-Robles, Normala Abdul Latip, and Khuriah Abdul Hamid. 2022. "Design and Development of Levodopa Loaded Polymeric Nanoparticles for Intranasal Delivery" Pharmaceuticals 15, no. 3: 370. https://doi.org/10.3390/ph15030370
APA StyleAhmad, M. Z., Sabri, A. H. B., Anjani, Q. K., Domínguez-Robles, J., Abdul Latip, N., & Hamid, K. A. (2022). Design and Development of Levodopa Loaded Polymeric Nanoparticles for Intranasal Delivery. Pharmaceuticals, 15(3), 370. https://doi.org/10.3390/ph15030370