
Identifying HSV-1 Inhibitors from Natural Compounds via Virtual Screening Targeting Surface Glycoprotein D

 , , , , ,
, , , , ,  and
and 
Abstract
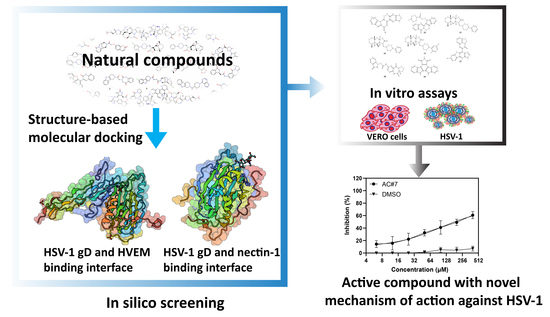
1. Introduction
2. Results
2.1. Virtual Screening in Search of Potential gD Inhibitors
2.2. Investigating the Antiviral Activity of Selected Compounds by In Vitro Assays
2.2.1. Determination of Cytotoxicity of ACs
2.2.2. Investigating the Mechanism of Action of ACs by Time of Addition Assay
2.2.3. Estimating the Efficacy of ACs by Plaque Reduction Assay
3. Discussion
4. Materials and Methods
4.1. In Silico Screening
4.1.1. Preparation of Natural Compound Library
4.1.2. Preparation of Receptor Proteins
4.1.3. Virtual Screening on the HVEM Binding Site of gD
4.1.4. ADMET Analysis
4.1.5. Redocking Selected Compounds on the Nectin-1 Binding Site of gD
4.2. In Vitro Validation
4.2.1. Chemicals
4.2.2. Cells and Virus
4.2.3. Cytotoxicity of ACs
4.2.4. Time of Addition Assay
4.2.5. Plaque Reduction Dose–Response Assay
4.2.6. Toxicity of AC#7 on VERO Cells at High Concentrations
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Massive Proportion of World’s Population Are Living with Herpes Infection. 2020. Available online: https://www.who.int/news/item/01-05-2020-massive-proportion-world-population-living-with-herpes-infection (accessed on 8 March 2021).
- Diefenbach, R.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2007, 18, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Kawada, J.-I. Neurological Disorders Associated with Human Alphaherpesviruses. In Human Herpesviruses; Kawaguchi, Y., Mori, Y., Kimura, H., Eds.; Springer Singapore: Singapore, 2018; pp. 85–102. [Google Scholar]
- Tyler, K.L. Acute Viral Encephalitis. N. Engl. J. Med. 2018, 379, 557–566. [Google Scholar] [CrossRef]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, B.A.; Kousoulas, K.G.; Fernandez, A.; Gershburg, E. Rational Design of Live-Attenuated Vaccines against Herpes Simplex Viruses. Viruses 2021, 13, 1637. [Google Scholar] [CrossRef] [PubMed]
- Zanjani, N.T.; Miranda-Saksena, M.; Valtchev, P.; Diefenbach, R.J.; Hueston, L.; Diefenbach, E.; Sairi, F.; Gomes, V.G.; Cunningham, A.L.; Dehghani, F. Abalone Hemocyanin Blocks the Entry of Herpes Simplex Virus 1 into Cells: A Potential New Antiviral Strategy. Antimicrob. Agents Chemother. 2016, 60, 1003–1012. [Google Scholar] [CrossRef]
- De Clercq, E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2002, 1, 13–25. [Google Scholar] [CrossRef]
- Sadowski, L.; Upadhyay, R.; Greeley, Z.; Margulies, B. Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2. Viruses 2021, 13, 1228. [Google Scholar] [CrossRef]
- Levin, M.J.; Bacon, T.H.; Leary, J.J. Resistance of Herpes Simplex Virus Infections to Nucleoside Analogues in HIV-Infected Patients. Clin. Infect. Dis. 2004, 39, S248–S257. [Google Scholar] [CrossRef]
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Thymidine kinase and protein kinase in drug-resistant herpesvirtises: Heads of a Lernaean Hydra. Drug Resist. Updates 2018, 37, 1–16. [Google Scholar] [CrossRef]
- Van de Sand, L.; Bormann, M.; Schmitz, Y.; Heilingloh, C.S.; Witzke, O.; Krawczyk, A. Antiviral Active Compounds Derived from Natural Sources against Herpes Simplex Viruses. Viruses 2021, 13, 1386. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, R.J.; Atanasiu, D.; Cairns, T.M.; Gallagher, J.R.; Krummenacher, C.; Cohen, G.H. Herpes Virus Fusion and Entry: A Story with Many Characters. Viruses 2012, 4, 800–832. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Genet. 2020, 19, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Lazear, E.; Whitbeck, J.C.; Zuo, Y.; Carfí, A.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Induction of conformational changes at the N-terminus of herpes simplex virus glycoprotein D upon binding to HVEM and nectin-1. Virology 2013, 448, 185–195. [Google Scholar] [CrossRef]
- Vallbracht, M.; Backovic, M.; Klupp, B.G.; Rey, F.A.; Mettenleiter, T.C. Common characteristics and unique features: A comparison of the fusion machinery of the alphaherpesviruses Pseudorabies virus and Herpes simplex virus. In Advances in Virus Research, 1st ed.; Kielian, M., Mettenleiter, T.C., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 104, pp. 225–281. ISBN 978-0-12-818395-3. [Google Scholar]
- Carfi, A.; Willis, S.H.; Whitbeck, J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Wiley, D.C. Herpes Simplex Virus Glycoprotein D Bound to the Human Receptor HveA. Mol. Cell 2001, 8, 169–179. [Google Scholar] [CrossRef]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfi, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar] [CrossRef]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral Lead Compounds from Marine Sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef]
- Hsiang, C.-Y.; Ho, T.-Y. Emodin is a novel alkaline nuclease inhibitor that suppresses herpes simplex virus type 1 yields in cell cultures. J. Cereb. Blood Flow Metab. 2008, 155, 227–235. [Google Scholar] [CrossRef]
- Huang, Y.; Li, X.; Pan, C.; Cheng, W.; Wang, X.; Yang, Z.; Zheng, L. The intervention mechanism of emodin on TLR3 pathway in the process of central nervous system injury caused by herpes virus infection. Neurol. Res. 2020, 43, 307–313. [Google Scholar] [CrossRef]
- Xiong, H.-R.; Luo, J.; Hou, W.; Xiao, H.; Yang, Z.-Q. The effect of emodin, an anthraquinone derivative extracted from the roots of Rheum tanguticum, against herpes simplex virus in vitro and in vivo. J. Ethnopharmacol. 2010, 133, 718–723. [Google Scholar] [CrossRef]
- De Oliveira, A.; Adams, S.D.; Lee, L.H.; Murray, S.R.; Hsu, S.D.; Hammond, J.R.; Dickinson, D.; Chen, P.; Chu, T.-C. Inhibition of herpes simplex virus type 1 with the modified green tea polyphenol palmitoyl-epigallocatechin gallate. Food Chem. Toxicol. 2012, 52, 207–215. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Yu, Z.-Y.; Chen, Y.-C.; Hung, S.-L. Effects of epigallocatechin-3-gallate and acyclovir on herpes simplex virus type 1 infection in oral epithelial cells. J. Formos. Med. Assoc. 2020, 120, 2136–2143. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, P.; Nguyen, M.L. Herpes simplex virus virucidal activity of MST-312 and epigallocatechin gallate. Virus Res. 2018, 249, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, C.E.; Wen, G.Y.; Xu, W.; Jia, J.H.; Rohan, L.; Corbo, C.; Di Maggio, V.; Jenkins, E.C.; Hillier, S. Epigallocatechin Gallate Inactivates Clinical Isolates of Herpes Simplex Virus. Antimicrob. Agents Chemother. 2008, 52, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Zandi, K.; Ramedani, E.; Mohammadi, K.; Tajbakhsh, S.; Deilami, I.; Rastian, Z.; Fouladvand, M.; Yousefi, F.; Farshadpour, F. Evaluation of Antiviral Activities of Curcumin Derivatives against HSV-1 in Vero Cell Line. Nat. Prod. Commun. 2010, 5, 1935–1938. [Google Scholar] [CrossRef] [PubMed]
- El-Halim, S.M.A.; Mamdouh, M.A.; El-Haddad, A.E.; Soliman, S.M. Fabrication of Anti-HSV-1 Curcumin Stabilized Nanostructured Proniosomal Gel: Molecular Docking Studies on Thymidine Kinase Proteins. Sci. Pharm. 2020, 88, 9. [Google Scholar] [CrossRef]
- Ferreira, V.H.; Nazli, A.; Dizzell, S.E.; Mueller, K.; Kaushic, C. The Anti-Inflammatory Activity of Curcumin Protects the Genital Mucosal Epithelial Barrier from Disruption and Blocks Replication of HIV-1 and HSV-2. PLoS ONE 2015, 10, e0124903. [Google Scholar] [CrossRef] [PubMed]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Smeriglio, A.; Mandalari, G.; Sciortino, M.T. In Vitro Anti-HSV-1 Activity of Polyphenol-Rich Extracts and Pure Polyphenol Compounds Derived from Pistachios Kernels (Pistacia vera L.). Plants 2020, 9, 267. [Google Scholar] [CrossRef]
- Pospisil, P.; Pilger, B.D.; Marveggio, S.; Schelling, P.; Wurth, C.; Scapozza, L.; Folkers, G.; Pongracic, M.; Mintas, M.; Malic, S.R. Synthesis, Kinetics, and Molecular Docking of Novel 9-(2-Hydroxypropyl)purine Nucleoside Analogs as Ligands of Herpesviral Thymidine Kinases. Helvetica Chim. Acta 2002, 85, 3237–3250. [Google Scholar] [CrossRef]
- Krištafor, S.; Novaković, I.; Kraljević, T.G.; Pavelić, S.K.; Lučin, P.; Westermaier, Y.; Pernot, L.; Scapozza, L.; Ametamey, S.M.; Raić-Malić, S. A new N-methyl thymine derivative comprising a dihydroxyisobutenyl unit as ligand for thymidine kinase of herpes simplex virus type 1 (HSV-1 TK). Bioorg. Med. Chem. Lett. 2011, 21, 6161–6165. [Google Scholar] [CrossRef]
- Kant, K.; Lal, U.R.; Kumar, A.; Ghosh, M. A merged molecular docking, ADME-T and dynamics approaches towards the genus of Arisaema as herpes simplex virus type 1 and type 2 inhibitors. Comput. Biol. Chem. 2018, 78, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, J.; Albuquerque, M.; Leal, K.Z.; Santos, F.D.C.; Batalha, P.N.; Brozeguini, L.; Seidl, P.R.; de Alencastro, R.B.; Cunha, A.; de Souza, M.C.B.V.; et al. Docking of anti-HIV-1 oxoquinoline-acylhydrazone derivatives as potential HSV-1 DNA polymerase inhibitors. J. Mol. Struct. 2014, 1074, 263–270. [Google Scholar] [CrossRef]
- Hassan, S.T.S.; Šudomová, M.; Berchová-Bímová, K.; Šmejkal, K.; Echeverría, J. Psoromic Acid, a Lichen-Derived Molecule, Inhibits the Replication of HSV-1 and HSV-2, and Inactivates HSV-1 DNA Polymerase: Shedding Light on Antiherpetic Properties. Molecules 2019, 24, 2912. [Google Scholar] [CrossRef] [PubMed]
- Mello, J.F.R.; Botelho, N.C.; Souza, A.M.T.; Oliveira, R.; Brito, M.A.; Abrahim-Vieira, B.D.A.; Sodero, A.C.R.; Castro, H.C.; Cabral, L.M.; Miceli, L.A.; et al. Computational Studies of Benzoxazinone Derivatives as Antiviral Agents against Herpes Virus Type 1 Protease. Molecules 2015, 20, 10689–10704. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, E.M.D.S.; Lima, T.L.C.; Boff, L.; Lima, S.G.M.; Lourenço, E.M.G.; Ferreira, É.G.; Barbosa, E.G.; Machado, P.R.L.; Farias, K.J.S.; Ferreira, L.D.S.; et al. Antiviral Potential of Spondias mombin L. Leaves Extract Against Herpes Simplex Virus Type-1 Replication Using In Vitro and In Silico Approaches. Planta Med. 2020, 86, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Shamsizadeh, F.; Tavakoli, A.; Baghban, N.; Khoradmehr, A.; Kameli, A.; Rasekh, P.; Daneshi, A.; Nabipour, I.; Vahdat, K.; et al. Chemical compositions and experimental and computational modeling activity of sea cucumber Holothuria parva ethanolic extract against herpes simplex virus type 1. Biomed. Pharmacother. 2021, 141, 111936. [Google Scholar] [CrossRef]
- Petermann, P.; Rahn, E.; Thier, K.; Hsu, M.-J.; Rixon, F.J.; Kopp, S.J.; Knebel-Mörsdorf, D. Role of Nectin-1 and Herpesvirus Entry Mediator as Cellular Receptors for Herpes Simplex Virus 1 on Primary Murine Dermal Fibroblasts. J. Virol. 2015, 89, 9407–9416. [Google Scholar] [CrossRef][Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Organic Chemistry Portal. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 10 November 2020).
- Zanjani, N.T.; Sairi, F.; Marshall, G.; Saksena, M.M.; Valtchev, P.; Gomes, V.G.; Cunningham, A.; Dehghani, F. Formulation of abalone hemocyanin with high antiviral activity and stability. Eur. J. Pharm. Sci. 2014, 53, 77–85. [Google Scholar] [CrossRef]
- Lyu, S.Y.; Rhim, J.Y.; Park, W.B. Antiherpetic activities of flavonoids against herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2)in vitro. Arch. Pharm. Res. 2005, 28, 1293–1301. [Google Scholar] [CrossRef]
- Safrin, S.; Elbeik, T.; Phan, L.; Robinson, D.; Rush, J.; Elbaggari, A.; Mills, J. Correlation between response to acyclovir and foscarnet therapy and in vitro susceptibility result for isolates of herpes simplex virus from human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 1994, 38, 1246–1250. [Google Scholar] [CrossRef] [PubMed]
- Sangdara, A.; Bhattarakosol, P. Acyclovir susceptibility of herpes simplex virus isolates at King Chulalongkorn Memorial Hospital, Bangkok. J. Med. Assoc. Thail. 2008, 91, 908–912. [Google Scholar]
- Knebel-Mörsdorf, D. Nectin-1 and HVEM: Cellular receptors for HSV-1 in skin. Oncotarget 2016, 7, 19087–19088. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.A.; Manchak, M.D.; Hager, E.J.; Krummenacher, C.; Whitbeck, J.C.; Levin, M.J.; Freed, C.R.; Wilcox, C.L.; Cohen, G.H.; Eisenberg, R.J.; et al. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibro-blasts. J. Neurovirol. 2005, 11, 208–218. [Google Scholar] [CrossRef]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef]
- Gopinath, S.C.B.; Hayashi, K.; Kumar, P.K.R. Aptamer That Binds to the gD Protein of Herpes Simplex Virus 1 and Efficiently Inhibits Viral Entry. J. Virol. 2012, 86, 6732–6744. [Google Scholar] [CrossRef]
- Spear, P.G.; Manoj, S.; Yoon, M.; Jogger, C.R.; Zago, A.; Myscofski, D. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology 2006, 344, 17–24. [Google Scholar] [CrossRef]
- Di Giovine, P.; Settembre, E.C.; Bhargava, A.K.; Luftig, M.A.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C.; Carfi, A. Structure of Herpes Simplex Virus Glycoprotein D Bound to the Human Receptor Nectin-1. PLoS Pathog. 2011, 7, e1002277. [Google Scholar] [CrossRef]
- Connolly, S.A.; Landsburg, D.J.; Carfi, A.; Whitbeck, J.C.; Zuo, Y.; Wiley, D.C.; Cohen, G.H.; Eisenberg, R.J. Potential Nectin-1 Binding Site on Herpes Simplex Virus Glycoprotein D. J. Virol. 2005, 79, 1282–1295. [Google Scholar] [CrossRef]
- Elion, G. Acyclovir: Discovery, mechanism of action, and selectivity. J. Med. Virol. 1993, 41, 2–6. [Google Scholar] [CrossRef]
- Kłysik, K.; Pietraszek, A.; Karewicz, A.; Nowakowska, M. Acyclovir in the treatment of herpes viruses–A review. Curr. Med. Chem. 2020, 27, 4118–4137. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.T.; Sacks, S.L. Docosanol: A topical antiviral for herpes labialis. Expert Opin. Pharmacother. 2004, 5, 2567–2571. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, M.-J.; Saiz, J.-C.; Priego, E.-M.; Martín-Acebes, M.A. Antivirals against (Re)emerging Flaviviruses: Should We Target the Virus or the Host? ACS Med. Chem. Lett. 2022, 13, 5–10. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Valeur, E.; Guéret, S.M.; Adihou, H.; Gopalakrishnan, R.; Lemurell, M.; Waldmann, H.; Grossmann, T.; Plowright, A.T. New Modalities for Challenging Targets in Drug Discovery. Angew. Chem. Int. Ed. 2017, 56, 10294–10323. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, K.T.; Bietz, S.; Briem, H.; Henzler, A.M.; Urbaczek, S.; Rarey, M. Facing the Challenges of Structure-Based Target Prediction by Inverse Virtual Screening. J. Chem. Inf. Model. 2014, 54, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Liuzzi, M.; Paris, W.; Lambert, M.; Lawetz, C.; Moss, N.; Jaramillo, J.; Gauthier, J.; Deéziel, R.; Cordingley, M.G. Antiviral Activity of a Selective Ribonucleotide Reductase Inhibitor against Acyclovir-Resistant Herpes Simplex Virus Type 1 In Vivo. Antimicrob. Agents Chemother. 1998, 42, 1629–1635. [Google Scholar] [CrossRef]
- Moss, N.; Beaulieu, P.; Duceppe, J.-S.; Ferland, J.-M.; Garneau, M.; Gauthier, J.; Ghiro, E.; Goulet, S.; Guse, I.; Jaramillo, J.; et al. Peptidomimetic Inhibitors of Herpes Simplex Virus Ribonucleotide Reductase with Improved in Vivo Antiviral Activity. J. Med. Chem. 1996, 39, 4173–4180. [Google Scholar] [CrossRef]
- Moss, N.; Deziel, R.; Adams, J.; Aubry, N.; Bailey, M.; Baillet, M.; Beaulieu, P.; DiMaio, J.; Duceppe, J.S. Inhibition of herpes simplex virus type 1 ribonucleotide reductase by substituted tetrapeptide derivatives. J. Med. Chem. 1993, 36, 3005–3009. [Google Scholar] [CrossRef]
- Gaudreau, P.; Paradis, H.; Langelier, Y.; Brazeau, P. Synthesis and inhibitory potency of peptides corresponding to the subunit 2 C-terminal region of herpes virus ribonucleotide reductases. J. Med. Chem. 1990, 33, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A.; et al. A comparative analysis of SARS-CoV-2 antivirals in human airway models characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hassan, S.T.S.; Masarčíková, R.; Berchová-Bímová, K. Bioactive natural products with anti-herpes simplex virus properties. J. Pharm. Pharmacol. 2015, 67, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Treml, J.; Gazdová, M.; Šmejkal, K.; Šudomová, M.; Kubatka, P.; Hassan, S.T.S. Natural Products-Derived Chemicals: Breaking Barriers to Novel Anti-HSV Drug Development. Viruses 2020, 12, 154. [Google Scholar] [CrossRef]
- Vijayalakshmi, U.; Shourie, A. Gas chromatography-mass spectrometric analysis of ethanolic extracts of Glycyrrhiza glabra Linn. roots. Int. J. Pharma Bio Sci. 2013, 4, 741–755. [Google Scholar]
- Lim, T.K. Glycyrrhiza glabra. In Edible Medicinal and Non-Medicinal Plants: Modified Stems, Roots, Bulbs; Lim, T.K., Ed.; Springer: Dordrecht, The Netherlands, 2016; Volume 10, pp. 354–457. [Google Scholar]
- Gomaa, A.A.; Abdel-Wadood, Y.A. The potential of glycyrrhizin and licorice extract in combating COVID-19 and associated conditions. Phytomed. Plus 2021, 1, 100043. [Google Scholar] [CrossRef]
- Huan, C.; Xu, Y.; Zhang, W.; Guo, T.; Pan, H.; Gao, S. Research Progress on the Antiviral Activity of Glycyrrhizin and its Derivatives in Liquorice. Front. Pharmacol. 2021, 12, 1706. [Google Scholar] [CrossRef]
- Wang, L.; Song, J.; Liu, A.; Xiao, B.; Li, S.; Wen, Z.; Lu, Y.; Du, G. Research progress of the antiviral bioactivities of natural flavonoids. Nat. Prod. Bioprospect. 2020, 10, 271–283. [Google Scholar] [CrossRef]
- Montana, M.; Montero, V.; Khoumeri, O.; Vanelle, P. Quinoxaline Derivatives as Antiviral Agents: A Systematic Review. Molecules 2020, 25, 2784. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, Q.; Rao, Z.; Fang, Y.; Jiang, X.; Liu, W.; Luan, F.; Zeng, N. Inhibition of herpes simplex virus 1 by cepharanthine via promoting cellular autophagy through up-regulation of STING/TBK1/P62 pathway. Antivir. Res. 2021, 193, 105143. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Erehman, J.; Gohlke, B.O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II—A database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, J.A.; Pérez-Jiménez, J.; Neveu, V.; Medina-Remón, A.; M'Hiri, N.; García-Lobato, P.; Manach, C.; Knox, C.; Eisner, R.; Wishart, D.S.; et al. Phenol-Explorer 3.0: A major update of the Phenol-Explorer database to incorporate data on the effects of food processing on polyphenol content. Database 2013, 2013, bat070. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2008, 37, D603–D610. [Google Scholar] [CrossRef]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-López, M.; Pérez-Sánchez, A.; Galiano, V.; Barrajón-Catalán, E.; Micol, V. An Updated Review on Marine Anticancer Compounds: The Use of Virtual Screening for the Discovery of Small-Molecule Cancer Drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Hernandez University Molecular Docking Database Site. Available online: http://docking.umh.es (accessed on 22 May 2021).
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 2.3.5. 2019. Available online: http://www.pymol.org/pymol (accessed on 15 September 2021).
- Chemaxon: Software Solutions and Services for Chemistry & Biology. Available online: https://www.chemaxon.com (accessed on 19 August 2020).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with pyrx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Zhang, N.; Yan, J.; Lu, G.; Guo, Z.; Fan, Z.; Wang, J.; Shi, Y.; Qi, J.; Gao, G.F. Binding of herpes simplex virus glycoprotein D to nectin-1 exploits host cell adhesion. Nat. Commun. 2011, 2, 577. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Chiang, L.; Chiang, W.; Chang, M.; Ng, L.; Lin, C. Antiviral activity of Plantago major extracts and related compounds in vitro. Antivir. Res. 2002, 55, 53–62. [Google Scholar] [CrossRef]
- Wang, Z.; Jia, J.; Wang, L.; Li, F.; Wang, Y.; Jiang, Y.; Song, X.; Qin, S.; Zheng, K.; Ye, J.; et al. Anti-HSV-1 activity of Aspergillipeptide D, a cyclic pentapeptide isolated from fungus Aspergillus sp. SCSIO 41501. Virol. J. 2020, 17, 41. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Database ID | Name | Empirical Formular | Molecular Weight | Docking Score on | Drug Score | |
|---|---|---|---|---|---|---|---|
| HVEM Site | Nectin-1 Site | ||||||
| 7 | Sn00074072 | 1-(1-benzofuran-2-yl)-2-[(5Z)-2H,6H,7H,8H-[1,3]dioxolo [4,5-g]isoquinoline-5-ylidene]ethenone | C20H15NO4 | 333.34 | −8.2 | −8.6 | 0.58 |
| 10 | Sn00115356 | 13-[3-(4-methylpiperazin-1-yl)-3-oxopropyl]-8,13-dihydroindolo[2′,3′:3,4] pyrido [2,1-b]quinazolin-5(7H)-one | C26H27N5O2 | 441.53 | −8.2 | −8.5 | 0.69 |
| 12 | Sn00099520 | (2S,5Ar,6Ar,9S,9Ar)-2,5a-dimethyl-9-((4-(isoquino-2-yl)piperazin-1-yl)methyl)octahydro-2H-oxireno[2′,3′:4,4a]naphtho[2,3-b]furan-8(9Bh)-one | C24H33N3O3 | 411.54 | −8.5 | −7.6 | 0.77 |
| 16 | Sn00104387 | (1Ar,2S,5Ar,6Ar,9S,9Ar,9Bs)-2,5a-dimethyl-9-((4-phenylpiperazin-1-yl)methyl)octahydro-2H-oxireno[2′,3′:4,4a]naphtho[2,3-b]furan-8(9Bh)-one | C25H34N2O3 | 410.56 | −8.3 | −7.6 | 0.74 |
| 17 | Sn00104404 | (1Ar,2S,5Ar,6Ar,9S,9Ar,9Bs)-9-( (4-( 5-chloro-2-methylphenyl)piperazin-1-yl)methyl)-2,5a-dimethyloctahydro-2H-oxireno[2′,3′:4,4a]naphtho[2,3-b]furan-8(9Bh)-one | C26H35ClN2O3 | 459.03 | −8.4 | −7.9 | 0.58 |
| 27 | Zinc96221711 | 5-(7-Hydroxy-1H-benzofuro[3,2-b]pyrazolo[4,3-e]isoquino-4-yl)-1H-pyrrolo[3,2,1-ij]isoquinol-4(2H)-one | C23H14N4O3 | 394.39 | −9.3 | −9.4 | 0.53 |
| 28 | Zinc96115494 | N-((S)-5,11-dioxo-2,3,5,10,11,11a-hexahydro-1H-benzo[e]pyrrolo[1,2-a][1,4]diazepin-7-yl)-2-(3-oxoisoindolin-1-yl)acetamide | C22H20N4O4 | 404.43 | −9.5 | −8.6 | 0.76 |
| 29 | Sn00346605 | Arcyriaflavin A | C20H11N3O2 | 325.3 | −9.1 | −9.1 | 0.89 |
| ID | HVEM Binding Interface | Nectin-1 Binding Interface | ||||
|---|---|---|---|---|---|---|
| No. of Interacting Residues | No. of H-bonds | Interacting Residues | No. of Interacting Residues | No. of H-bonds | Interacting Residues | |
| 7 | 9 | 0 | M11, A12, P14, F17, L22, P23, V24, L25, Y234 | 9 | 2 | Y38, H39, R134, D215, L220, P221, I296, P297, A303 |
| 10 | 11 | 1 | M11, A12, P14, F17, L22, P23, V24, L25, D26, Q27, Y234 | 12 | 2 | Y38, H39, R134, D215, M219, L220, P221, I296, P297, S298, I299, A303 |
| 12 | 9 | 0 | M11, A12, P14, F17, P23, V24, L25, Q27, Y234 | 9 | 0 | Y38, H39, R134, T213, D215, I299, D301, A302, A303 |
| 16 | 9 | 0 | M11, A12, P14, F17, P23, V24, L25, Q27, Y234 | 9 | 0 | Y38, H39, R134, T213, D215, I299, D301, A302, A303 |
| 17 | 11 | 0 | M11, A12, D13, P14, F17, P23, V24, L25, D26, Q27, Y234 | 10 | 1 | Y38, R134, D215, L220, P221, I296, P297, S298, I299, A303 |
| 27 | 9 | 2 | A12, P14, N15, F17, R18, G19, L22, V24, L25 | 8 | 2 | Y38, R134, D215, L220, P221, R222, I296, P297 |
| 28 | 13 | 3 | M11, A12, D13, P14, F17, R18, G19, L22, P23, V24, L25, Q27, Y234 | 8 | 3 | Y38, H39, R134, T213, D215, P221, A303, T304 |
| 29 | 9 | 1 | M11, A12, P14, F17, L22, V24, L25, Q27, Y234 | 6 | 2 | Y38, R134, D215, G218, L220, P221 |
| ID | Highest Concentration with Cell Viability above 75% | Relative Cell Viability * | Test Concentration |
|---|---|---|---|
| 7 | >100 µg/mL | 123.7% ± 4.8% | 10 µg/mL |
| 10 | 1 µg/mL | 73.2% ± 5.9% | 1 µg/mL |
| 12 | 1 µg/mL | 77.4% ± 4.2% | 1 µg/mL |
| 16 | 1 µg/mL | 102.1% ± 16.7% | 1 µg/mL |
| 17 | 1 µg/mL | 96.8% ± 7.1% | 1 µg/mL |
| 27 | >100 µg/mL | 105.9% ± 0.2% | 10 µg/mL |
| 28 | >100 µg/mL | 105.0% ± 0.5% | 10 µg/mL |
| 29 | 1 µg/mL | 86.2% ± 14.8% | 1 µg/mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Power, H.; Miranda-Saksena, M.; Valtchev, P.; Schindeler, A.; Cunningham, A.L.; Dehghani, F. Identifying HSV-1 Inhibitors from Natural Compounds via Virtual Screening Targeting Surface Glycoprotein D. Pharmaceuticals 2022, 15, 361. https://doi.org/10.3390/ph15030361
Wu J, Power H, Miranda-Saksena M, Valtchev P, Schindeler A, Cunningham AL, Dehghani F. Identifying HSV-1 Inhibitors from Natural Compounds via Virtual Screening Targeting Surface Glycoprotein D. Pharmaceuticals. 2022; 15(3):361. https://doi.org/10.3390/ph15030361
Chicago/Turabian StyleWu, Jiadai, Helen Power, Monica Miranda-Saksena, Peter Valtchev, Aaron Schindeler, Anthony L. Cunningham, and Fariba Dehghani. 2022. "Identifying HSV-1 Inhibitors from Natural Compounds via Virtual Screening Targeting Surface Glycoprotein D" Pharmaceuticals 15, no. 3: 361. https://doi.org/10.3390/ph15030361
APA StyleWu, J., Power, H., Miranda-Saksena, M., Valtchev, P., Schindeler, A., Cunningham, A. L., & Dehghani, F. (2022). Identifying HSV-1 Inhibitors from Natural Compounds via Virtual Screening Targeting Surface Glycoprotein D. Pharmaceuticals, 15(3), 361. https://doi.org/10.3390/ph15030361