
Cannabidiol on the Path from the Lab to the Cancer Patient: Opportunities and Challenges

 , , ,
, , , 
 and
and 
Abstract
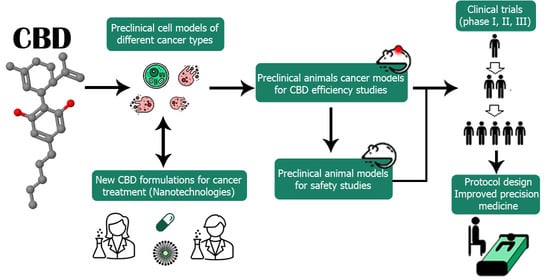
1. Introduction
2. CBD Shows Anticancer Properties in Pre-Clinical Studies In Vitro and In Vivo
3. Synergism: CBD Improves the Effect of Conventional Anticancer Therapy
4. CBD in Palliative Care
4.1. CBD in Chemotherapy-Induced Pain
4.2. CBD for Healthy Cells’ Protection
4.3. CBD against Opportunistic Infections
4.4. CBD in Anorexia-Cachexia Syndrome
5. Evidence of Anticancer Activity of CBD from Clinical Trials and Case Reports
6. CBD Tolerability, Toxicity, and Adverse Effects
7. Concerning Better CBD Delivery for Cancer Therapy
7.1. Free CBD Delivery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Participants | Delivery Method Doses | Plasma Concentration, ng/mL | Reference |
|---|---|---|---|
| Young healthy male volunteers (n = 5) | Smoking 20 mg | Max at 3 min: 110 ± 55 Max at 1 h: 10.2 ± 6.6 | [142] |
| i.v. 20 mg | Max at 3 min: 686 ± 239 Max at 1 h: 48.4 ± 10.7 | ||
| Male ICR mice (n = 3) | p.o. 20 mg/kg | Max at 2 h: 111 ± 52 Max at 4 h: 60 ± 58 | [145] |
| i.v. 10 mg/kg | Max at 10 min: 3343 ± 1048 Max at 1 h: 376 ± 229 | ||
| Healthy male/female volunteers (n = 8/8) | p.o. 25 mg | Max at 3 h: 3.05: range: 1.57–4.54 Max at 8 h: 1 | [139] |
| p.o., SEDDS 25 mg | Max at 1 h: 13.53, range: 7.9–19.1 4 h: 2.5 | ||
| Healthy male/female volunteers | inhalation, THC/CBD 20/20 mg | 5 min (max): 2–17 | [143] |
| i.v., THC/CBD 10/10 mg | Max at 5 min: 14–26 | ||
| Healthy male/female volunteers | p.o., single dose 1500 mg 3000 mg 6000 mg | Max at 5 h: 292.4 ± 87.9 533.0 ± 35.1 782.0 ± 83.0 | [144] |
| p.o., multiple dose 2 × 750 mg or 2 × 1500 mg daily | Max at 7 d: 330 541 |
7.2. Nanotechnology May Improve CBD Delivery for Cancer Therapy: General Considerations and Experimental Evidence
8. Regulation Issues
9. General Conclusions and Further Considerations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
| Time | Model | Effect | CBD Concentration | Cellular Target Involved | FBS (%) | References |
|---|---|---|---|---|---|---|
| 200 s | MDA-MB-231 (breast cancer, human) | ↑ [Ca2+]i | EC50 0.7 ± 0.1 µM | N.E. | 10 | [9] |
| Jurkat (acute lymphoblastic leukemia, human) | ↑ [Ca2+]i | 30 µM | CB1 (−) CB2 (−) GPR55 (−) | 5 | [25] | |
| ↑ [Ca2+]m | ||||||
| 10 min | SH-SY5Y (neuroblastoma, human) | ↑ p-p42/44 MAPK | 10 µM | N.E. | 10 | [44] |
| Jurkat (acute lymphoblastic leukemia, human) | ↓ ΔΨm | 10–100 µM | N.E. | 5 | [25] | |
| 20 min | Jurkat (acute lymphoblastic leukemia, human) | Cyt-c release | 30 µM | N.E. | ||
| 30 min | SH-SY5Y (neuroblastoma, human) | ↓ p-AKT | 10 µM | N.E. | 10 | [44] |
| EL-4 (thymoma, murine) | ↑ ROS | 16 µM | N.E. | 5 | [33] | |
| U87MG (glioblastoma, human) | ↓ migration | IC50 5.05 µM | CB1 (−) CB2 (−) TRPV1 (−) | 10 | [13] | |
| 1 h | MDA-MB-231 (breast cancer, human) | ↑ ROS | 10–25 µM | N.E. | 10 | [9] |
| Jurkat (acute lymphoblastic leukemia, human) | 30 µM | N.E. | 5 | [25] | ||
| EL-4 (thymoma, murine) | ↑ apoptosis (sub-G0/G1) | 16 µM | N.E. | [33] | ||
| 2 h | SH-SY5Y (neuroblastoma, human) | ↑ LC3-II | 10 µM | CB1 (+) CB2 (+) TRPV1 (+) | 10 | [44] |
| HeLa (cervical cancer, human) | ↑ p-p38 MAPK | 10 µM | CB1 (+) CB2 (+) TRPV1 (+) | Serum-free | [39] | |
| ↑ p-p42/44 MAPK | ||||||
| EL-4 (thymoma, murine) | ↑ ROS | 8–16 µM | N.E. | 5 | [33] | |
| Jurkat (acute lymphoblastic leukemia, human) | ↓ migration | 10–30 µM | N.E. | [25] | ||
| ↑ apoptosis | 30–100 µM | N.E. | ||||
| ↓ p-mTOR | 10 µM | N.E. | 10 | [32] | ||
| ↓ p-AKT | 10 µM | N.E. | [32] | |||
| 4 h | MDA-MB-231 (breast cancer, human) | ↓ p-AKT | 5 µM | N.E. | 10 | [22] |
| T98G (glioblastoma, human) | 20 µM | N.E. | [19] | |||
| U87MG, U118MG (glioblastoma, human) | 10 µM | N.E. | [18] | |||
| T98G (glioblastoma, human) | ↑ p-p38 MAPK | 20 µM | N.E. | [19] | ||
| U87MG, U118MG (glioblastoma, human) | 10 µM | N.E. | [18] | |||
| HeLa (cervical cancer, human) | N.E. | Serum-free | [39] | |||
| ↑ p-p42/44 MAPK | N.E. | |||||
| Jurkat (acute lymphoblastic leukemia, human) | ↑ apoptosis | 30–100 µM | N.E. | 5 | [25] | |
| ↑ LC3-II | 30 µM | N.E. | ||||
| 6 h | Jurkat (acute lymphoblastic leukemia, human) | ↑ apoptosis | 30–100 µM | N.E. | 5 | [25] |
| U87MG (glioblastoma, human) | ↑ ROS | 25 µM | N.E. | Serum-free | [14] | |
| Glioma stem-like cells (human) | ↑ ULK2 ↑ BECN1 ↑ ATGs | 10 µM | N.E. | Serum-free | [16] | |
| ↑ BAX ↑ BAD ↓ BCL2 | N.E. | |||||
| N.E. | ||||||
| MDA-MB-231 (breast cancer, human) | ↓ p-AKT | 5 µM | N.E. | 10 | [22] | |
| T98G (glioblastoma, human) | ↓ ΔΨm | 10 µM | N.E. | 10 | [19] | |
| 10 h | U87MG (glioblastoma, human) | Cyt-c release | 25 µM | N.E. | Serum-free | [14] |
| ↑ Caspase-8 activity | N.E. | |||||
| 12 h | HeLa (cervical cancer, human) | ↑ p-p38 MAPK | 10 µM | N.E. | Serum-free | [39] |
| ↑ p-p42/44 MAPK | N.E. | |||||
| Jurkat (acute lymphoblastic leukemia, human) | ↑ Caspase-9 | 30 µM | N.E. | 5 | [25] | |
| ↑ Cleaved/activated Caspase-3 | 15 µM | CB1 (+) CB2 (+) | 10 | [27] | ||
| LNCaP (prostate carcinoma, human) | ||||||
| SW480 (colon carcinoma, human) | CB1 (+) CB2 (+) | 5 | ||||
| EL-4 (thymoma, murine) | ↑ apoptosis (sub-G0/G1) | 12–16 µM | N.E. | 5 | [33] | |
| MDA-MB-231 (breast cancer, human) | ↑ ROS | 5 µM | N.E. | 10 | [22] | |
| ↓ ΔΨm | N.E. | |||||
| LNCaP (prostate carcinoma, human) | ↑ Cleaved PARP | 15 µM | CB1 (−) CB2 (+) | 10 | [27] | |
| SW480 (colon carcinoma, human) | CB1 (+) CB2 (+) | 5 | ||||
| 16 h | Jurkat (acute lymphoblastic leukemia, human) | ↓ p-p38 MAPK | 5 µM | CB1 (−) CB2 (+) | 10 | [31] |
| MDA-MB-231 (breast cancer, human) | ↑ apoptosis | 7.5 µM | N.E. | 10 | [22] | |
| ↑ autophagy | N.E. | |||||
| T24 (bladder urothelial carcinoma, human) | ↓ migration/invasion | 32 µM | N.E. | 10 | [49] | |
| U87MG (glioblastoma, human) | ↑ Caspase-9 ↑ Caspase-8 ↑ Caspase-3 activity | 25 µM | N.E. | Serum-free | [14] | |
| 20 h | MDA-MB-231 (breast cancer, human) | ↓ invasion | 0.1–1.5 µM | N.E. | 0.1 | [20] |
| U87MG (glioblastoma, human) | ↑ Caspase-8 ↑ Caspase-3 activity | 25 µM | N.E. | Serum-free | [14] | |
| 24 h | U87MG, U373MG (glioblastoma, human) | ↓ viability/proliferation | 25 µM | CB1 (−) CB2 (+) TRPV1 (−) | Serum-free | [12] |
| U87MG (glioblastoma, human) | 20–50 µM | N.E. | [14] | |||
| MDA-MB-231 (breast cancer, human) | 5–10 µM | CB1 (−) CB2 (−) TRPV1 (−) | [22] | |||
| EL-4 (thymoma, murine) | 5 µM | CB1 (−) CB2 (+) TRPV1 (−) | [31] | |||
| Jurkat (acute lymphoblastic leukemia, human) | 5 µM | CB1 (−) CB2 (+) TRPV1 (−) | ||||
| HT-29 (colorectal adenocarcinoma, human) | 10 µM | N.E. | 0.5 | [38] | ||
| SK-N-SH (neuroblastoma, human) | 32 µM | N.E. | 10 | [43] | ||
| Jurkat, MOLT-3, CCRF-CEM, RS4;11, Reh (acute lymphoblastic leukemia, human) | 12–50 µM | CB2 (−) | 5 | [25] | ||
| Glioma stem-like cells (human) | 10 µM | CB1 (−) CB2 (−) TRPV1 (−) TRPV2 (+) | Serum-free | [16] | ||
| DU-145, LNCaP (prostate cancer, human) | 5–6 µM | N.E. | [29] | |||
| SGC-7901 (gastric cancer, human) | 74 µM | N.E. | 10 | [34] | ||
| Caco-2, HCT116 (colon adenocarcinoma, human) | 10 µM | CB1 (+) CB2 (−) TRPV1 (+) | 10 | [36] | ||
| Gastric cancer cell lines (human) | 6–10 µM | N.E. | [35] | |||
| Colorectal cancer cell lines (human) | 6–8 µM | N.E. | Serum-free | [37] | ||
| U87MG, T98G (glioblastoma, human) | IC50 11–13 µM | N.E. | [17] | |||
| T-47D, MDA-MB-231 (breast cancer, human) | 2.2–5 µM | N.E. | [24] | |||
| FaDu, SCC15, Hep2 (head and neck squamous cell carcinoma, human) | 6–6.5 µM | N.E. | [50] | |||
| SiHa, HeLa, ME-180 (cervical cancer, human) | 5–10 µM | N.E. | [42] | |||
| SK-N-SH (neuroblastoma, human) | ↓ migration/invasion | 32 µM | N.E. | 10 | [43] | |
| U87MG, T98G (glioblastoma, human) | 1–9 µM | N.E. | Serum-free | [17] | ||
| Ishikawa, PCEM004b (endometrial cancer, human) | 12–25 µM | N.E. | Low | [48] | ||
| SCC15 (head and neck squamous cell carcinoma, human) | 4–8 µM | N.E. | 10 | [50] | ||
| HeLa (cervical cancer, human) | 10 µM | N.E. | Serum-free | [39] | ||
| SGC-7901 (gastric cancer, human) | (G0/G1) cell cycle arrest | 63–127 µM | N.E. | 10 | [34] | |
| ASPC1 (pancreatic cancer, human) | 40 µM | N.E. | 10 | [47] | ||
| Glioma stem-like cells (human) | 10 µM | N.E. | Serum-free | [16] | ||
| MDA-MB-231 (breast cancer, human) | ↓ Cyclin D1 | 7.5–10 µM | N.E. | 10 | [22] | |
| T-47D, MDA-MB-231 (breast cancer, human) | 5 µM | N.E. | 10 | [24] | ||
| SGC-7901 (gastric cancer, human) | ↓ Cyclin E | 31.79–127.2 µM | N.E. | 10 | [34] | |
| U87MG (glioblastoma, human) | ↑ necrosis (PI staining) | 25 µM | N.E. | Serum-free | [12] | |
| MDA-MB-231 (breast cancer, human) | ↑ apoptosis (Annexin V) | 5–10 µM | N.E. | 10 | [22] | |
| Jurkat (acute lymphoblastic leukemia, human) | ↑ apoptosis (TUNEL) | 5 µM | CB2 (+) | Serum-free | [31] | |
| SH-SY5Y (neuroblastoma, human) | ↑ apoptosis (sub-G0/G1) | 50–100 µM | N.E. | 10 | [44] | |
| SGC-7901 (gastric cancer, human) | ↑ apoptosis–necrosis (Annexin V-PI) | 32–127 µM | N.E. | 10 | [34] | |
| ASPC1 (pancreatic cancer, human) | 40 µM | N.E. | 10 | [47] | ||
| HL-60 (acute myeloblastic leukemia, human) | ↑ apoptosis | 25 µM | N.E. | 5 | [30] | |
| HCT116, DLD-1 (colorectal cancer, human) | ↑ apoptosis–necrosis (Annexin V-PI) | 6 µM | N.E. | 10 | [37] | |
| AGS, MKN45 (gastric cancer, human) | 4–10 µM | N.E. | [35] | |||
| T-47D, MDA-MB-231 (breast cancer, human) | ↑ apoptosis | 3–5 µM | N.E. | [24] | ||
| FaDu, SCC15, Hep2 (head and neck squamous cell carcinoma, human) | ↑ apoptosis–necrosis (Annexin V-PI) | 6–10 µM | N.E. | [50] | ||
| SiHa, HeLa, ME-180 (cervical cancer, human) | ↑ apoptosis (sub-G0/G1, Annexin V) | 10 µM | N.E. | 10 | [42] | |
| SK-N-SH (neuroblastoma, human) | apoptosis–necrosis (Annexin V-7AAD) | 32 µM | N.E. | 10 | [43] | |
| MDA-MB-231 (breast cancer, human) | ↑ Cleaved Caspase-7 | 7.5–10 µM | N.E. | 10 | [22] | |
| SCC15 (head and neck squamous cell carcinoma, human) | 10 µM | N.E. | 10 | [50] | ||
| Jurkat (acute lymphoblastic leukemia, human) | ↑ Cleaved/activated Caspase-8 | 5 µM | CB2 (+) | 10 | [31] | |
| MDA-MB-231 (breast cancer, human) | 7.5–10 µM | N.E. | 10 | [22] | ||
| U87MG (glioblastoma, human) | 25 µM | N.E. | Serum-free | [14] | ||
| HCT116, DLD-1 (colorectal cancer, human) | 6 µM | N.E. | 10 | [37] | ||
| AGS, MKN45 (gastric cancer, human) | 4–10 µM | N.E. | 10 | [35] | ||
| SGC-7901 (gastric cancer, human) | ↑ Cleaved/activated Caspase-3 | 32–127 µM | N.E. | 10 | [34] | |
| MDA-MB-231 (breast cancer, human) | 7.5–10 µM | N.E. | 10 | [22] | ||
| U87MG (glioblastoma, human) | 25 µM | N.E. | Serum-free | [14] | ||
| HCT116, DLD-1 (colorectal cancer, human) | 6 µM | N.E. | 10 | [37] | ||
| AGS, MKN45 (gastric cancer, human) | 4–10 µM | N.E. | [35] | |||
| SiHa, HeLa, ME-180 (cervical cancer, human) | 10.2 µM | N.E. | [42] | |||
| ASPC1 (pancreatic cancer, human) | 40 µM | N.E. | 10 | [47] | ||
| SGC-7901 (gastric cancer, human) | ↑ Cleaved Caspase-9, | 32–127 µM | N.E. | 10 | [34] | |
| MDA-MB-231 (breast cancer, human) | 7.5–10 µM | N.E. | 10 | [22] | ||
| HCT116, DLD-1 (colorectal cancer, human) | 6 µM | N.E. | [37] | |||
| AGS, MKN45 (gastric cancer, human) | 4–10 µM | N.E. | [35] | |||
| MDA-MB-231 (breast cancer, human) | ↑ t-Bid | 7.5–10 µM | N.E. | [22] | ||
| AGS, MKN45 (gastric cancer, human | 4–10 µM | N.E. | [35] | |||
| MDA-MB-231 (breast cancer, human) | ↑ Cleaved Beclin1 | 10 µM | N.E. | [22] | ||
| MDA-MB-231 (breast cancer, human) | ↑ Bax | N.E. | [22] | |||
| SCC15 (head and neck squamous cell carcinoma, human) | N.E. | [50] | ||||
| SGC-7901 (gastric cancer, human) | 32 µM | N.E. | 10 | [34] | ||
| SGC-7901 (gastric cancer, human) | ↑ Bad | N.E. | ||||
| SGC-7901 (gastric cancer, human) | ↓ Bcl-2 | N.E. | ||||
| MDA-MB-231 (breast cancer, human) | 10 µM | N.E. | 10 | [22] | ||
| SCC15 (head and neck squamous cell carcinoma, human) | N.E. | [50] | ||||
| MDA-MB-231 (breast cancer, human) | Cyt-C release | 2.5–5 µM | N.E. | [22] | ||
| Jurkat (acute lymphoblastic leukemia, human) | CB2 (+) | 10 | [31] | |||
| SGC-7901 (gastric cancer, human) | 63–127 µM | N.E. | [34] | |||
| MDA-MB-231 (breast cancer, human) | ↑ Cleaved PARP | 5–10 µM | N.E. | 10 | [22] | |
| Jurkat (acute lymphoblastic leukemia, human) | 2.5–5 µM | CB2 (+) | 10 | [31] | ||
| HCT116, DLD-1 (colorectal cancer, human) | 6 µM | N.E. | 5 | [37] | ||
| AGS, MKN45 (gastric cancer, human) | 4–10 µM | N.E. | [35] | |||
| T-47D, MDA-MB-231 (breast cancer, human) | 3 µM | N.E. | [24] | |||
| SCC15 (head and neck squamous cell carcinoma, human) | 10 µM | N.E. | [50] | |||
| LNCaP (prostate carcinoma, human) | 15 µM | N.E. | [27] | |||
| SW480 (colon carcinoma, human) | N.E. | |||||
| Jurkat, MOLT-4 (acute lymphoblastic leukemia, human) | ↑ ROS | 2.5–10 µM | N.E. | Serum-free | [31] | |
| SGC-7901 (gastric cancer, human) | 32 µM | N.E. | 10 | [34] | ||
| MCF-7 (breast cancer, human) | ↑ mitochondrial ROS | 20 µM | N.E. | 10 | [26] | |
| ↑ [Ca2+]m | N.E. | |||||
| Jurkat (acute lymphoblastic leukemia, human) | ↓ ΔΨm | 2.5–5 µM | N.E. | 10 | [31] | |
| SGC-7901 (gastric cancer, human) | 32 µM | N.E. | 10 | [34] | ||
| AGS (gastric cancer, human) | 4 µM | N.E. | 10 | [35] | ||
| MDA-MB-231 (breast cancer, human) | ↑ LC3-II | 5 µM | N.E. | 10 | [22] | |
| Jurkat (acute lymphoblastic leukemia, human) | 10 µM | N.E. | 5 | [25] | ||
| Glioma stem-like cells (human) | TRPV2 (+) | Serum-free | [16] | |||
| SCC15 (head and neck squamous cell carcinoma, human) | N.E. | 10 | [50] | |||
| Glioma stem-like cells (human) | ↑ Beclin1 | 10 µM | N.E. | Serum-free | [16] | |
| SCC15 (head and neck squamous cell carcinoma, human) | N.E. | 10 | [50] | |||
| MDA-MB-231 (breast cancer, human) | ↓ p-mTOR | 5 µM | N.E. | [22] | ||
| T-47D, MDA-MB-231 (breast cancer, human) | 5 µM | N.E. | [24] | |||
| MDA-MB-231 (breast cancer, human) | ↓ p-AKT | 5 µM | N.E. | [22] | ||
| U87MG, T98G (glioblastoma, human) | 5–9 µM | N.E. | Serum-free | [17] | ||
| U87MG, T98G (glioblastoma, human) | ↓ p-p42/44 MAPK | 5–9 µM | N.E. | |||
| AGS (gastric cancer, human) | ↑ p-p42/44 MAPK | 4 µM | N.E. | 10 % FBS | [35] | |
| 48 h | U87MG, U373MG (glioblastoma, human) | ↓ viability/proliferation | 25 µM (daily) | N.E. | Serum-free | [12] |
| FaDu, SCC15, Hep2 (head and neck squamous cell carcinoma, human) | 0.1–2 µM | N.E. | Serum-free | [50] | ||
| LNCaP (prostate carcinoma, human) | IC50 10 µM | N.E. | 2.5 | [27] | ||
| SW480 (colon carcinoma, human) | IC50 9.4 µM | N.E. | ||||
| U87MG (glioblastoma, human) | 10 µM | N.E. | 10 | [19] | ||
| Jurkat (acute lymphoblastic leukemia, human) | 30–100 µM (daily) | N.E. | 5 | [25] | ||
| A549, H460, H1792 (lung cancer, human) | N.E. | 5–10 | [41] | |||
| A549, H460 (lung cancer, human) | IC50 14.2–15.9 µM | N.E | Serum-free | [56] | ||
| CCRF-CEM (acute lymphoblastic leukemia, human) | 7.8 ± 0.2 μM | N.E. | 10 | [10] | ||
| A549, H460, H1792 (lung cancer, human) | ↓ migration/invasion | 30 µM | N.E. | Serum-free | [41] | |
| HeLa (cervical cancer, human) | 10 µM | N.E. | Serum-free | [39] | ||
| PCEM004b, PCEM004a (endometrial cancer, human) | (G0/G1) cell cycle arrest | 25–50 µM (daily) | N.E. | 10 | [48] | |
| MCF-7 (breast cancer, human) | (G1/S) cell cycle arrest | 10 µM (daily) | N.E. | 10 | [9] | |
| T24 (bladder urothelial carcinoma, human) | ↑ apoptosis-necrosis (Annexin V-PI) | 47.7 µM | N.E. | 10 | [49] | |
| U87MG (glioblastoma, human) | 20 µM | N.E. | 10 | [19] | ||
| ASPC1 (pancreatic cancer, human) | 40 µM | N.E. | 10 | [47] | ||
| U87MG, U118MG (glioblastoma, human) | ↑ apoptosis (sub-G0/G1) | 5–20 µM | CB1 (+) CB2 (+) | 10 | [18] | |
| SH-SY5Y (neuroblastoma, human) | 25–100 µM | N.E. | 10 | [44] | ||
| MDA-MB-231 (breast cancer, human) | ↑ Caspase-3 | 10 µM (daily) | N.E. | 10 | [9] | |
| U87MG (glioblastoma, human) | ↑ Cleaved PARP | 20 µM | N.E. | 10 | [19] | |
| D425 (medulloblastoma, human) | 5 µM | N.E. | 1.5 | [45] | ||
| ↑ LC3-II | N.E. | |||||
| ↑ p-p42/44 MAPK | N.E. | |||||
| MDA-MB-231 (breast cancer, human) | 1.5 µM (daily) | N.E. | 0.1 | [21] | ||
| D283 (medulloblastoma, human) | ↓ p-p42/44 MAPK | 6.5 µM | N.E. | 1.5 | [45] | |
| 72 h | FaDu, SCC15, Hep2 (head and neck squamous cell carcinoma, human) | ↓ viability/proliferation | 0.03–0.8 µM | N.E. | Serum-free | [50] |
| U87MG, U373MG (glioblastoma, human) | 25 µM (daily) | N.E. | Serum-free | [12] | ||
| LNCaP (prostate carcinoma, human) | IC50 5.95 µM | N.E. | 2.5 | [27] | ||
| SW480 (colon carcinoma, human) | IC50 5.06µM | N.E. | 2.5 | [27] | ||
| SiHa, HeLa, ME-180 (cervical cancer, human) | 10 µM | N.E. | Serum-free | [42] | ||
| Endometrial cancer cell lines (human) | 7–45 µM (daily) | N.E. | 10 | [48] | ||
| 19–75 µM | N.E. | [48] | ||||
| U87MG (glioblastoma, human) | 10 µM | N.E. | [19] | |||
| D283, D425 (medulloblastoma, human) | 3–7.5 µM | N.E. | 1.5 | [45] | ||
| IC-1425EPN, DKFZ-EP1NS (ependymoma, human) | 8–10 µM | N.E. | 1.5 | [45] | ||
| DU-145, LNCaP (prostate cancer, human) | 25 µM | N.E. | 10 | [29] | ||
| Jurkat (acute lymphoblastic leukemia, human) | 30–100 µM (daily) | N.E. | 5 | [25] | ||
| Jurkat (acute lymphoblastic leukemia, human) | 10–100 µM | N.E. | 5 | [25] | ||
| 2.5 µM | N.E. | 1 | [32] | |||
| 6.4 µM | N.E. | 5 | [32] | |||
| U251 (glioblastoma, human) | 0.6–1.2 µM | N.E. | 0.1 | [15] | ||
| MDA-MB-231 (breast cancer, human) | 1.5 µM (daily) | N.E. | 0.1 | [21] | ||
| U87MG, T98G, HG19 (glioblastoma, human) | ↓ migration/invasion | 2–3 µM | N.E. | Serum-free | [11] | |
| U251 (glioblastoma, human) | 0.1 µM | N.E. | 0.1 | [15] | ||
| HeLa, C33A (cervical cancer, human) | 10 µM | CB1 (+) CB2 (+) TRPV1 (+) | Serum-free | [39] | ||
| A549 (lung cancer, human) | CB1 (+) CB2 (+) TRPV1 (+) | |||||
| 0.1–1 µM | CB1 (+) CB2 (+) TRPV1 (+) | Serum-free | [40] | |||
| MDA-MB-231 (breast cancer, human) | 1.5 µM (daily) | N.E. | 0.1 | [21] | ||
| Endometrial cancer cell lines (human) | ↑ apoptosis (Annexin V-PI) | 7–45 µM (daily) | N.E. | 10 | [48] | |
| U87MG, U118MG (glioblastoma, human) | ↑ apoptosis (sub-G0/G1) | 5–20 µM | CB1 (+) CB2 (+) | 10 | [18] | |
| U251 (glioblastoma, human) | ↑ apoptosis (Annexin V-PI) | 2 µM | CB1 (−) CB2 (−) | 0.1 | [15] | |
| U251 (glioblastoma, human) | (G0/G1) cell cycle arrest | 0.4 µM | N.E. | 0.1 | [15] | |
| Jurkat (acute lymphoblastic leukemia, human) | 10 µM | N.E. | Serum-free | [32] | ||
| HPAFII, ASPC1 (pancreatic cancer, human) | GPR55 (+) | 10 | [46] | |||
| PCEM004b, PCEM004a (endometrial cancer, human) | ↑ LC3-II | 12–25 µM | N.E. | 10 | [48] | |
| 96 h | U87MG (glioblastoma, human) | ↓ viability/proliferation | 10 µM | N.E. | 10 | [19] |
| U87MG, U373MG (glioblastoma, human) | 25 µM (daily) | N.E. | Serum-free | [12] | ||
| MCF-7 (breast cancer, human) | IC50 8.2 µM (daily) | N.E. | 10 | [9] | ||
| MDA-MB-231 (breast cancer, human) | 10 µM (daily) | CB2 (+) TRPV1 (+) | ||||
| DU-145 (prostate cancer, human) | IC50 20.2 µM (daily) | N.E. | ||||
| Caco-2 (colon adenocarcinoma, human) | IC50 7.5 µM (daily) | N.E. | ||||
| AGS (gastric cancer, human) | IC50 7.5 µM (daily) | N.E. |
| Organ or System Involved | Acute Adverse Effects Reported | Species/Route/Range of CBD Dose | Chronic Adverse Effects Reported | Species/Route/Range of CBD Dose | References |
|---|---|---|---|---|---|
| Systemic | Organ weight elevation |
| Organ weight elevation | Rhesus monkeys/oral/ 30–300 mg/kg/day | [112,113,114,115] |
| Changes in THC metabolism | Rats/subcutaneous/10 mg/kg + 10 mg/kg THC | Decreased growth | Pregnant rats/oral/75–250 mg/kg/day | ||
| Cardiovascular | Bradycardia | Rhesus monkeys/intravenous/150–300 mg/kg/day | [113,116] | ||
| Hypopnea | |||||
| Cardiac failure (higher doses) | |||||
| Hypotension | Piglets/intravenous/10–50 mg/kg | ||||
| Cardiac arrest | |||||
| Nervous system | Tremors | Rhesus monkeys/intravenous/ 150–300 mg/kg/day | Anxiogenic-like effect | Rats/intraperitoneal/10 mg/kg | [113,114,117,118] |
| Central nervous system inhibition | Decreased brain-derived neurotrophic factor (BDNF) expression and related signaling proteins in the hippocampus and frontal cortex | ||||
| Convulsions | Decreased cell proliferation and neurogenesis in the hippocampus and in subgranular zone | Mice/intraperitoneal/30 mg/kg | |||
| Hypolocomotion | Rats/subcutaneous/10 mg/kg + 10mg/kg THC | Neurobehavioral changes | Pregnant rats/oral/75–250 mg/kg/day | ||
| Reproductive system, fertility, and alterations in development and growth of the descendants | Seminiferous tubule degeneration | Rat/inhaled/0.6–1.2 mg/kg | Decreased circulating testosterone | Mice/oral/15–30 mg/kg | [112,113,119,120,121,122] |
| Decrease in testicular size | Rhesus monkeys/oral/ 30–300 mg/kg/day | ||||
| Spermatogenesis inhibition | |||||
| Decrease in number of spermatozoa in the epididymis tail | Mice/oral/15–30 mg/kg | ||||
| Changes in normal cell stage in sperm formation | |||||
| Interference in sperm maturation | Rat/inhaled/0.6–1.2 mg/kg | Head abnormalities in sperm | |||
| Cytoplasmic droplets in the flagella medial region | |||||
| Testicular weight decrease | Rhesus monkeys/intravenous/150–300 mg/kg/day | Increased embryofetal mortality | Pregnant rats/oral/75–250 mg/kg/day | ||
| Inhibition of spermatogenesis | Rhesus monkeys/intravenous/ 150–300 mg/kg/day | Developmental toxicity | |||
| Dose-dependent decreased fertility of eggs and sperms | Sea urchin eggs and sperm/incubation in CBD-enriched sea water/0.1–10 µM | Decreased fetal body weight | |||
| Fertilization inhibition | Increased fetal structural variations | ||||
| Decrease in testosterone metabolism | Rats/intraperitoneal/10 mg/kg | Delayed sexual maturation | |||
| Dose- and time-dependent acrosome reaction inhibition without reduced motility | Sea urchin sperm/incubation in CBD-enriched sea water/0.1–100 µM | Alterations in male reproductive organ development and fertility in offspring | |||
| Hepatic | Decrease in CYP aniline hydroxylation and p-nitroanisole demethylation | Rats/intraperitoneal/10 mg/kg | [121] | ||
| Alteration of CYP content |
References
- Zuardi, A.W.; Crippa, J.A.; Hallak, J.E.; Bhattacharyya, S.; Atakan, Z.; Martin-Santos, R.; McGuire, P.K.; Guimarães, F.S. A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Curr. Pharm. Des. 2012, 18, 5131–5140. [Google Scholar] [CrossRef] [PubMed]
- Massi, P.; Solinas, M.; Cinquina, V.; Parolaro, D. Cannabidiol as potential anticancer drug. Br. J. Clin. Pharmacol. 2012, 75, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Yeshurun, M.; Shpilberg, O.; Herscovici, C.; Shargian, L.; Dreyer, J.; Peck, A.; Israeli, M.; Levy-Assaraf, M.; Gruenewald, T.; Mechoulam, R.; et al. Cannabidiol for the prevention of graft-versus-host-disease after allogeneic hematopoietic cell transplantation: Results of a phase II Study. Biol. Blood Marrow Transplant. 2015, 21, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.; Pruskowski, J. The role of cannabidiol in palliative care #370. J. Palliat. Med. 2019, 22, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Sunda, F.; Arowolo, A. A molecular basis for the anti-inflammatory and anti-fibrosis properties of cannabidiol. FASEB J. 2020, 34, 14083–14092. [Google Scholar] [CrossRef]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, D., Jr.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a promising anti-cancer drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Mangal, N.; Erridge, S.; Habib, N.; Sadanandam, A.; Reebye, V.; Sodergren, M.H. Cannabinoids in the landscape of cancer. J. Cancer Res. Clin. Oncol. 2021, 147, 2507–2534. [Google Scholar] [CrossRef]
- Mlost, J.; Bryk, M.; Starowicz, K. Cannabidiol for pain treatment: Focus on pharmacology and mechanism of action. Int. J. Mol. Sci. 2020, 21, 8870. [Google Scholar] [CrossRef]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef]
- Scott, K.A.; Dalgleish, A.G.; Liu, W.M. Anticancer effects of phytocannabinoids used with chemotherapy in leukaemia cells can be improved by altering the sequence of their administration. Int. J. Oncol. 2017, 51, 369–377. [Google Scholar] [CrossRef]
- Torres, S.; Lorente, M.; Rodríguez-Fornés, F.; Hernández-Tiedra, S.; Salazar, M.; García-Taboada, E.; Barcia, J.; Guzmán, M.; Velasco, G. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Ceruti, S.; Colombo, A.; Abbracchio, M.P.; Parolaro, D. Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J. Pharmacol. Exp. Ther. 2004, 308, 838–845. [Google Scholar] [CrossRef]
- Vaccani, A.; Massi, P.; Colombo, A.; Rubino, T.; Parolaro, D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br. J. Pharmacol. 2005, 144, 1032–1036. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Bianchessi, S.; Costa, B.; Macchi, P.; Parolaro, D. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell. Mol. Life Sci. 2006, 63, 2057–2066. [Google Scholar] [CrossRef]
- Marcu, J.P.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Horowitz, M.P.; Lee, J.; Pakdel, A.; Allison, J.; Limbad, C.; Moore, D.H.; et al. Cannabidiol enhances the inhibitory effects of delta9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol. Cancer Ther. 2010, 9, 180–189. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef]
- Solinas, M.; Massi, P.; Cinquina, V.; Valenti, M.; Bolognini, D.; Gariboldi, M.; Monti, E.; Rubino, T.; Parolaro, D. Cannabidiol, a non-psychoactive cannabinoid compound, inhibits proliferation and invasion in U87-MG and T98G glioma cells through a multitarget effect. PLoS ONE 2013, 8, e76918. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Wu, J.; Hei, T.K. Regulation of human glioblastoma cell death by combined treatment of cannabidiol, γ-radiation and small molecule inhibitors of cell signaling pathways. Oncotarget 2017, 8, 74068–74095. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Wu, J.; Wang, T.J.C.; Hei, T.K. Correction: Inhibition of ATM kinase upregulates levels of cell death induced by cannabidiol and γ-irradiation in human glioblastoma cells. Oncotarget 2019, 10, 7012–7013. [Google Scholar] [CrossRef]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer Ther. 2007, 6, 2921–2927. [Google Scholar] [CrossRef]
- McAllister, S.D.; Murase, R.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Allison, J.; Almanza, C.; Pakdel, A.; Lee, J.; Limbad, C.; et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res. Treat. 2011, 129, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E., III; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.S.; Marie, M.A.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Mould, R.R.; Botchway, S.W.; Parkinson, J.R.C.; Thomas, E.L.; Guy, G.W.; Bell, J.D.; Nunn, A.V.W. Cannabidiol modulates mitochondrial redox and dynamics in MCF7 cancer cells: A study using fluorescence lifetime imaging microscopy of NAD(P)H. Front. Mol. Biosci. 2021, 8, 630107. [Google Scholar] [CrossRef]
- Sreevalsan, S.; Joseph, S.; Jutooru, I.; Chadalapaka, G.; Safe, S.H. Induction of apoptosis by cannabinoids in prostate and colon cancer cells is phosphatase dependent. Anticancer Res. 2011, 31, 3799–3807. [Google Scholar]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2011, 30, 142–152. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Schiano Moriello, A.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: Pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2013, 168, 79–102. [Google Scholar] [CrossRef]
- Gallily, R.; Even-Chen, T.; Katzavian, G.; Lehmann, D.; Dagan, A.; Mechoulam, R. γ-irradiation enhances apoptosis induced by cannabidiol, a non-psychotropic cannabinoid, in cultured HL-60 myeloblastic leukemia cells. Leuk. Lymphoma 2003, 44, 1767–1773. [Google Scholar] [CrossRef]
- McKallip, R.J.; Jia, W.; Schlomer, J.; Warren, J.W.; Nagarkatti, P.S.; Nagarkatti, M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol. Pharmacol. 2006, 70, 897–908. [Google Scholar] [CrossRef]
- Kalenderoglou, N.; Macpherson, T.; Wright, K.L. Cannabidiol reduces leukemic cell size—But is it important? Front. Pharmacol. 2017, 8, 144. [Google Scholar] [CrossRef]
- Lee, C.Y.; Wey, S.P.; Liao, M.H.; Hsu, W.L.; Wu, H.Y.; Jan, T.R. A comparative study on cannabidiol-induced apoptosis in murine thymocytes and EL-4 thymoma cells. Int. Immunopharmacol. 2008, 8, 732–740. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, Y.; Pan, Z.; Li, M.; Liu, X.; Chen, X.; Qu, G.; Zhou, L.; Xu, M.; Zheng, Q.; et al. Cannabidiol induces cell cycle arrest and cell apoptosis in human gastric cancer SGC-7901 cells. Biomolecules 2019, 9, 302. [Google Scholar] [CrossRef]
- Jeong, S.; Jo, M.J.; Yun, H.K.; Kim, D.Y.; Kim, B.R.; Kim, J.L.; Park, S.H.; Na, Y.J.; Jeong, Y.A.; Kim, B.G.; et al. Cannabidiol promotes apoptosis via regulation of XIAP/Smac in gastric cancer. Cell Death Dis. 2019, 10, 846. [Google Scholar] [CrossRef]
- Aviello, G.; Romano, B.; Borrelli, F.; Capasso, R.; Gallo, L.; Piscitelli, F.; Di Marzo, V.; Izzo, A.A. Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. J. Mol. Med. 2012, 90, 925–934. [Google Scholar] [CrossRef]
- Jeong, S.; Yun, H.K.; Jeong, Y.A.; Jo, M.J.; Kang, S.H.; Kim, J.L.; Kim, D.Y.; Park, S.H.; Kim, B.R.; Na, Y.J.; et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer Lett. 2019, 447, 12–23. [Google Scholar] [CrossRef]
- Sainz-Cort, A.; Müller-Sánchez, C.; Espel, E. Anti-proliferative and cytotoxic effect of cannabidiol on human cancer cell lines in presence of serum. BMC Res. Notes 2020, 13, 389. [Google Scholar] [CrossRef]
- Ramer, R.; Merkord, J.; Rohde, H.; Hinz, B. Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases-1. Biochem. Pharmacol. 2010, 79, 955–966. [Google Scholar] [CrossRef]
- Ramer, R.; Rohde, A.; Merkord, J.; Rohde, H.; Hinz, B. Decrease of plasminogen activator inhibitor-1 may contribute to the anti-invasive action of cannabidiol on human lung cancer cells. Pharm. Res. 2010, 27, 2162–2174. [Google Scholar] [CrossRef]
- Milian, L.; Mata, M.; Alcacer, J.; Oliver, M.; Sancho-Tello, M.; de Llano, J.J.M.; Camps, C.; Galbis, J.; Carretero, J.; Carda, C. Cannabinoid receptor expression in non-small cell lung cancer. Effectiveness of tetrahydrocannabinol and cannabidiol inhibiting cell proliferation and epithelial-mesenchymal transition in vitro. PLoS ONE 2020, 15, e0228909. [Google Scholar] [CrossRef] [PubMed]
- Lukhele, S.T.; Motadi, L.R. Cannabidiol rather than Cannabis sativa extracts inhibit cell growth and induce apoptosis in cervical cancer cells. BMC Complement. Altern. Med. 2016, 16, 335. [Google Scholar] [CrossRef] [PubMed]
- Fisher, T.; Golan, H.; Schiby, G.; PriChen, S.; Smoum, R.; Moshe, I.; Peshes-Yaloz, N.; Castiel, A.; Waldman, D.; Gallily, R.; et al. In vitro and in vivo efficacy of non-psychoactive cannabidiol in neuroblastoma. Curr. Oncol. 2016, 23, S15–S22. [Google Scholar] [CrossRef] [PubMed]
- Vrechi, T.A.M.; Leão, A.H.F.F.; Morais, I.B.M.; Abílio, V.C.; Zuardi, A.W.; Hallak, J.E.C.; Crippa, J.A.; Bincoletto, C.; Ureshino, R.P.; Smaili, S.S.; et al. Cannabidiol induces autophagy via ERK1/2 activation in neural cells. Sci. Rep. 2021, 11, 5434. [Google Scholar] [CrossRef]
- Andradas, C.; Byrne, J.; Kuchibhotla, M.; Ancliffe, M.; Jones, A.C.; Carline, B.; Hii, H.; Truong, A.; Storer, L.C.D.; Ritzmann, T.A.; et al. Assessment of cannabidiol and Δ9-tetrahydrocannabiol in mouse models of medulloblastoma and ependymoma. Cancers 2021, 13, 330. [Google Scholar] [CrossRef]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef]
- Emhemmed, F.; Zhao, M.; Yorulmaz, S.; Steyer, D.; Leitao, C.; Alignan, M.; Cerny, M.; Paillard, A.; Delacourt, F.M.; Julien-David, D.; et al. Cannabis sativa extract induces apoptosis in human pancreatic 3D cancer models: Importance of major antioxidant molecules present therein. Molecules 2022, 27, 1214. [Google Scholar] [CrossRef]
- Marinelli, O.; Morelli, M.B.; Annibali, D.; Aguzzi, C.; Zeppa, L.; Tuyaerts, S.; Amantini, C.; Amant, F.; Ferretti, B.; Maggi, F.; et al. The effects of cannabidiol and prognostic role of TRPV2 in human endometrial cancer. Int. J. Mol. Sci. 2020, 21, 5409. [Google Scholar] [CrossRef]
- Anis, O.; Vinayaka, A.C.; Shalev, N.; Namdar, D.; Nadarajan, S.; Anil, S.M.; Cohen, O.; Belausov, E.; Ramon, J.; Mayzlish Gati, E.; et al. Cannabis-derived compounds cannabichromene and Δ9-tetrahydrocannabinol interact and exhibit cytotoxic activity against urothelial cell carcinoma correlated with inhibition of cell migration and cytoskeleton organization. Molecules 2021, 26, 465. [Google Scholar] [CrossRef]
- Go, Y.Y.; Kim, S.R.; Kim, D.Y.; Chae, S.W.; Song, J.J. Cannabidiol enhances cytotoxicity of anti-cancer drugs in human head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 20622. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Bergamaschi, M.M.; Queiroz, R.H.C.; Zuardi, A.W.; Crippa, J.A.S. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr. Drug Saf. 2011, 6, 237–249. [Google Scholar] [CrossRef]
- Rimmerman, N.; Ben-Hail, D.; Porat, Z.; Juknat, A.; Kozela, E.; Daniels, M.P.; Connelly, P.S.; Leishman, E.; Bradshaw, H.B.; Shoshan-Barmatz, V.; et al. Direct modulation of the outer mitochondrial membrane channel, voltage-dependent anion channel 1 (VDAC1) by cannabidiol: A novel mechanism for cannabinoid-induced cell death. Cell Death Dis. 2013, 4, e949. [Google Scholar] [CrossRef]
- Elbaz, M.; Ahirwar, D.; Zhang, X.; Zhou, X.; Lustberg, M.; Nasser, M.W.; Shilo, K.; Ganju, R.K. TRPV2 is a novel biomarker and therapeutic target in triple negative breast cancer. Oncotarget 2018, 9, 33459–33470. [Google Scholar] [CrossRef]
- Misri, S.; Kaul, K.; Mishra, S.; Charan, M.; Verma, A.K.; Barr, M.P.; Ahirwar, D.K.; Ganju, R.K. Cannabidiol inhibits tumorigenesis in cisplatin-resistant non-small cell lung cancer via TRPV2. Cancers 2022, 14, 1181. [Google Scholar] [CrossRef]
- Jan, T.R.; Su, S.T.; Wu, H.Y.; Liao, M.H. Suppressive effects of cannabidiol on antigen-specific antibody production and functional activity of splenocytes in ovalbumin-sensitized BALB/c mice. Int. Immunopharmacol. 2007, 7, 773–780. [Google Scholar] [CrossRef]
- Liu, D.Z.; Hu, C.M.; Huang, C.H.; Wey, S.P.; Jan, T.R. Cannabidiol attenuates delayed-type hypersensitivity reactions via suppressing T-cell and macrophage reactivity. Acta Pharmacol. Sin. 2010, 31, 1611–1617. [Google Scholar] [CrossRef]
- Kaplan, B.L.F.; Springs, A.E.B.; Kaminski, N.E. The profile of immune modulation by cannabidiol (CBD) involves deregulation of nuclear factor of activated T cells (NFAT). Biochem. Pharmacol. 2008, 76, 726–737. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Fride, E.; Sheskin, T.; Tamiri, T.; Rhee, M.H.; Vogel, Z.; Bisogno, T.; De Petrocellis, L.; Di Marzo, V.; Mechoulam, R. An entourage effect: Inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur. J. Pharmacol. 1998, 353, 23–31. [Google Scholar] [CrossRef]
- Ferber, S.G.; Namdar, D.; Hen-Shoval, D.; Eger, G.; Koltai, H.; Shoval, G.; Shbiro, L.; Weller, A. The “entourage effect”: Terpenes coupled with cannabinoids for the treatment of mood disorders and anxiety disorders. Curr. Neuropharmacol. 2020, 18, 87–96. [Google Scholar] [CrossRef]
- Tomko, A.M.; Whynot, E.G.; Dupré, D.J. Anti-cancer properties of cannabidiol and Δ9-tetrahydrocannabinol and potential synergistic effects with gemcitabine, cisplatin and other cannabinoids in bladder cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Fernández-Carballido, A.; Simancas-Herbada, R.; Martin-Sabroso, C.; Torres-Suárez, A.I. CBD loaded microparticles as a potential formulation to improve paclitaxel and doxorubicin-based chemotherapy in breast cancer. Int. J. Pharm. 2020, 574, 118916. [Google Scholar] [CrossRef]
- Ward, S.J.; McAllister, S.D.; Kawamura, R.; Murase, R.; Neelakantan, H.; Walker, E.A. Cannabidiol inhibits paclitaxel-induced neuropathic pain through 5-HT(1A) receptors without diminishing nervous system function or chemotherapy efficacy. Br. J. Pharmacol. 2014, 171, 636–645. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ng, L.; Ozawa, T.; Stella, N. Quantitative analyses of synergistic responses between cannabidiol and DNA-damaging agents on the proliferation and viability of glioblastoma and neural progenitor cells in culture. J. Pharmacol. Exp. Ther. 2017, 360, 215–224. [Google Scholar] [CrossRef]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer 2014, 134, 2534–2546. [Google Scholar] [CrossRef]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Offidani, M.; Amantini, C.; Gentili, S.; Soriani, A.; Cardinali, C.; Leoni, P.; Santoni, G. Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 2016, 7, 77543–77557. [Google Scholar] [CrossRef]
- Scott, K.A.; Dalgleish, A.G.; Liu, W.M. The combination of cannabidiol and Δ9-tetrahydrocannabinol enhances the anticancer effects of radiation in an orthotopic murine glioma model. Mol. Cancer Ther. 2014, 13, 2955–2967. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships; the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Gómez-Sandoval, Z.; Villatoro-Gómez, K.; Pottosin, I.; Dobrovinskaya, O. Tamoxifen sensitizes acute lymphoblastic leukemia cells to cannabidiol by targeting cyclophilin-D and altering mitochondrial Ca2+ homeostasis. Int. J. Mol. Sci. 2021, 22, 8688. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.H.; Back, A.; Benedetti, C.; Billings, J.A.; Block, S.; Boston, B.; Bruera, E.; Dy, S.; Eberle, C.; Foley, K.M.; et al. NCCN clinical practice guidelines in oncology: Palliative care. J. Natl. Comp. Canc. Netw. 2009, 7, 436–473. [Google Scholar] [CrossRef] [PubMed]
- Bar-Lev Schleider, L.; Mechoulam, R.; Lederman, V.; Hilou, M.; Lencovsky, O.; Betzalel, O.; Shbiro, L.; Novack, V. Prospective analysis of safety and efficacy of medical cannabis in large unselected population of patients with cancer. Eur. J. Intern. Med. 2018, 49, 37–43. [Google Scholar] [CrossRef]
- Highet, B.H.; Lesser, E.R.; Johnson, P.W.; Kaur, J.S. Tetrahydrocannabinol and cannabidiol use in an outpatient palliative medicine population. Am. J. Hosp. Palliat. Care 2020, 37, 589–593. [Google Scholar] [CrossRef]
- Zenone, M.A.; Snyder, J.; Crooks, V.A. What are the informational pathways that shape people’s use of cannabidiol for medical purposes? J. Cannabis Res. 2021, 3, 13. [Google Scholar] [CrossRef]
- Bonn-Miller, M.O.; Loflin, M.J.E.; Thomas, B.F.; Marcu, J.P.; Hyke, T.; Vandrey, R. Labeling accuracy of cannabidiol extracts sold online. JAMA 2017, 318, 1708–1709. [Google Scholar] [CrossRef]
- Good, P.; Haywood, A.; Gogna, G.; Martin, J.; Yates, P.; Greer, R.; Hardy, J. Oral medicinal cannabinoids to relieve symptom burden in the palliative care of patients with advanced cancer: A double-blind, placebo controlled, randomised clinical trial of efficacy and safety of cannabidiol (CBD). BMC Palliat. Care 2019, 18, 110. [Google Scholar] [CrossRef]
- Scripture, C.D.; Figg, W.D.; Sparreboom, A. Peripheral neuropathy induced by paclitaxel: Recent insights and future perspectives. Curr. Neuropharmacol. 2006, 4, 165–172. [Google Scholar] [CrossRef]
- Hu, L.Y.; Mi, W.L.; Wu, G.C.; Wang, Y.Q.; Mao-Ying, Q.L. Prevention and treatment for chemotherapy-induced peripheral neuropathy: Therapies based on CIPN mechanisms. Curr. Neuropharmacol. 2019, 17, 184–196. [Google Scholar] [CrossRef]
- Harris, H.M.; Gul, W.; ElSohly, M.A.; Sufka, K.J. Effects of cannabidiol and a novel cannabidiol analog against tactile allodynia in a murine model of cisplatin-induced neuropathy: Enhanced effects of sub-analgesic doses of m orphine. Med. Cannabis Cannabinoids 2018, 1, 54–59. [Google Scholar] [CrossRef]
- Portenoy, R.K.; Ganae-Motan, E.D.; Allende, S.; Yanagihara, R.; Shaiova, L.; Weinstein, S.; McQuade, R.; Wright, S.; Fallon, M.T. Nabiximols for opioid-treated cancer patients with poorly-controlled chronic pain: A randomized, placebo-controlled, graded-dose trial. J. Pain 2012, 13, 438–449. [Google Scholar] [CrossRef]
- Johnson, J.R.; Burnell-Nugent, M.; Lossignol, D.; Ganae-Motan, E.D.; Potts, R.; Fallon, M.T. Multicenter, double-blind, randomized, placebo-controlled, parallel-group study of the efficacy, safety, and tolerability of THC: CBD extract and THC extract in patients with intractable cancer-related pain. J. Pain Symptom Manag. 2010, 39, 167–179. [Google Scholar] [CrossRef]
- Johnson, J.R.; Lossignol, D.; Burnell-Nugent, M.; Fallon, M.T. An open-label extension study to investigate the long-term safety and tolerability of THC/CBD oromucosal spray and oromucosal THC spray in patients with terminal cancer-related pain refractory to strong opioid analgesics. J. Pain Symptom Manag. 2013, 46, 207–218. [Google Scholar] [CrossRef]
- Fallon, M.T.; Albert Lux, E.; McQuade, R.; Rossetti, S.; Sanchez, R.; Sun, W.; Wright, S.; Lichtman, A.H.; Kornyeyeva, E. Sativex oromucosal spray as adjunctive therapy in advanced cancer patients with chronic pain unalleviated by optimized opioid therapy: Two double-blind, randomized, placebo-controlled phase 3 studies. Br. J. Pain 2017, 11, 119–133. [Google Scholar] [CrossRef]
- Lichtman, A.H.; Lux, E.A.; McQuade, R.; Rossetti, S.; Sanchez, R.; Sun, W.; Wright, S.; Kornyeyeva, E.; Fallon, M.T. Results of a double-blind, randomized, placebo-controlled study of nabiximols oromucosal spray as an adjunctive therapy in advanced cancer patients with chronic uncontrolled pain. J. Pain Symptom Manag. 2018, 55, 179–188. [Google Scholar] [CrossRef]
- King, K.M.; Myers, A.M.; Soroka-Monzo, A.J.; Tuma, R.F.; Tallarida, R.J.; Walker, E.A.; Ward, S.J. Single and combined effects of Δ9-tetrahydrocannabinol and cannabidiol in a mouse model of chemotherapy-induced neuropathic pain. Br. J. Pharmacol. 2017, 174, 2832–2841. [Google Scholar] [CrossRef]
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.J.; Lu, L.; Klein, C.L.; Dinarello, C.A.; et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 2007, 322, 8–15. [Google Scholar] [CrossRef]
- Pan, H.; Mukhopadhyay, P.; Rajesh, M.; Patel, V.; Mukhopadhyay, B.; Gao, B.; Haskó, G.; Pacher, P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J. Pharmacol. Exp. Ther. 2009, 328, 708–714. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [PubMed]
- Hao, E.; Mukhopadhyay, P.; Cao, Z.; Erdélyi, K.; Holovac, E.; Liaudet, L.; Lee, W.S.; Haskó, G.; Mechoulam, R.; Pacher, P. Cannabidiol protects against doxorubicin-induced cardiomyopathy by modulating mitochondrial function and biogenesis. Mol. Med. 2015, 21, 38–45. [Google Scholar] [CrossRef]
- Friedman, H.S.; Mahaley, M.S., Jr.; Schold, S.C., Jr.; Vick, N.A.; Falletta, J.M.; Bullard, D.E.; D’Souza, B.J.; Khandekar, J.D.; Lew, S.; Oakes, W.J.; et al. Efficacy of vincristine and cyclophosphamide in the therapy of recurrent medulloblastoma. Neurosurgery 1986, 18, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Bodey, G.P. Infections in cancer patients. Cancer Treat. Rev. 1975, 2, 89–128. [Google Scholar] [CrossRef]
- Freifeld, A.G.; Bow, E.J.; Sepkowitz, K.A.; Boeckh, M.J.; Ito, J.I.; Mullen, C.A.; Raad, I.I.; Rolston, K.V.; Young, J.A.; Wingard, J.R.; et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2011, 52, e56–e93. [Google Scholar] [CrossRef]
- Alshaarawy, O. Total and differential white blood cell count in cannabis users: Results from the cross-sectional National Health and Nutrition Examination Survey, 2005–2016. J. Cannabis Res. 2019, 1, 6. [Google Scholar] [CrossRef]
- Iseppi, R.; Brighenti, V.; Licata, M.; Lambertini, A.; Sabia, C.; Messi, P.; Pellati, F.; Benvenuti, S. Chemical characterization and evaluation of the antibacterial activity of essential oils from fibre-type Cannabis sativa L. (Hemp). Molecules 2019, 24, 2302. [Google Scholar] [CrossRef]
- Karas, J.A.; Wong, L.J.M.; Paulin, O.K.A.; Mazeh, A.C.; Hussein, M.H.; Li, J.; Velkov, T. The antimicrobial activity of cannabinoids. Antibiotics 2020, 9, 406. [Google Scholar] [CrossRef]
- Martinenghi, L.D.; Jønsson, R.; Lund, T.; Jenssen, H. Isolation, purification, and antimicrobial characterization of cannabidiolic acid and cannabidiol from Cannabis sativa L. Biomolecules 2020, 10, 900. [Google Scholar] [CrossRef]
- Wassmann, C.S.; Højrup, P.; Klitgaard, J.K. Cannabidiol is an effective helper compound in combination with bacitracin to kill Gram-positive bacteria. Sci. Rep. 2020, 10, 4112. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T.; Kavanagh, A.M.; Elliott, A.G.; Zhang, B.; Ramu, S.; Amado, M.; Lowe, G.J.; Hinton, A.O.; Pham, D.M.T.; Zuegg, J.; et al. The antimicrobial potential of cannabidiol. Commun. Biol. 2021, 4, 7. [Google Scholar] [CrossRef]
- Borrelli, F.; Aviello, G.; Romano, B.; Orlando, P.; Capasso, R.; Maiello, F.; Guadagno, F.; Petrosino, S.; Capasso, F.; Di Marzo, V.; et al. Cannabidiol, a safe and non-psychotropic ingredient of the marijuana plant Cannabis sativa, is protective in a murine model of colitis. J. Mol. Med. 2009, 87, 1111–1121. [Google Scholar] [CrossRef]
- Harvey, B.S.; Sia, T.C.; Wattchow, D.A.; Smid, S.D. Interleukin 17A evoked mucosal damage is attenuated by cannabidiol and anandamide in a human colonic explant model. Cytokine 2014, 65, 236–244. [Google Scholar] [CrossRef]
- Gigli, S.; Seguella, L.; Pesce, M.; Bruzzese, E.; D’Alessandro, A.; Cuomo, R.; Steardo, L.; Sarnelli, G.; Esposito, G. Cannabidiol restores intestinal barrier dysfunction and inhibits the apoptotic process induced by Clostridium difficile toxin A in Caco-2 cells. United Eur. Gastroenterol. J. 2017, 5, 1108–1115. [Google Scholar] [CrossRef]
- Abdel-Salam, O. Gastric acid inhibitory and gastric protective effects of Cannabis and cannabinoids. Asian Pac. J. Trop. Med. 2016, 9, 413–419. [Google Scholar] [CrossRef]
- Holland, J.C.; Rowland, J.; Plumb, M. Psychological aspects of anorexia in cancer patients. Cancer Res. 1977, 37, 2425–2428. [Google Scholar]
- Wang, J.; Wang, Y.; Tong, M.; Pan, H.; Li, D. New prospect for cancer cachexia: Medical cannabinoid. J. Cancer 2019, 10, 716–720. [Google Scholar] [CrossRef]
- Kenyon, J.; Liu, W.; Dalgleish, A. Report of objective clinical responses of cancer patients to pharmaceutical-grade synthetic cannabidiol. Anticancer Res. 2018, 38, 5831–5835. [Google Scholar] [CrossRef]
- Guggisberg, J.; Schumacher, M.; Gilmore, G.; Zylla, D.M. Cannabis as an anticancer agent: A review of clinical data and assessment of case reports. Cannabis Cannabinoid Res. 2022, 7, 24–33. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Warning Letters and Test Results for Cannabidiol-Related Products. Available online: https://www.fda.gov/newsevents/publichealthfocus/ucm484109.htm (accessed on 15 August 2021).
- Rosenkrantz, H.; Hayden, D.W. Acute and subacute inhalation toxicity of Turkish marihuana, cannabichromene, and cannabidiol in rats. Toxicol. Appl. Pharmacol. 1979, 48, 375–386. [Google Scholar] [CrossRef]
- Rosenkrantz, H.; Fleischman, R.W.; Grant, R.J. Toxicity of short-term administration of cannabinoids to rhesus monkeys. Toxicol. Appl. Pharmacol. 1981, 58, 118–131. [Google Scholar] [CrossRef]
- Hložek, T.; Uttl, L.; Kadeřábek, L.; Balíková, M.; Lhotková, E.; Horsley, R.R.; Nováková, P.; Šíchová, K.; Štefková, K.; Tylš, F.; et al. Pharmacokinetic and behavioural profile of THC, CBD, and THC+CBD combination after pulmonary, oral, and subcutaneous administration in rats and confirmation of con- version in vivo of CBD to THC. Eur. Neuropsychopharmacol. 2017, 27, 1223–1237. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research. Non-Clinical Reviews. US FDA Report. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000PharmR.pdf (accessed on 24 January 2022).
- Garberg, H.T.; Solberg, R.; Barlinn, J.; Martinez-Orgado, J.; Løberg, E.M.; Saugstad, O.D. High-dose cannabidiol induced hypotension after global hypoxia-ischemia in piglets. Neonatology 2017, 112, 143–149. [Google Scholar] [CrossRef]
- ElBatsh, M.M.; Assareh, N.; Marsden, C.A.; Kendall, D.A. Anxiogenic-like effects of chronic cannabidiol administration in rats. Psychopharmacology 2012, 221, 239–247. [Google Scholar] [CrossRef]
- Schiavon, A.P.; Bonato, J.M.; Milani, H.; Guimarães, F.S.; Weffort de Oliveira, R.M. Influence of single and repeated cannabidiol administration on emotional behavior and markers of cell proliferation and neurogenesis in non-stressed mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 27–34. [Google Scholar] [CrossRef]
- Schuel, H.; Schuel, R.; Zimmerman, A.M.; Zimmerman, S. Cannabinoids reduce fertility of sea urchin sperm. Biochem. Cell Biol. 1987, 65, 130–136. [Google Scholar] [CrossRef]
- Schuel, H.; Berkery, D.; Schuel, R.; Chang, M.C.; Zimmerman, A.M.; Zimmerman, S. Reduction of the fertilizing capacity of sea urchin sperm by cannabinoids derived from marihuana. I. Inhibition of the acrosome reaction induced by egg jelly. Mol. Reprod. Dev. 1991, 29, 51–59. [Google Scholar] [CrossRef]
- Narimatsu, S.; Watanabe, K.; Matsunaga, T.; Yamamoto, I.; Imaoka, S.; Funae, Y.; Yoshimura, H. Inhibition of hepatic microsomal cytochrome P450 by cannabidiol in adult male rats. Chem. Pharm. Bull. 1990, 38, 1365–1368. [Google Scholar] [CrossRef]
- Carvalho, R.K.; Santos, M.L.; Souza, M.R.; Rocha, T.L.; Guimarães, F.S.; Anselmo-Franci, J.A.; Mazaro-Costa, R. Chronic exposure to cannabidiol induces reproductive toxicity in male Swiss mice. J. Appl. Toxicol. 2018, 38, 1545. [Google Scholar] [CrossRef] [PubMed]
- Iffland, K.; Grotenhermen, F. An update on safety and side effects of cannabidiol: A review of clinical data and relevant animal studies. Cannabis Cannabinoid Res. 2017, 2, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, R.K.; Andersen, M.L.; Mazaro-Costa, R. The effects of cannabidiol on male reproductive system: A literature review. J. Appl. Toxicol. 2020, 40, 132–150. [Google Scholar] [CrossRef] [PubMed]
- Huestis, M.A.; Solimini, R.; Pichini, S.; Pacifici, R.; Carlier, J.; Busardò, F.P. Cannabidiol adverse effects and toxicity. Curr. Neuropharmacol. 2019, 17, 974–989. [Google Scholar] [CrossRef]
- Černe, K. Toxicological properties of Δ9-tetrahydrocannabinol and cannabidiol. Arch. Ind. Hyg. Toxicol. 2020, 71, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Bisson, J.; Singh, G.; Graham, J.G.; Chen, S.N.; Friesen, J.B.; Dahlin, J.L.; Niemitz, M.; Walters, M.A.; Pauli, G.F. The essential medicinal chemistry of cannabidiol (CBD). J. Med. Chem. 2020, 63, 12137–12155. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S.; Cannabidiol in Dravet Syndrome Study Group. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef]
- Koo, C.M.; Kang, H.C. Could cannabidiol be a treatment option for intractable childhood and adolescent epilepsy? J. Epilepsy Res. 2017, 7, 16–20. [Google Scholar] [CrossRef][Green Version]
- Neale, M. Efficacy and safety of cannabis for treating children with refractory epilepsy. Nurs. Child. Young People 2017, 29, 32–37. [Google Scholar] [CrossRef]
- Greenwich Biosciences Inc. Epidiolex [Package Insert]. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210365lbl.pdf (accessed on 11 June 2021).
- Thiele, E.A.; Marsh, E.D.; French, J.A.; Mazurkiewicz-Beldzinska, M.; Benbadis, S.R.; Joshi, C.; Lyons, P.D.; Taylor, A.; Roberts, C.; Sommerville, K.; et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018, 391, 1085–1096. [Google Scholar] [CrossRef]
- Jiang, R.; Yamaori, S.; Takeda, S.; Yamamoto, I.; Watanabe, K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011, 89, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Parihar, V.; Rogers, A.; Blain, A.M.; Zacharias, S.R.K.; Patterson, L.L.; Siyam, M.A.M. Reduction in tamoxifen metabolites endoxifen and N-desmethyltamoxifen with chronic administration of low dose cannabidiol: A CYP3A4 and CYP2D6 drug interaction. J. Pharm. Pract. 2020, 897190020972208. [Google Scholar] [CrossRef] [PubMed]
- Millar, S.A.; Stone, N.L.; Yates, A.S.; O’Sullivan, S.E. A systematic review on the pharmacokinetics of cannabidiol in humans. Front. Pharmacol. 2018, 9, 1365. [Google Scholar] [CrossRef] [PubMed]
- Millar, S.A.; Maguire, R.F.; Yates, A.S.; O’Sullivan, S.E. Towards better delivery of cannabidiol (CBD). Pharmaceuticals 2020, 13, 219. [Google Scholar] [CrossRef]
- Bruni, N.; Della Pepa, C.; Oliaro-Bosso, S.; Pessione, E.; Gastaldi, D.; Dosio, F. Cannabinoid delivery systems for pain and inflammation treatment. Molecules 2018, 23, 2478. [Google Scholar] [CrossRef]
- Knaub, K.; Sartorius, T.; Dharsono, T.; Wacker, R.; Wilhelm, M.; Schön, C. A novel self-emulsifying drug delivery system (SEDDS) based on VESIsorb® formulation technology improving the oral bioavailability of cannabidiol in healthy subjects. Molecules 2019, 24, 2967. [Google Scholar] [CrossRef]
- Cherniakov, I.; Izgelov, D.; Barasch, D.; Davidson, E.; Domb, A.J.; Hoffman, A. Piperine-pro-nanolipospheres as a novel oral delivery system of cannabinoids: Pharmacokinetic evaluation in healthy volunteers in comparison to buccal spray administration. J. Control. Release 2017, 266, 1–7. [Google Scholar] [CrossRef]
- Atsmon, J.; Cherniakov, I.; Izgelov, D.; Hoffman, A.; Domb, A.J.; Deutsch, L.; Deutsch, F.; Heffetz, D.; Sacks, H. PTL401, a new formulation based on pro-nano dispersion technology, improves oral cannabinoids bioavailability in healthy volunteers. J. Pharm. Sci. 2018, 107, 1423–1429. [Google Scholar] [CrossRef]
- Ohlsson, A.; Lindgren, J.E.; Andersson, S.; Agurell, S.; Gillespie, H.; Hollister, L.E. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed. Environ. Mass Spectrom. 1986, 13, 77–83. [Google Scholar] [CrossRef]
- Meyer, P.; Langos, M.; Brenneisen, R. Human pharmacokinetics and adverse effects of pulmonary and intravenous THC-CBD formulations. Med. Cannabis Cannabinoids 2018, 1, 36–43. [Google Scholar] [CrossRef]
- Taylor, L.; Gidal, B.; Blakey, G.; Tayo, B.; Morrison, G.A. A phase I, Randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs 2018, 32, 1053–1067. [Google Scholar] [CrossRef]
- Xu, C.; Chang, T.; Du, Y.; Yu, C.; Tan, X.; Li, X. Pharmacokinetics of oral and intravenous cannabidiol and its antidepressant-like effects in chronic mild stress mouse model. Environ. Toxicol. Pharmacol. 2019, 70, 103202. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef]
- Alavi, M.; Hamidi, M. Passive and active targeting in cancer therapy by liposomes and lipid nanoparticles. Drug Metab. Pers. Ther. 2019, 34, 20180032. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Delgado, Y.; Castillo, B.; Figueroa, C.M.; Molina, A.M.; Torres, A.; Milián, M.; Griebenow, K. Smart targeting to improve cancer therapeutics. Drug Des. Devel. Ther. 2019, 13, 3753–3772. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef]
- Choi, I.K.; Strauss, R.; Richter, M.; Yun, C.O.; Lieber, A. Strategies to increase drug penetration in solid tumors. Front. Oncol. 2013, 3, 193. [Google Scholar] [CrossRef]
- Maeda, H.; Tsukigawa, K.; Fang, J. A retrospective 30 years after discovery of the enhanced permeability and retention effect of solid tumors: Next-generation chemotherapeutics and photodynamic therapy: Problems, solutions, and prospects. Microcirculation 2016, 23, 173–182. [Google Scholar] [CrossRef]
- Tardi, P.; Wan, C.P.L.; Mayer, L.J. Passive and semi-active targeting of bone marrow and leukemia cells using anionic low cholesterol liposomes. J. Drug Target 2016, 24, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug delivery: Is the enhanced permeability and retention effect sufficient for curing cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- McCallion, C.; Peters, A.D.; Booth, A.; Rees-Unwin, K.; Adams, J.; Rahi, R.; Pluen, A.; Hutchinson, C.V.; Webb, S.J.; Burthem, J. Dual-action CXCR4-targeting liposomes in leukemia: Function blocking and drug delivery. Blood Adv. 2019, 3, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; LoRusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. 64Cu-MM-302 positron emission tomography quantifies variability of enhanced permeability and retention of nanoparticles in relation to treatment response in patients with metastatic breast cancer. Clin. Cancer Res. 2017, 23, 4190–4202. [Google Scholar] [CrossRef]
- Schleich, N.; Po, C.; Jacobs, D.; Ucakar, B.; Gallez, B.; Danhier, F.; Préat, V. Comparison of active, passive and magnetic targeting to tumors of multifunctional paclitaxel/SPIO-loaded nanoparticles for tumor imaging and therapy. J. Control. Release 2014, 194, 82–91. [Google Scholar] [CrossRef]
- Ravar, F.; Saadat, E.; Gholami, M.; Dehghankelishadi, P.; Mahdavi, M.; Azami, S.; Dorkoosh, F.A. Hyaluronic acid-coated liposomes for targeted delivery of paclitaxel, in-vitro characterization and in-vivo evaluation. J. Control. Release 2016, 229, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, Y.; Gu, X.; Li, Q. Development of a hyaluronic acid-based nanocarrier incorporating doxorubicin and cisplatin as a pH-sensitive and CD44-targeted anti-breast cancer drug delivery system. Front. Pharmacol. 2020, 11, 532457. [Google Scholar] [CrossRef]
- Ngwa, W.; Kumar, R.; Moreau, M.; Dabney, R.; Herman, A. Nanoparticle drones to target lung cancer with radiosensitizers and cannabinoids. Front. Oncol. 2017, 7, 208. [Google Scholar] [CrossRef]
- Greish, K.; Mathur, A.; Al Zahrani, R.; Elkaissi, S.; Al Jishi, M.; Nazzal, O.; Taha, S.; Pittalà, V.; Taurin, S. Synthetic cannabinoids nano-micelles for the management of triple negative breast cancer. J. Control. Release 2018, 291, 184–195. [Google Scholar] [CrossRef]
- Hernán Pérez de la Ossa, D.; Lorente, M.; Gil-Alegre, M.E.; Torres, S.; García-Taboada, E.; Aberturas, M.; Molpeceres, J.; Velasco, G.; Torres-Suárez, A.I. Local delivery of cannabinoid-loaded microparticles inhibits tumor growth in a murine xenograft model of glioblastoma multiforme. PLoS ONE 2013, 8, e54795. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Fernández-Carballido, A.; Delie, F.; Cohen, M.; Martin-Sabroso, C.; Mezzanzanica, D.; Figini, M.; Satta, A.; Torres-Suárez, A.I. Enhancing ovarian cancer conventional chemotherapy through the combination with cannabidiol loaded microparticles. Eur. J. Pharm. Biopharm. 2020, 154, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.I.; Torres-Suárez, A.I.; Cohen, M.; Delie, F.; Bastida-Ruiz, D.; Yart, L.; Martin-Sabroso, C.; Fernández-Carballido, A. PLGA nanoparticles for the intraperitoneal administration of CBD in the treatment of ovarian cancer: In vitro and in ovo assessment. Pharmaceutics 2020, 12, 439. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Blanco, J.; Romero, I.A.; Male, D.K.; Slowing, K.; García-García, L.; Torres-Suárez, A.I. Cannabidiol enhances the passage of lipid nanocapsules across the blood-brain barrier both in vitro and in vivo. Mol. Pharm. 2019, 16, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Blanco, J.; Sebastián, V.; Benoit, J.P.; Torres-Suárez, A.I. Lipid nanocapsules decorated and loaded with cannabidiol as targeted prolonged release carriers for glioma therapy: In vitro screening of critical parameters. Eur. J. Pharm. Biopharm. 2019, 134, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Lugasi, L.; Grinberg, I.; Margel, S. Targeted delivery of CBD-targeted poly (RGD) protenoid nanoparticles for antitumor therapy. J. Nanomed. Nanotechnol. 2020, 11, 552. [Google Scholar] [CrossRef]
- Moosavian, S.A.; Bianconi, V.; Pirro, M.; Sahebkar, A. Challenges and pitfalls in the development of liposomal delivery systems for cancer therapy. Semin. Cancer Biol. 2021, 69, 337–348. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Warns 15 Companies for Illegally Selling Various Products Containing Cannabidiol as Agency Details Safety Concerns. Available online: https://www.fda.gov/news-events/press-announcements/fda-warns-15-companies-illegally-selling-various-products-containing-cannabidiol-agency-details (accessed on 10 September 2021).
- U.S. Food and Drug Administration. FDA and Cannabis: Research and Drug Approval Process. Available online: https://www.fda.gov/news-events/public-health-focus/fda-and-cannabis-research-and-drug-approval-process (accessed on 10 September 2021).



| Cancer Type | Experimental Model | Chemotherapeuticals Employed | Combination Index (CI) | Synergistic Effects | Proposed Mechanism | References |
|---|---|---|---|---|---|---|
| Bladder cancer | Cell lines: T24 | Gemcitabine (0–20 μM) Cysplatin (0–100 μM) CBD (0–30 μM) | ND | ↑ Cytotoxicity | ND | [62] |
| Breast cancer | Cell lines: MCF-7, MDA-MB-231 | Doxorubicin (0–20 μM) Paclitaxel (0–500 nM) CBD (0–20 μM) | MDA-MB-231 0.59–0.83 MCF-7: 0.54–0.63 | ↑ Cytotoxicity | ND | [63] |
| Breast cancer | Xenograft: MDA-MB-231, 4T1 | Paclitaxel (2.5–35 μM) CBD (2.7–4 μM) | MDA-MB-231: 4T1: 0.6–0.4 4T1: 0.9–0.8 | ↑ Cytotoxicity ↓ Tumor volume | 5HT1A receptors | [64] |
| Glioma | Xenograft: U87MG | Temozolomide (5 mg/kg) CBD (3.7 mg/kg) | 0.78–0.887 | ↓ Tumor volume ↓ Tumor weight | Autophagy-mediated cell death | [11] |
| Glioma | Cell lines: U87MG, MZC | Doxorubicin (0–200 nM) Temozolomide (0–400 μM) Carmustine (0–200 μM) CBD (10 μM) | ND | ↑ Cytotoxicity | TRPV2 overexpression TRPV2 activation | [65] |
| Glioma | Cell lines: T98G, U251, U87MG, | Temozolomide (1–1000 μM) Carmustine (3–1000 μM) Cisplatin (0–1000 μM) CBD (1–10 μM) | ND | ↓ Proliferation ↑ Cytotoxicity | ND | [66] |
| Leukemia | Cell lines: CCFR-CEM, HL60 | Cytarabine (5.4 μM) Vincristine (1.9 nM) CBD (4 μM) | CCFR:CEM: 0.92–0.61 HL60: 0.43-0.034 | ↑ Cytotoxicity | ND | [10] |
| Medulloblastoma Ependymoma | Cell lines: D283, D425, PER547 | Cyclophosphamide (0–20 μM) CBD (0–7 μM) | ND | ↑ Cytotoxicity ↑ Cell cycle arrest ↑ Apoptosis | ND | [45] |
| Multiple myeloma | Cell lines: U266, RPMI8226 | Carfilzomib (0–100 nM) CBD (12.5 μM) | Specified as CI < 1 | ↑ Cytotoxicity | Apoptosis induction | [69] |
| Synergism with other cytotoxic compounds | ||||||
| Glioma | Cell lines: U87MG Xenograft: U87MG | THC (0–3.5 μM) CBD (0–3.5 μM) | ND | ↑ Cytotoxicity ↓ Tumor volume ↓ Tumor weight | Autophagy-mediated cell death | [11] |
| Glioma | Cell lines: GSC387, GSC3832 | Erastin (2.5–10 μM) Piperazine erastin (10 μM) CBD (0–10 μM) | GSC387: 0.64 GSC387: 0.53 GSC3832: 0.52 | ↑ ROS ↓ Tumor cell ↓ Invasion | ROS-mediated SLC7A11 upregulation | [68] |
| Glioma | Cell lines: U251, SF126 | THC (0–5.4 μM) CBD (0–1.4 μM) | SF126: 0.22 U251: 0.29–0.27 | ↓ Cell growth ↑ Caspase activation ↑ Apoptosis | ERK inhibition | [15] |
| Glioma | Cell lines: U87MG, T98G Orthotopic tumor: GL261 in C57BL6 mice | CBD (0–20 μM) Irradiation (0–5 Gy) | U87MG: 0.9–08 T98G: 0.9–0.8 GL261: 0.9 | ↑ Cytotoxicity ↑ Autophagy ↓ Tumor volume ↓ Tumor progression | MAPK signaling | [70] |
| Leukemia | Cell lines: CCFR-CEM, HL60 | THC (0–50 μM) CBD (0–50 μM) | CCRF-CEM: 0.53–0.44 HL60: 0.34–0.29 | ↑ Cytotoxicity | ND | [10] |
| Multiple myeloma | Cell lines: U266, RPMI8226 | THC (12.5–50 μM) CBD (0–50 μM) | Specified as C < 1 | ↑ Cytotoxicity | Cell cycle arrest Autophagic cell death | [69] |
| Medulloblastoma Ependymoma | Cell lines: D283, PER547 | THC (0–10.5 μM) CBD (0–9.5 μM) | ND | ↑ Cytotoxicity ↑ Cell cycle arrest ↑ Autophagy | ROS-dependent mediated autophagy and apoptosis | [45] |
| Carrier System | Structural Details | Models Tested | Administration Route | Advantages | Concerns and Limitations | Reference |
|---|---|---|---|---|---|---|
| Inorganic nanoparticles | Gold drones loaded with CBD | In vivo: transgenic mouse model bearing lung adenocarcinoma | Inhalation i.v. | Improved: Stability Bioavailability Retention in tumors | Loading concentration Drone size for EPR | [161] |
| Nano-micelles | Poly(styrene-co-maleic anhydride), cumene-terminated (SMA) micelles loaded with WIN | In vitro: breast cancer cell lines | Added to growth medium | Improved: Stability Bioavailability Retention in tumors | Loading concentration Micelle size for EPR | [162] |
| In vivo: Female Balb/c mice bearing 4T1 mammary carcinoma | i.v. | |||||
| Polymeric microparticles | CBD-loaded poly-ε-caprolactone microparticles | In vivo: murine xenograft (glioblastoma) model | Local delivery | Long-lasting CBD delivery | Optimal particle size for better drug delivery | [163] |
| CBD-loaded PLGA microparticles (25 μM) | In vitro and in ovo: breast or ovarian cancer cell lines | Added to growth medium or inoculated in chicken embryos | PLGA is FDA-approved Long-lasting delivery Possibility for multi-drug codelivery | Particle sterilization caused polymer erosion Particle size should be optimized to be suitable for bloodstream circulation | [164,165] | |
| Lipid nanoparticles | CBD-loaded and CBD-decorated (functionalized) lipid nanoparticles | In vitro: glioma cell lines | Added to growth medium | Enhanced targeting and crossing of BBB Enhanced tumor targeting Biocompatible Biodegradable | Nanoparticle stability in organism | [166,167] |
| In vivo: murine xenograft (glioma) model | i.v. | |||||
| Proteinoid nanoparticles | CBD-loaded Poly(RGD) proteinoid nanoparticles | In vitro: Colon carcinoma and breast cancer Cell lines | Added to growth medium | Cancer tissue targeting | [168] | |
| In vivo: Athymic mice bearing colon and breast cancer xenografts | i.v. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivas-Aguirre, M.; Torres-López, L.; Villatoro-Gómez, K.; Perez-Tapia, S.M.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol on the Path from the Lab to the Cancer Patient: Opportunities and Challenges. Pharmaceuticals 2022, 15, 366. https://doi.org/10.3390/ph15030366
Olivas-Aguirre M, Torres-López L, Villatoro-Gómez K, Perez-Tapia SM, Pottosin I, Dobrovinskaya O. Cannabidiol on the Path from the Lab to the Cancer Patient: Opportunities and Challenges. Pharmaceuticals. 2022; 15(3):366. https://doi.org/10.3390/ph15030366
Chicago/Turabian StyleOlivas-Aguirre, Miguel, Liliana Torres-López, Kathya Villatoro-Gómez, Sonia Mayra Perez-Tapia, Igor Pottosin, and Oxana Dobrovinskaya. 2022. "Cannabidiol on the Path from the Lab to the Cancer Patient: Opportunities and Challenges" Pharmaceuticals 15, no. 3: 366. https://doi.org/10.3390/ph15030366
APA StyleOlivas-Aguirre, M., Torres-López, L., Villatoro-Gómez, K., Perez-Tapia, S. M., Pottosin, I., & Dobrovinskaya, O. (2022). Cannabidiol on the Path from the Lab to the Cancer Patient: Opportunities and Challenges. Pharmaceuticals, 15(3), 366. https://doi.org/10.3390/ph15030366