
Structural Insight of New Butyrylcholinesterase Inhibitors Based on Benzylbenzofuran Scaffold

 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
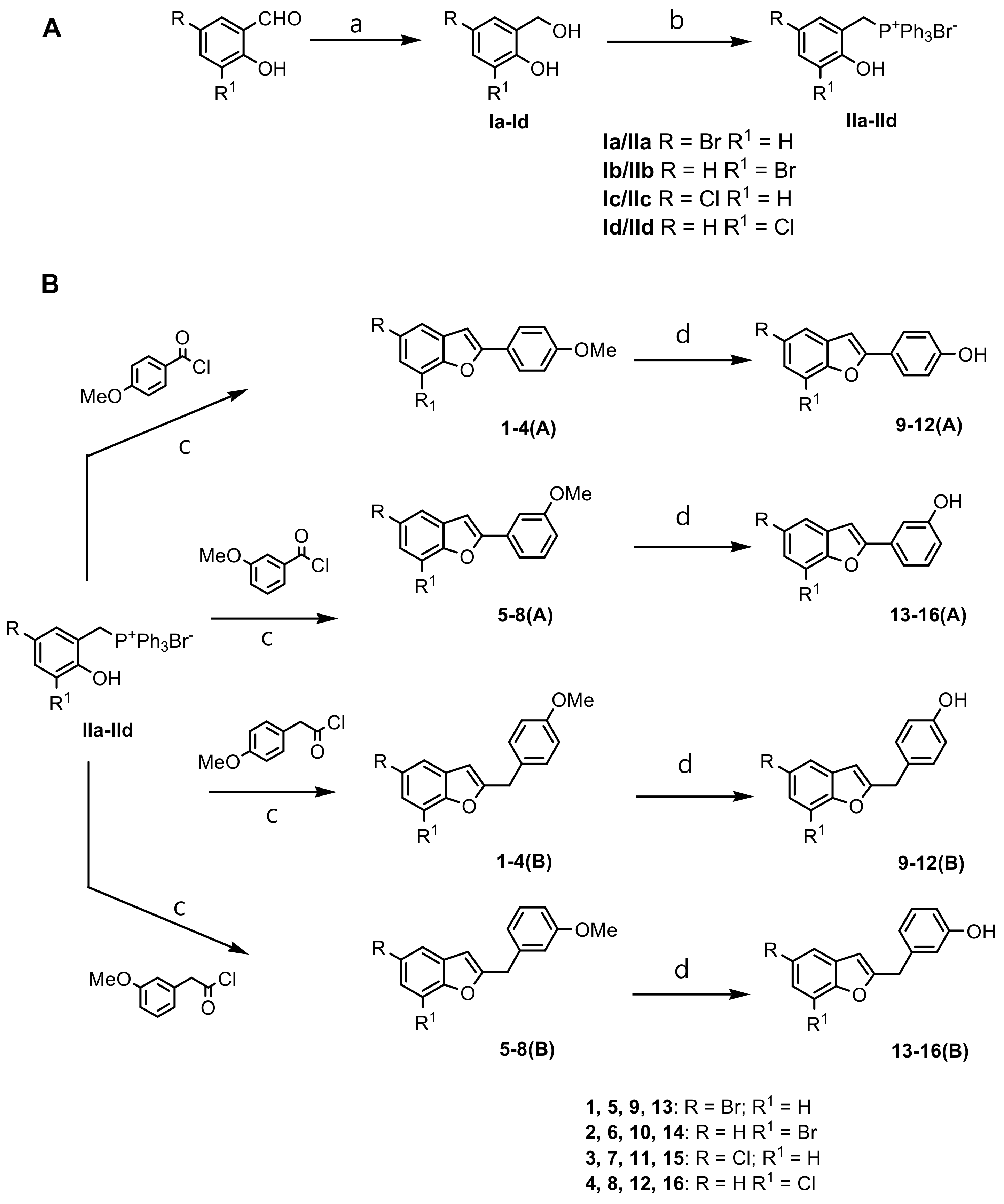
2.1. Chemistry
2.2. Biological Activity
2.3. Molecular and Physicochemical Properties of Compounds
3. Materials and Methods
3.1. Chemistry
3.2. Determination of Butyrylcholinesterase Inhibition
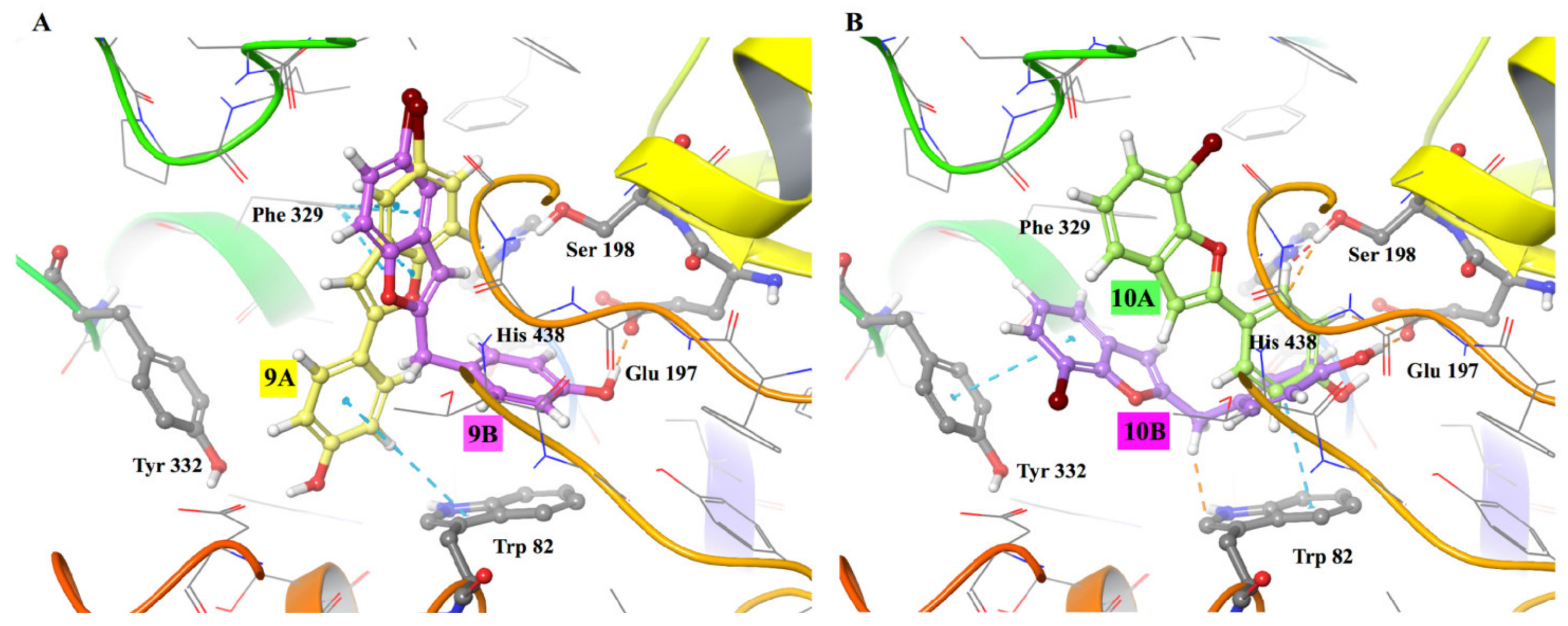
3.3. Molecular Modelling Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Xu, X.; Huang, M.; Zou, X. Docking-based inverse virtual screening: Methods, applications, and challenges. Biophys. Rep. 2018, 4, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Oku, Y.; Hano, Y.; Terada, S. Antimicrobial activities of hydrophobic 2-arylbenzofurans and an isoflavone against vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus. Planta Med. 2004, 70, 685–687. [Google Scholar] [CrossRef]
- Han, S.J.; Ryu, S.N.; Kang, S.S. A new 2-arylbenzofuran with antioxidant activity from the black colored rice (Oryza sativa L.) bran. Chem. Pharm. Bull. 2004, 52, 1365–1366. [Google Scholar] [CrossRef] [Green Version]
- Khodarahmi, G.A.; Asadi, P.; Hassanzadeh, F.; Khodarahmi, E. Benzofuran as a promising scaffold for the synthesis of antimicrobial and antibreast cancer agents: A review. J. Res. Med. Sci. 2015, 20, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gao, B.; Huang, W.; Yongmin, L.; Guosheng, H.; Yongxiang, M. Simple, Convenient, and Efficient Synthesis of 2-Aryl-substituted Benzo[b.furans. Synth. Commun. 2009, 39, 197–204. [Google Scholar] [CrossRef]
- Miao, Y.H.; Hu, Y.H.; Yang, J.; Liu, T.; Sun, J.; Wang, X.J. Natural source, bioactivity and synthesis of benzofuran derivatives. RSC Adv. 2019, 9, 27510–27540. [Google Scholar] [CrossRef] [Green Version]
- Delogu, G.L.; Matos, M.J.; Fanti, M.; Era, B.; Medda, R.; Pieroni, E.; Fais, A.; Kumar, A.; Pintus, F. 2-Phenylbenzofuran derivatives as butyrylcholinesterase inhibitors: Synthesis, biological activity and molecular modeling. Bioorganic Med. Chem. Lett. 2016, 26, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pintus, F.; Di Petrillo, A.; Medda, R.; Caria, P.; Matos, M.J.; Viña, D.; Pieroni, E.; Delogu, F.; Era, B.; et al. Novel 2-pheynlbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci. Rep. 2018, 8, 4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fais, A.; Kumar, A.; Medda, R.; Pintus, F.; Delogu, F.; Matos, M.J.; Era, B.; Delogu, G.L. Synthesis, molecular docking and cholinesterase inhibitory activity of hydroxylated 2-phenylbenzofuran derivatives. Bioorg. Chem. 2019, 84, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.L.; Pintus, F.; Mayán, L.; Matos, M.J.; Vilar, S.; Munín, J.; Fontenla, J.A.; Hripcsak, G.; Borges, F.; Viña, D. MAO inhibitory activity of bromo-2-phenylbenzofurans: Synthesis, in vitro study, and docking calculations. MedChemComm 2017, 8, 1788–1796. [Google Scholar] [CrossRef]
- Ferino, G.; Cadoni, E.; Matos, M.J.; Quezada, E.; Uriarte, E.; Santana, L.; Vilar, S.; Tatonetti, N.P.; Yáñez, M.; Viña, D.; et al. MAO Inhibitory Activity of 2-Arylbenzofurans versus 3-Arylcoumarins: Synthesis, in vitro Study, and Docking Calculations. ChemMedChem 2013, 8, 956–966. [Google Scholar] [CrossRef]
- Delogu, G.L.; Kumar, A.; Gatto, G.; Bustelo, F.; Saavedra, L.M.; Rodríguez-Franco, M.I.; Laguna, R.; Viña, D. Synthesis and in vitro study of nitro- and methoxy-2-phenylbenzofurans as human monoamine oxidase inhibitors. Bioorg. Chem. 2021, 107, 104616. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular pathogenesis of alzheimer’s disease: An update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and neurodegenerative diseases. A systematic review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef]
- Larik, F.A.; Shah, M.S.; Saeed, A.; Shah, H.S.; Channar, P.A.; Bolte, M.; Iqbal, J. New cholinesterase inhibitors for Alzheimer’s disease: Structure activity relationship, kinetics and molecular docking studies of 1–butanoyl–3–arylthiourea derivatives. Int. J. Biol. Macromol. 2018, 116, 144–150. [Google Scholar] [CrossRef]
- Wei, R.; Ma, Q.; Zhong, G. Anti-Ache Benzylbenzofuran Derivatives from Silene conoidea. Chem. Nat. Compd. 2019, 55, 654–657. [Google Scholar] [CrossRef]
- Assoah, B.; Vale, J.R.; Kalenius, E.; Veiros, L.F.; Candeias, N.R. Lewis Base Catalyzed Intramolecular Reduction of Salicylaldehydes by Pinacol-Derived Chlorohydrosilane. Eur. J. Org. Chem. 2018, 2018, 2910–2917. [Google Scholar] [CrossRef]
- Quezada, E.; Delogu, G.; Picciau, C.; Santana, L.; Podda, G.; Borges, F.; García-Morales, V.; Viña, D.; Orallo, F. Synthesis and vasorelaxant and platelet antiaggregatory activities of a new series of 6-Halo-3-phenylcoumarins. Molecules 2010, 15, 270–279. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Kumar, A.; Baccoli, R.; Fais, A.; Cincotti, A.; Pilia, L.; Gatto, G. Substitution effects on the optoelectronic properties of coumarin derivatives. Appl. Sci. 2020, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, A.D.M.J.K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Compounds 9–16(B) | Compounds 9–16(A) | ||
|---|---|---|---|
| IC50 (µM) | |||
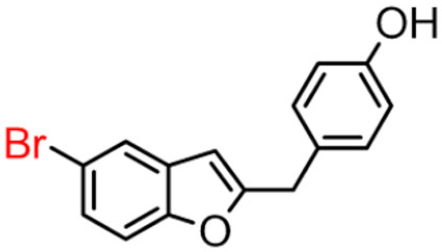 9B | 2.93 ± 0.38 | >100 | 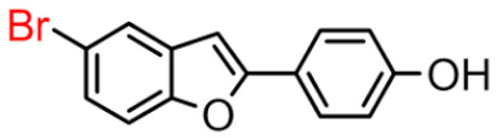 9A [8] |
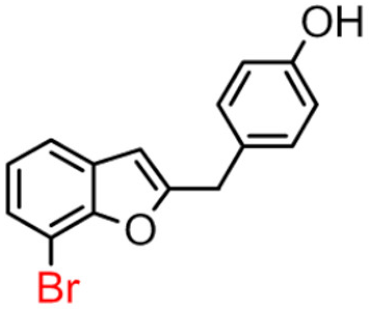 10B | 32.60 ± 2.04 | 82.5 ± 7.1 | 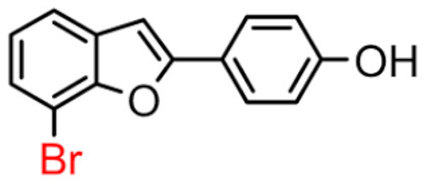 10A [8] |
 11B | 55.66 ± 0.80 | >100 | 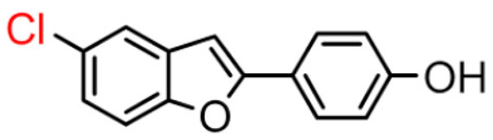 11A [8] |
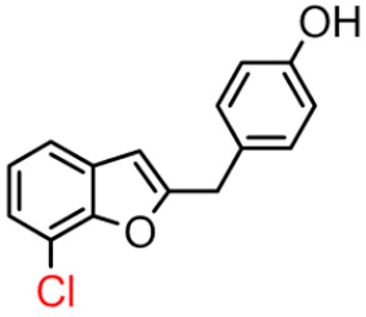 12B | 42.98 ± 5.65 | 30.3 ± 2.1 | 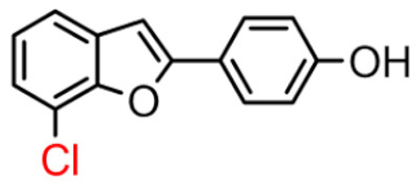 12A [8] |
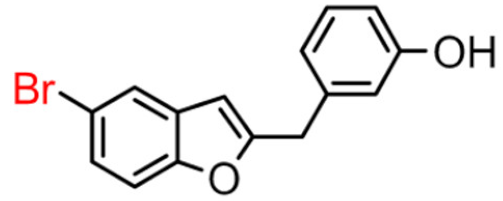 13B | 39.95 ± 4.31 | 10.60 ± 1.65 |  13A |
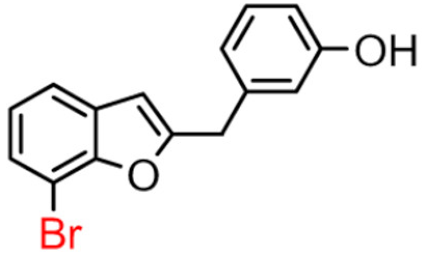 14B | 31.37 ± 0.15 | 7.96 ± 0.28 | 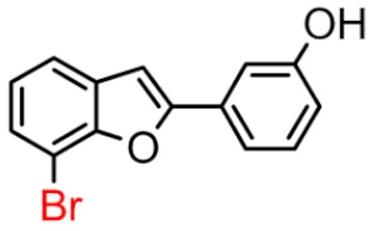 14A [10] |
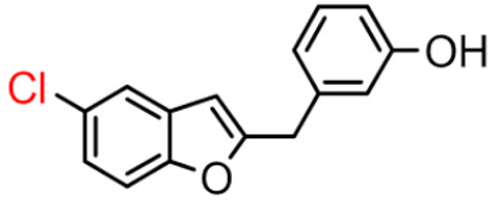 15B | 27.84 ± 3.07 | 10.52 ± 0.40 | 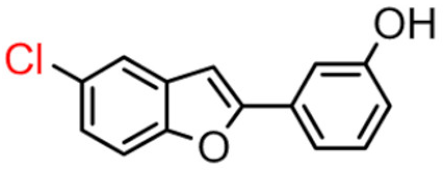 15A |
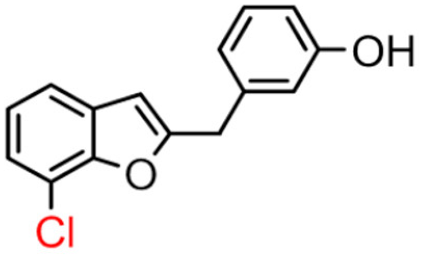 16B | 61.65 ± 5.90 | 13.42 ± 2.57 | 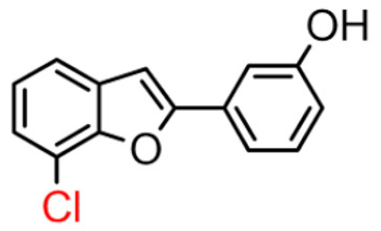 16A [10] |
| Galantamine | 28.06 ± 2.10 | ||
| Compound | Docking Energy (kcal/mol) | IC50 (µM) |
|---|---|---|
| 9A | −7.0 | >100 |
| 9B | −8.8 | 2.9 |
| 10A | −7.5 | 82.5 |
| 10B | −7.8 | 32.6 |
| Physicochemical Properties | Compound | |
|---|---|---|
| Without Spacer 9A/10A | With Spacer 9B/10B | |
| MW (g/mol) | 289.12 | 303.15 |
| Molecular volume (Å3) | 206.91 | 223.72 |
| Rotatable bonds | 1 | 2 |
| H-bond acceptor atoms | 2 | 2 |
| H-bond donor atoms | 1 | 1 |
| TPSA (Å2) | 33.37 | 33.37 |
| Lipophilicity (logPo/w) | 3.75/3.73 | 4.00/3.93 |
| Water solubility | Moderate | Moderate |
| Drug-likeness (Lipinski) | Yes; 0 viol | Yes; 0 viol |
| GI absorption | High | High |
| BBB permeability | Yes | Yes |
| PAINS | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delogu, G.L.; Fais, A.; Pintus, F.; Goyal, C.; Matos, M.J.; Era, B.; Kumar, A. Structural Insight of New Butyrylcholinesterase Inhibitors Based on Benzylbenzofuran Scaffold. Pharmaceuticals 2022, 15, 304. https://doi.org/10.3390/ph15030304
Delogu GL, Fais A, Pintus F, Goyal C, Matos MJ, Era B, Kumar A. Structural Insight of New Butyrylcholinesterase Inhibitors Based on Benzylbenzofuran Scaffold. Pharmaceuticals. 2022; 15(3):304. https://doi.org/10.3390/ph15030304
Chicago/Turabian StyleDelogu, Giovanna L., Antonella Fais, Francesca Pintus, Chinmayi Goyal, Maria J. Matos, Benedetta Era, and Amit Kumar. 2022. "Structural Insight of New Butyrylcholinesterase Inhibitors Based on Benzylbenzofuran Scaffold" Pharmaceuticals 15, no. 3: 304. https://doi.org/10.3390/ph15030304
APA StyleDelogu, G. L., Fais, A., Pintus, F., Goyal, C., Matos, M. J., Era, B., & Kumar, A. (2022). Structural Insight of New Butyrylcholinesterase Inhibitors Based on Benzylbenzofuran Scaffold. Pharmaceuticals, 15(3), 304. https://doi.org/10.3390/ph15030304