
Journey on VX-809-Based Hybrid Derivatives towards Drug-like F508del-CFTR Correctors: From Molecular Modeling to Chemical Synthesis and Biological Assays

 , , , , , , , ,
, , , , , , , , 

Abstract
:1. Introduction
2. Results
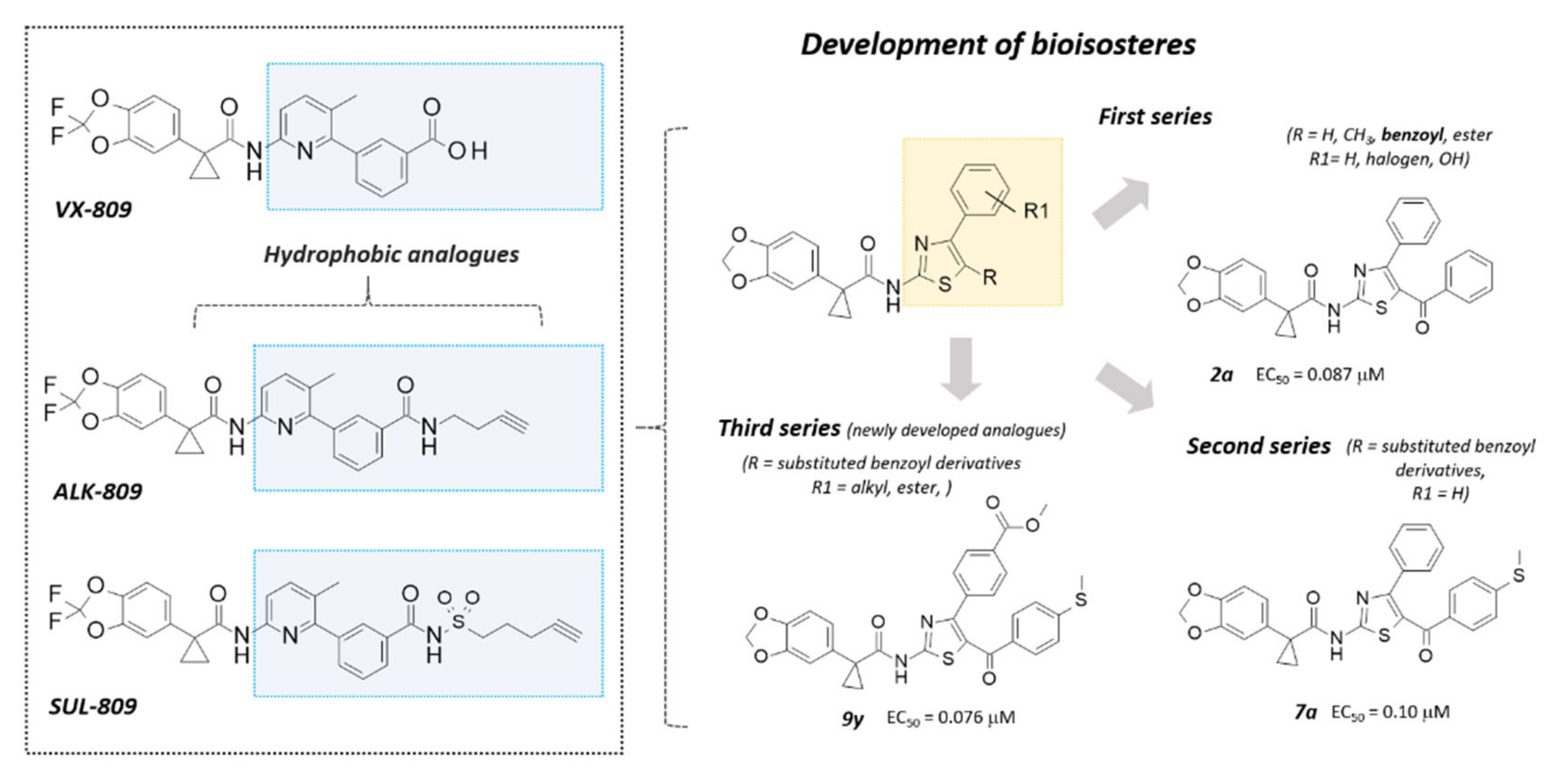
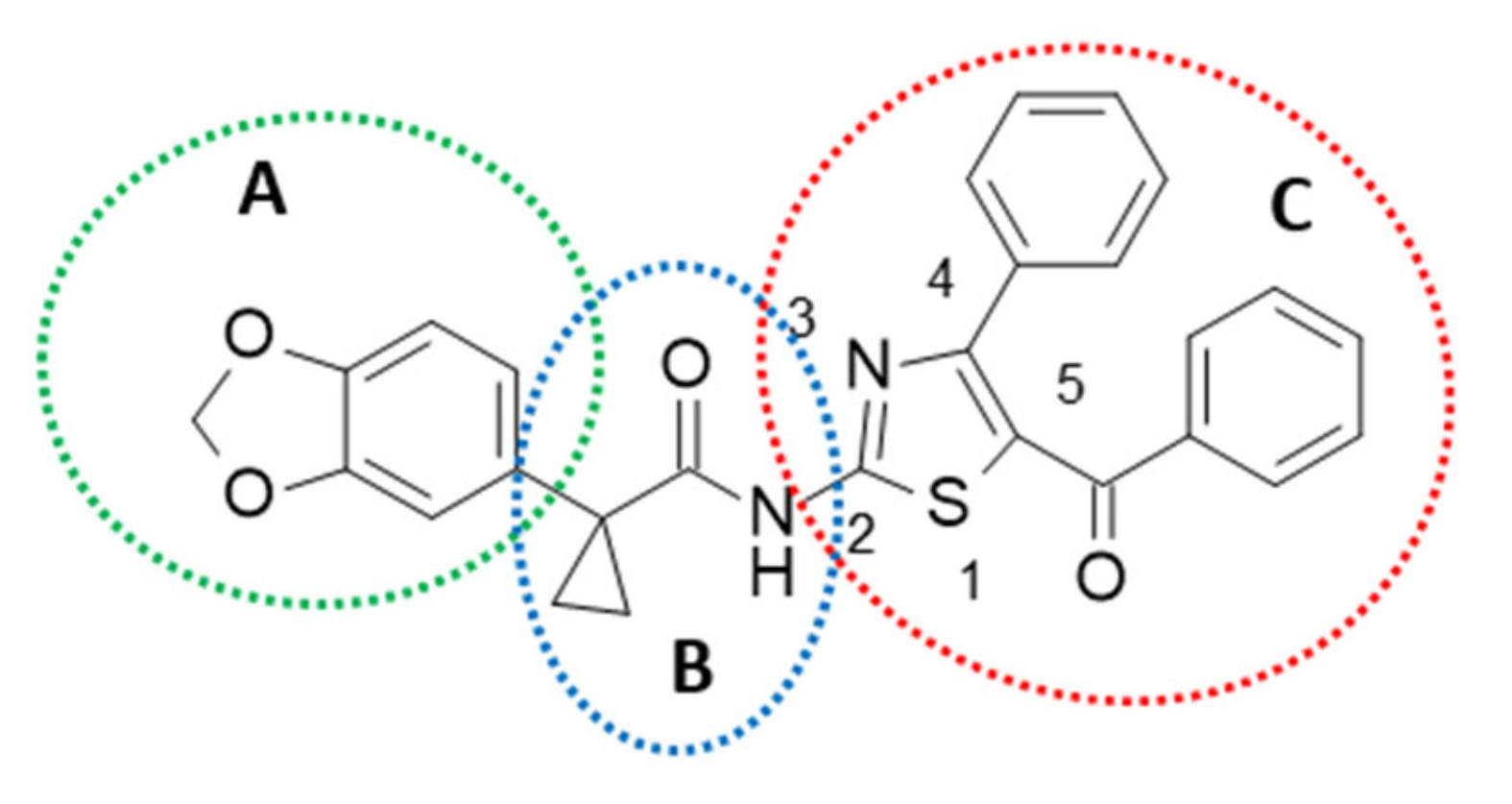
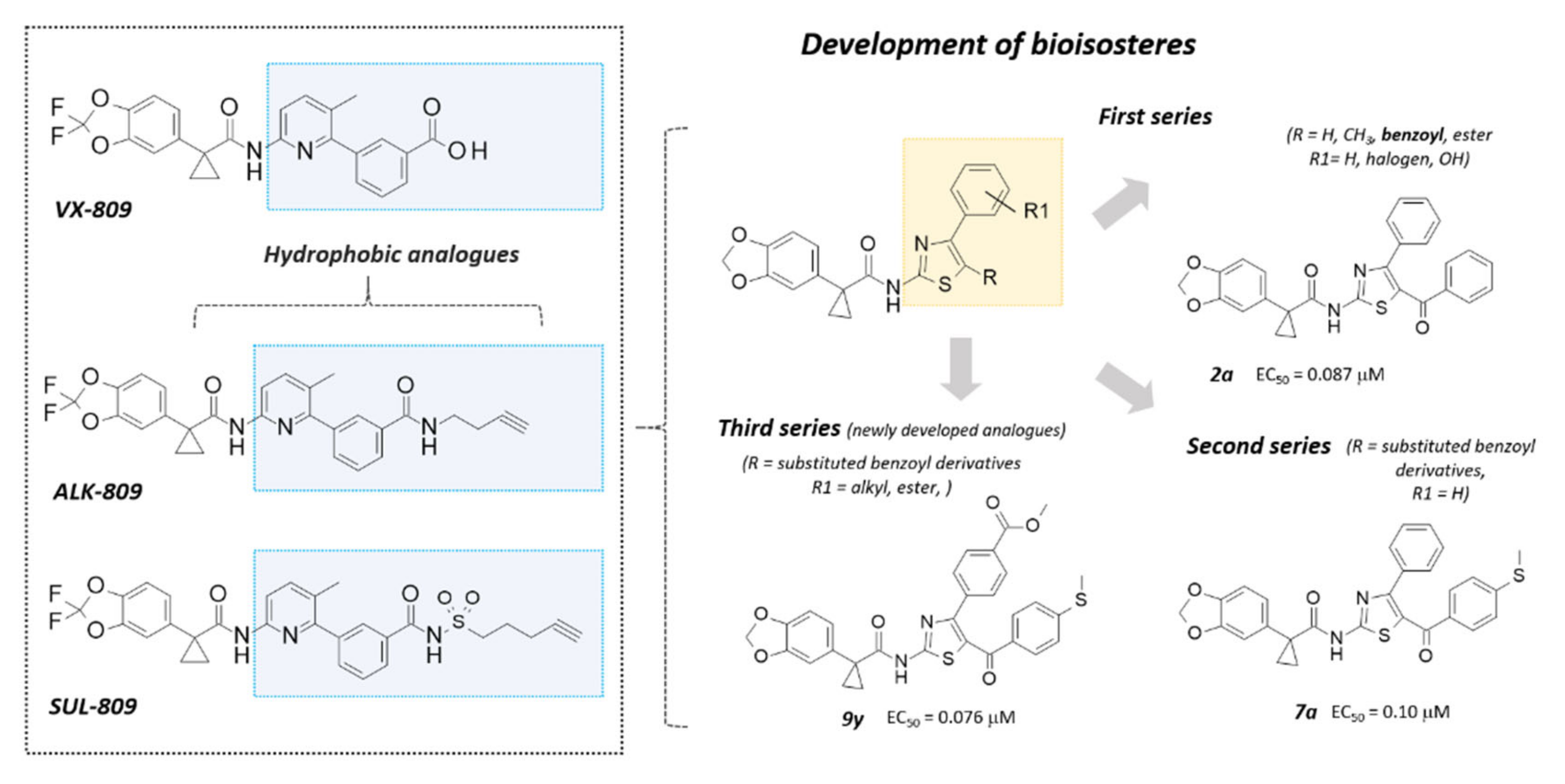
2.1. Structure-Based Studies Guiding the Rational Design of Novel Derivatives
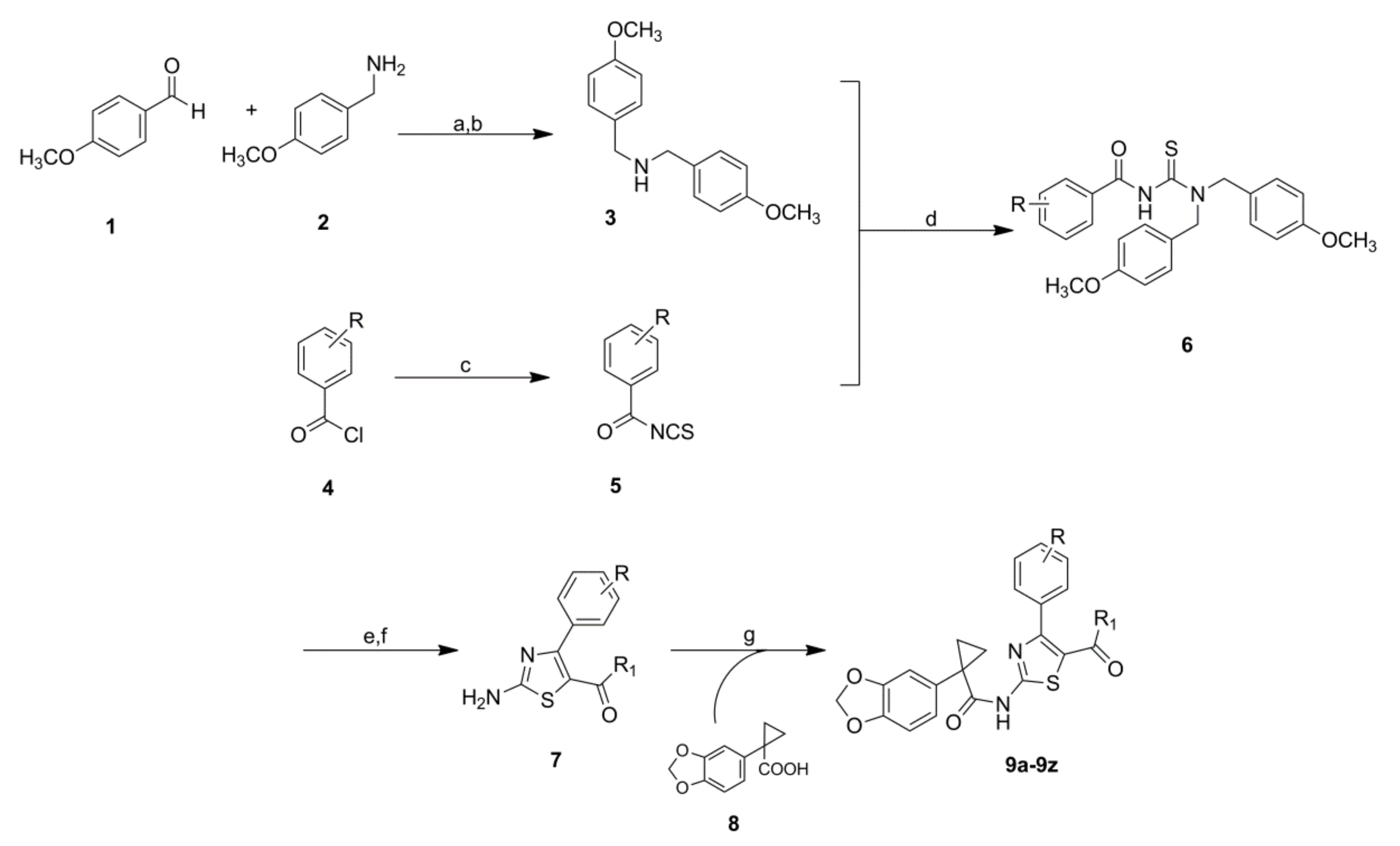
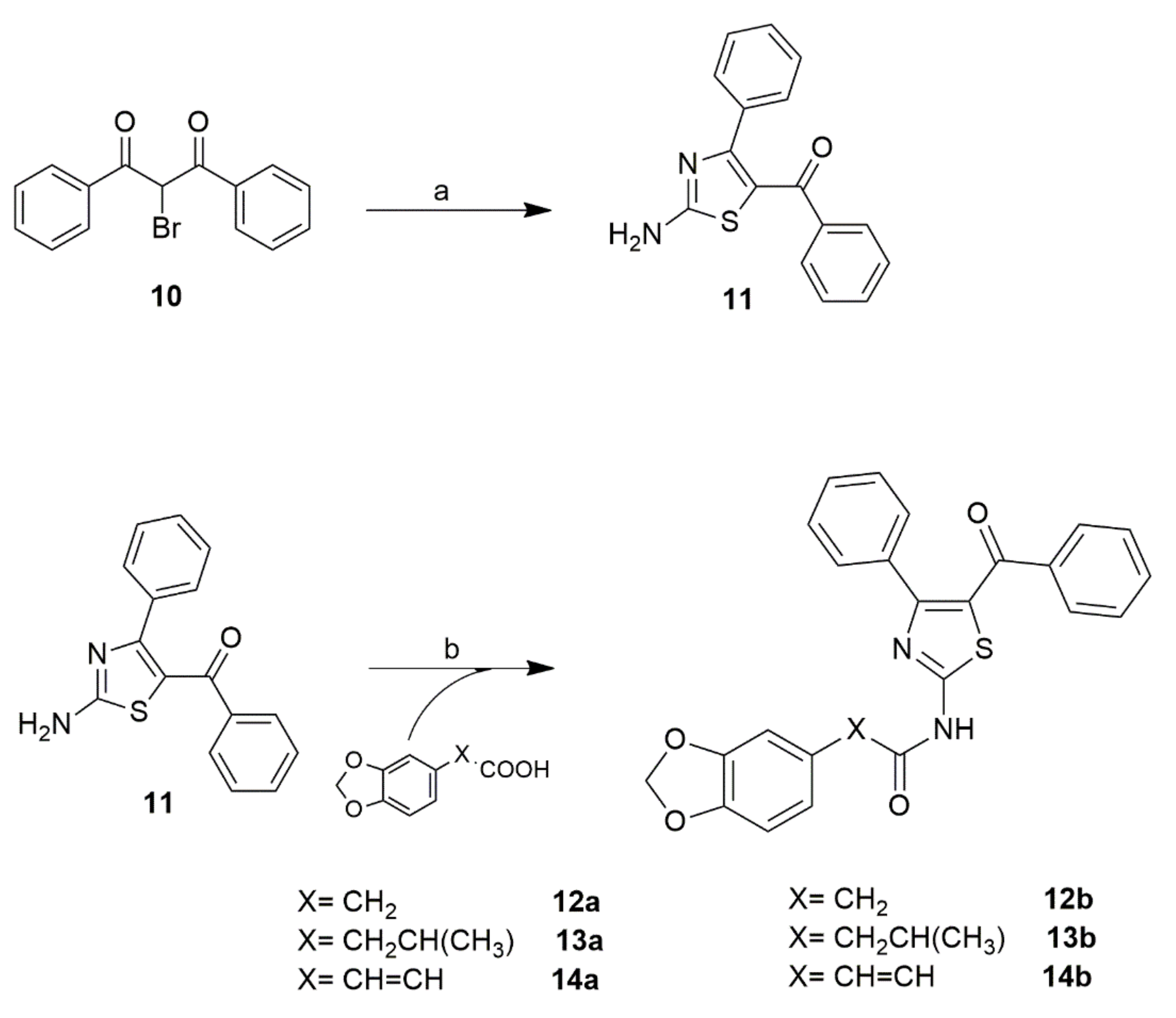
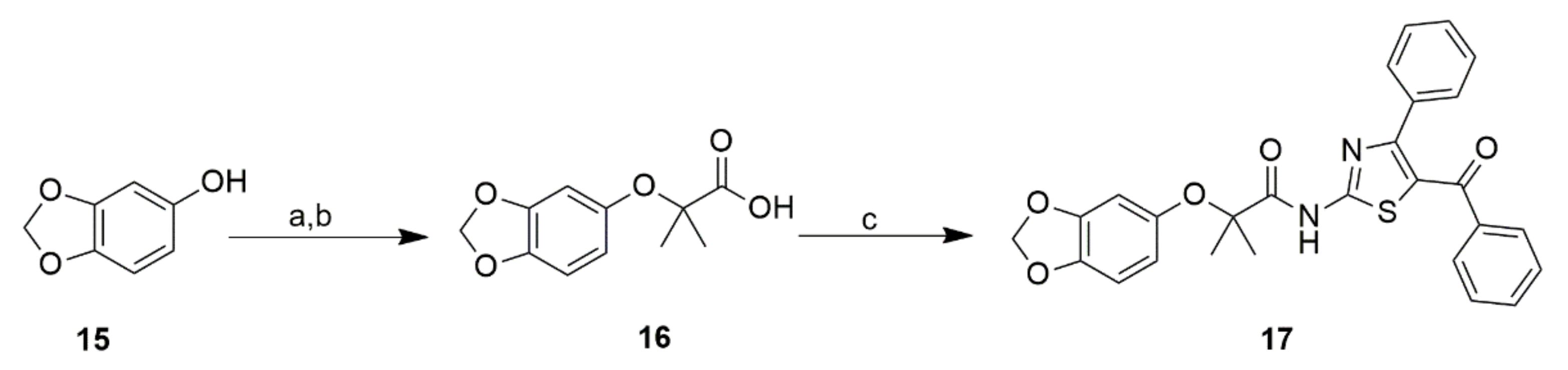
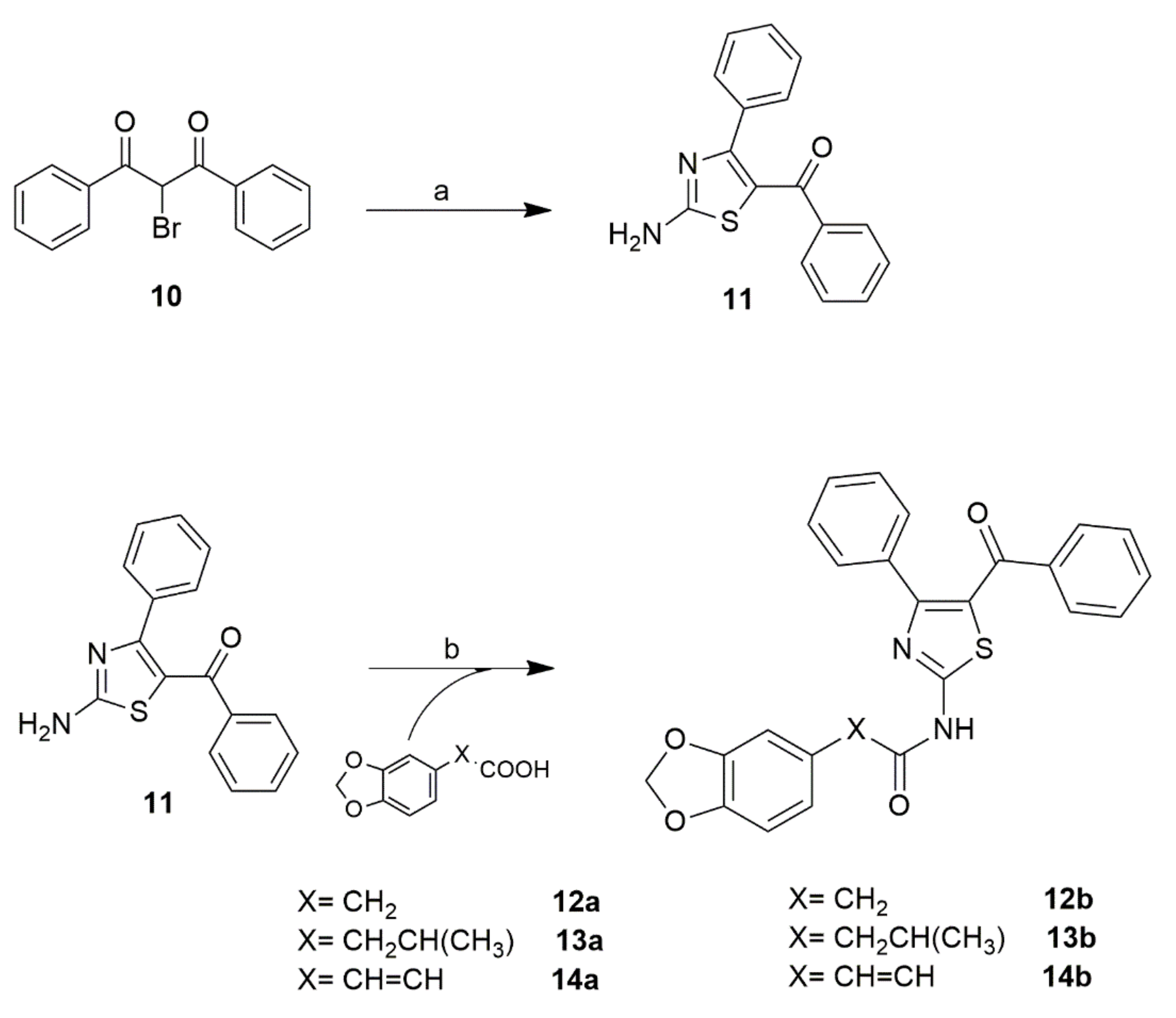
2.2. Chemistry
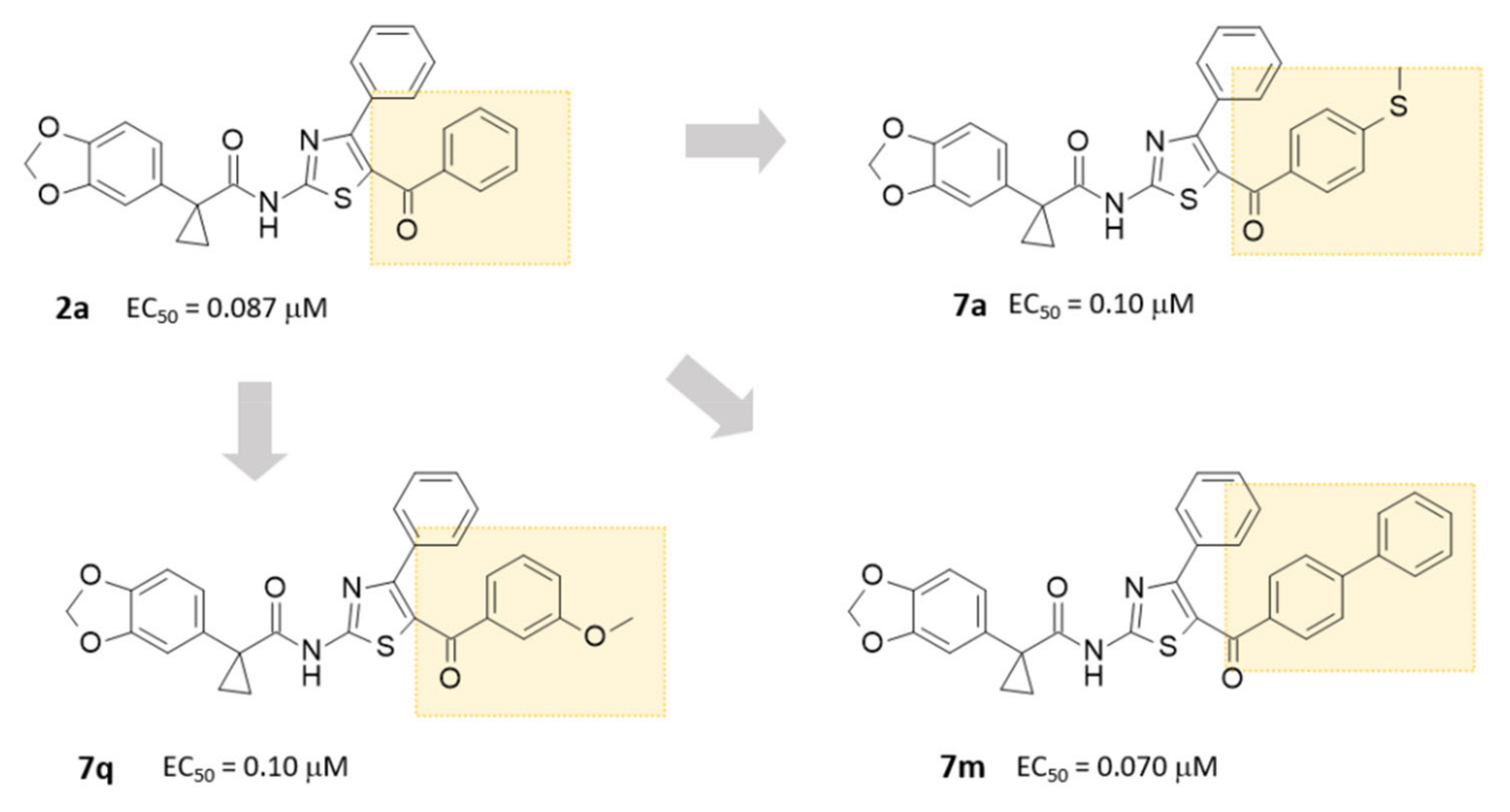
2.3. Structure-Activity Relationship of Hybrids Third Series
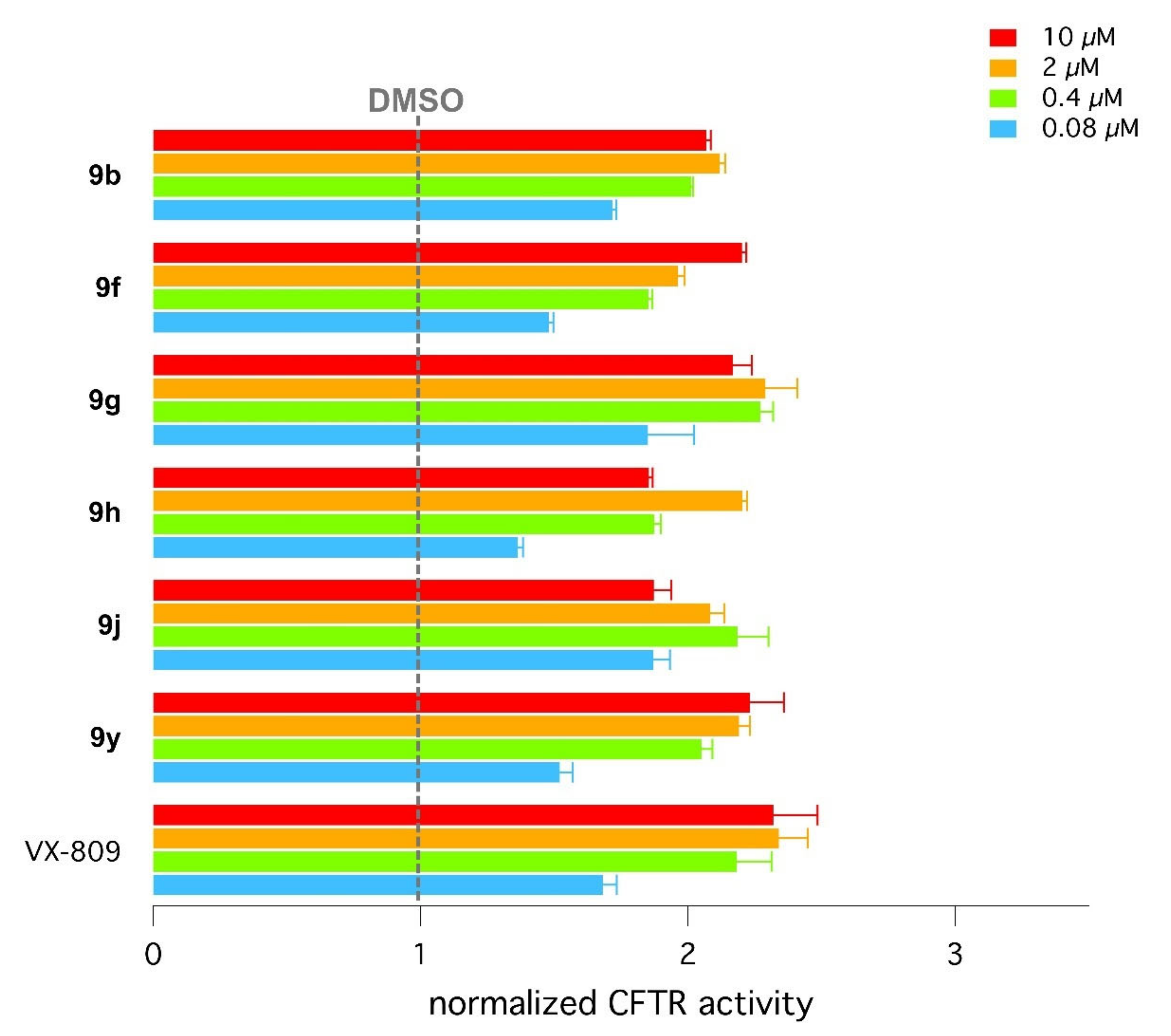
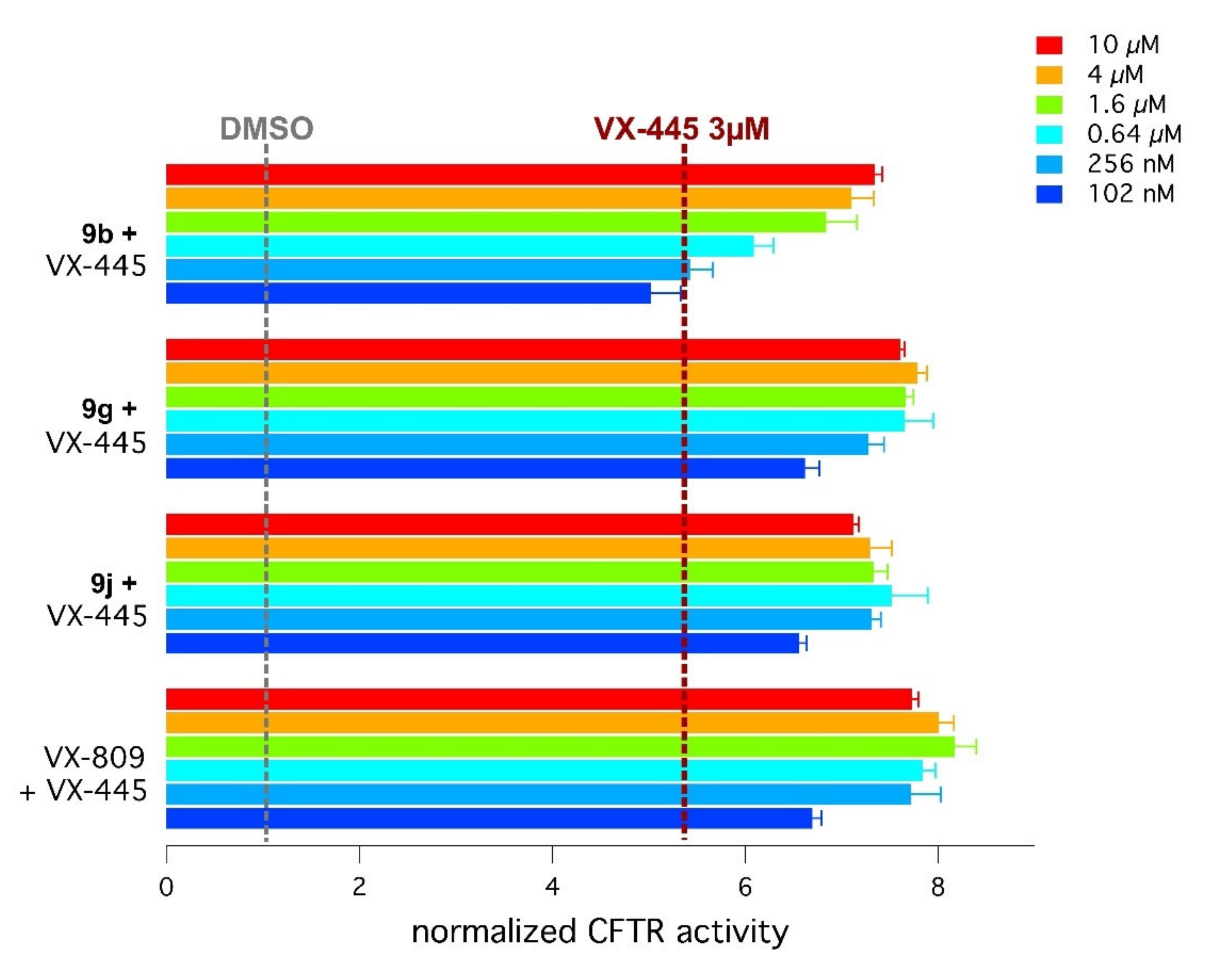
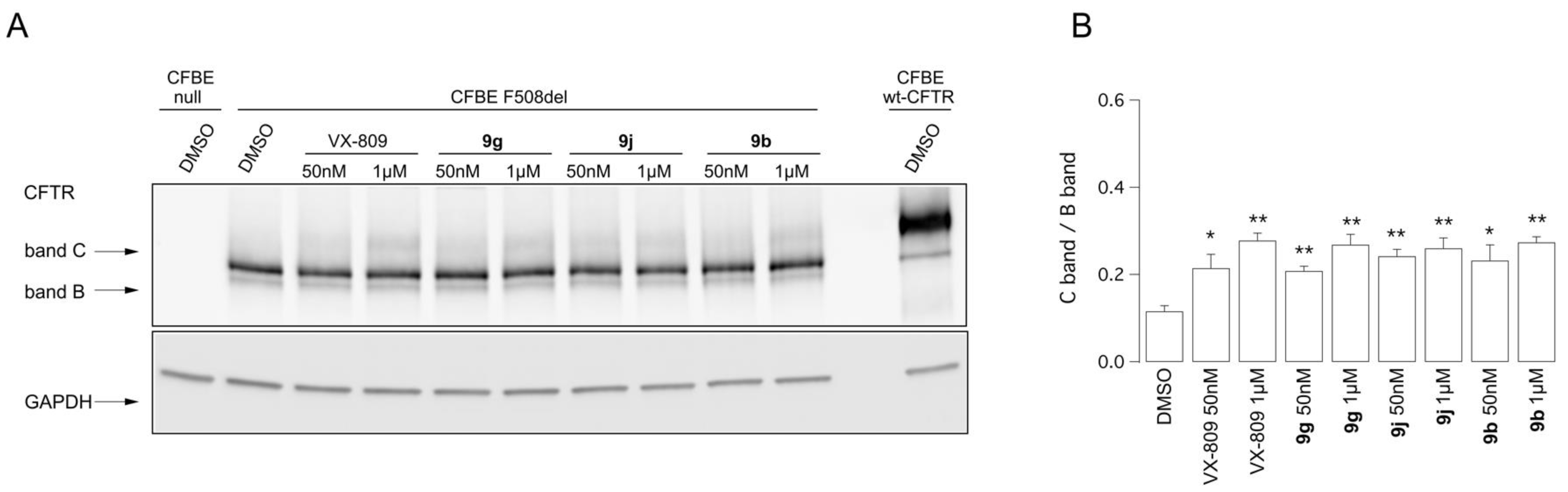
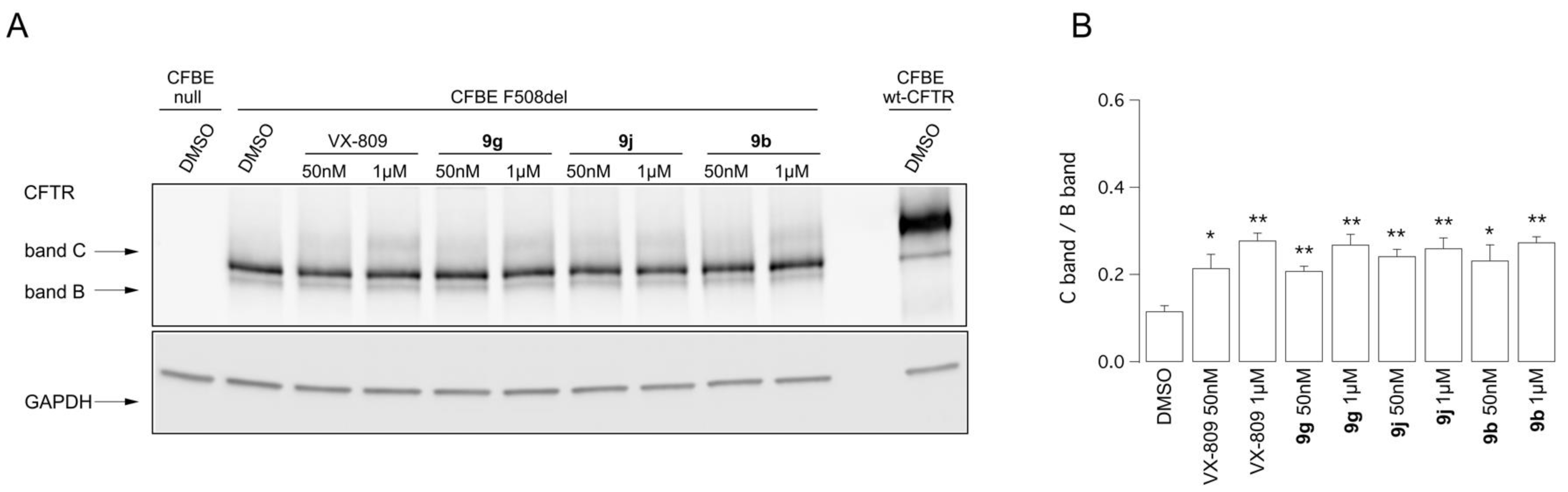
2.4. Biological Assays of Novel Hybrids
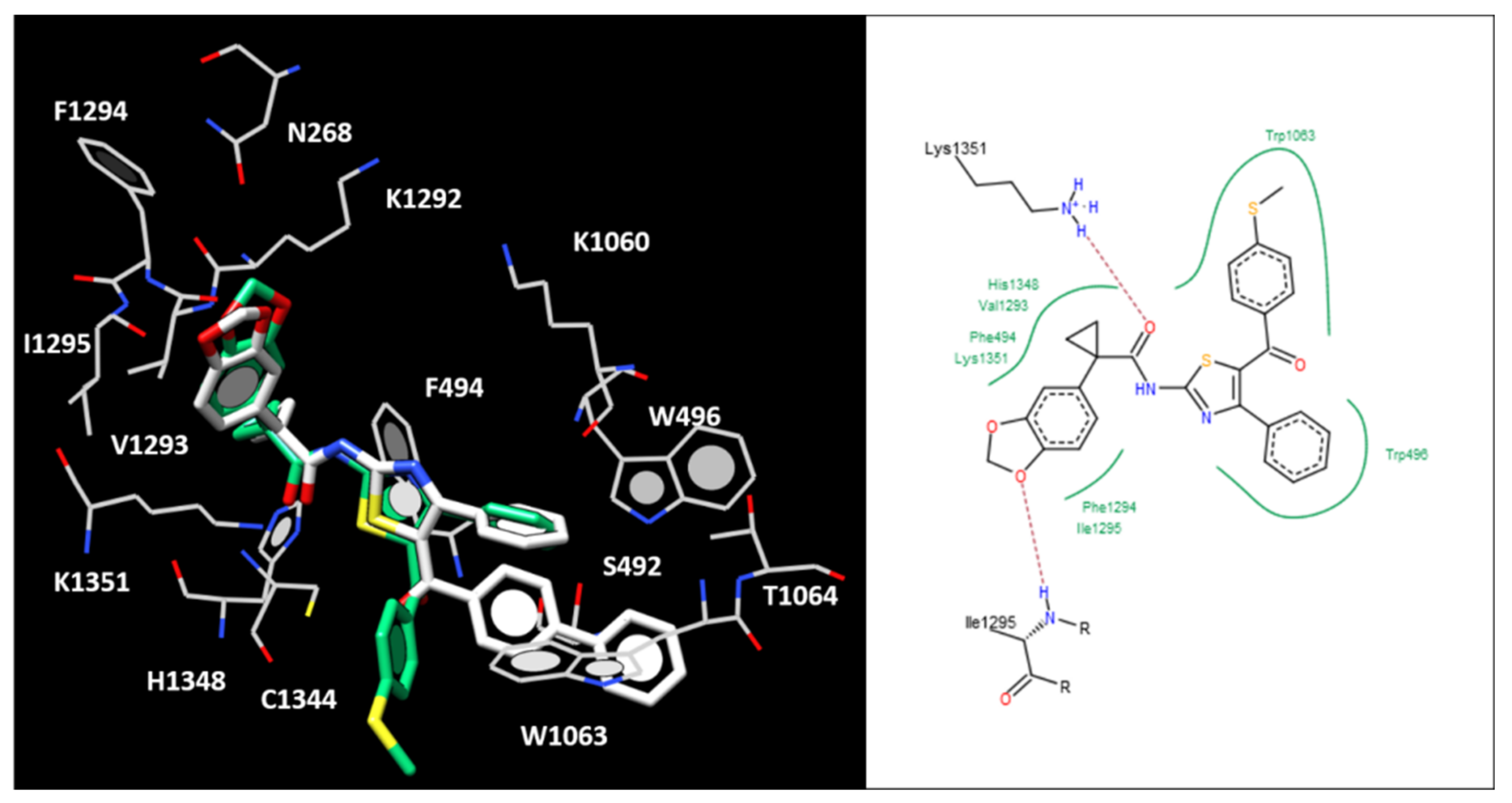
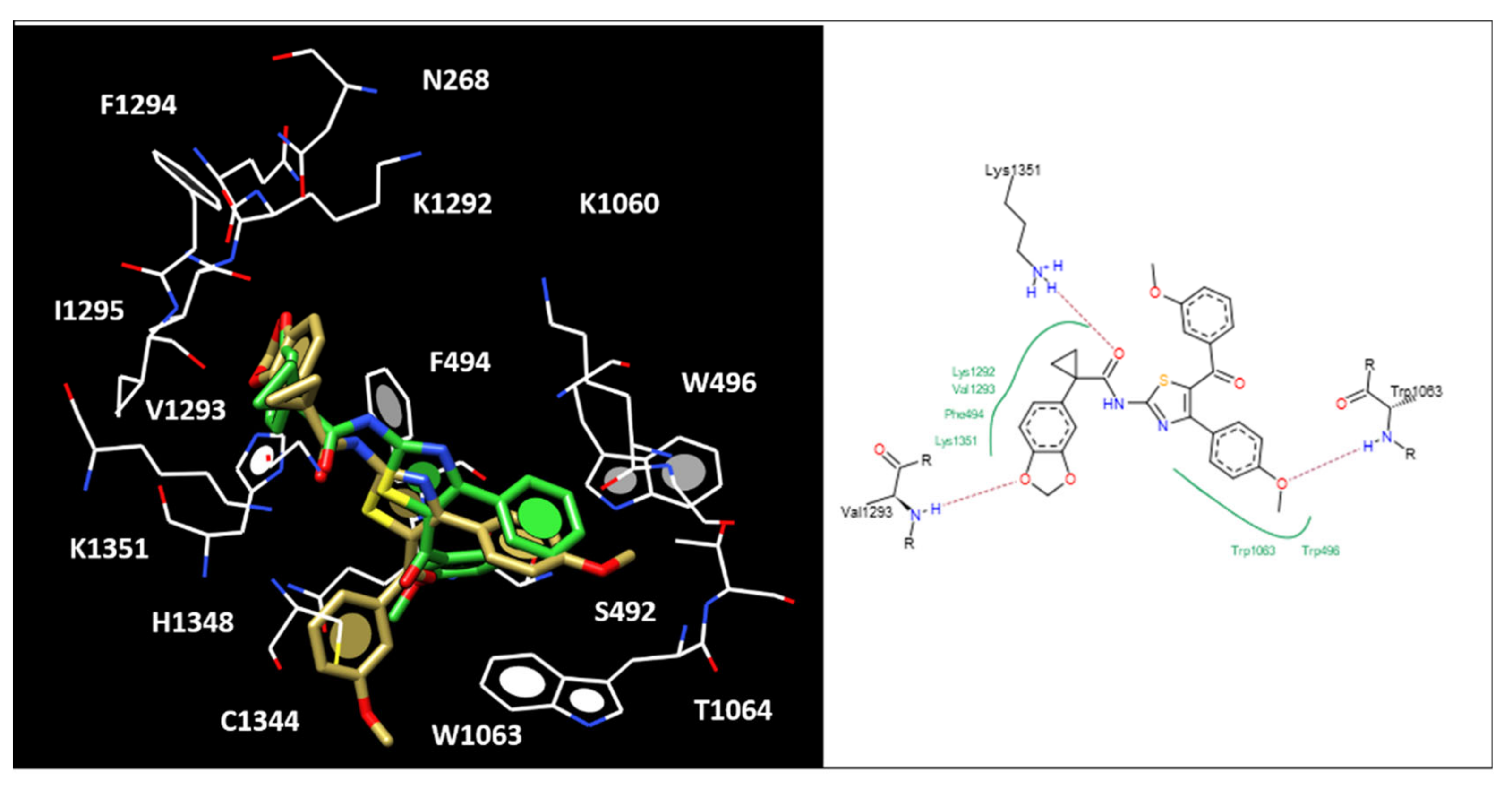
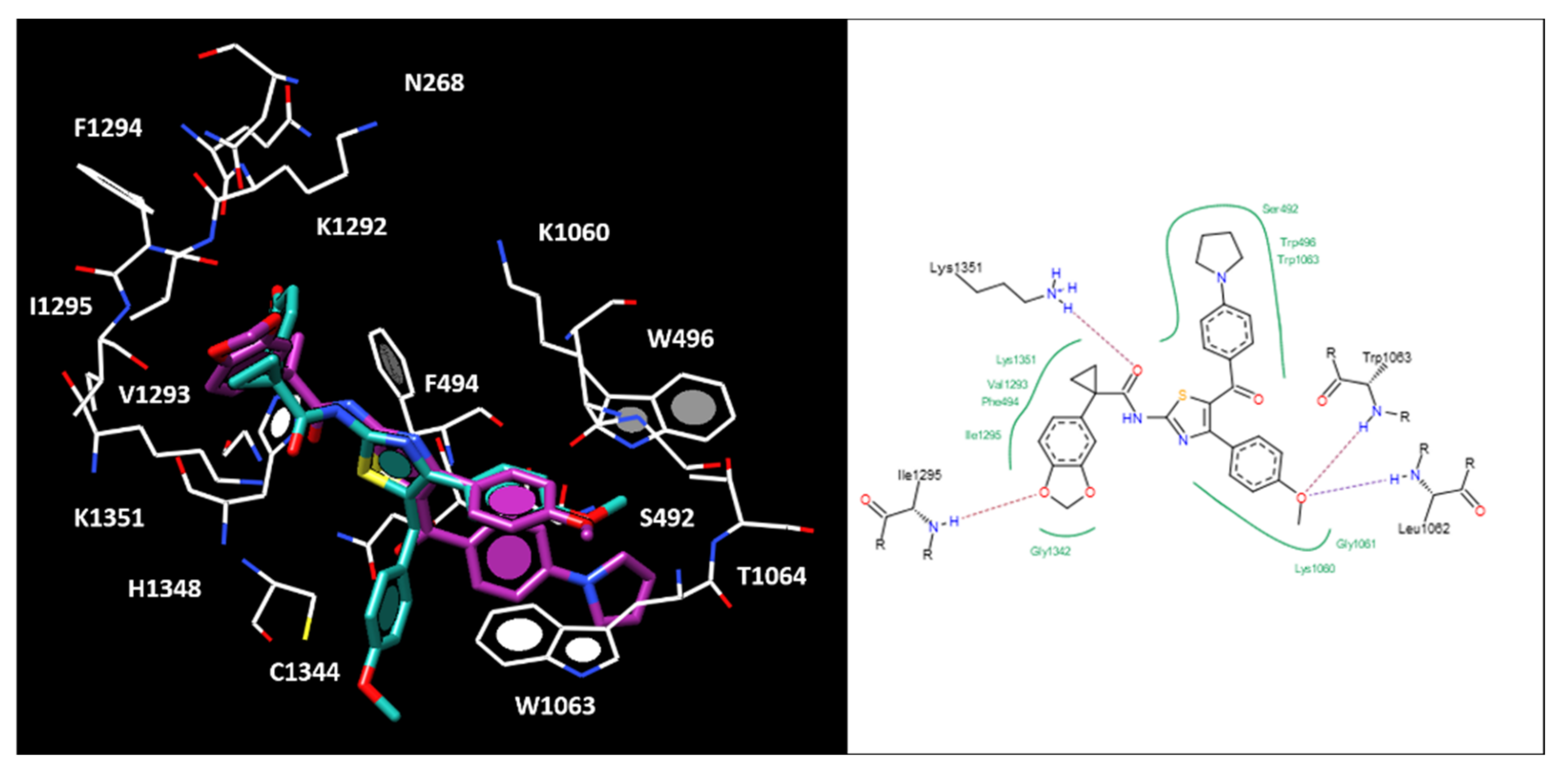
2.5. Molecular Docking of the Most Potent New Hybrids
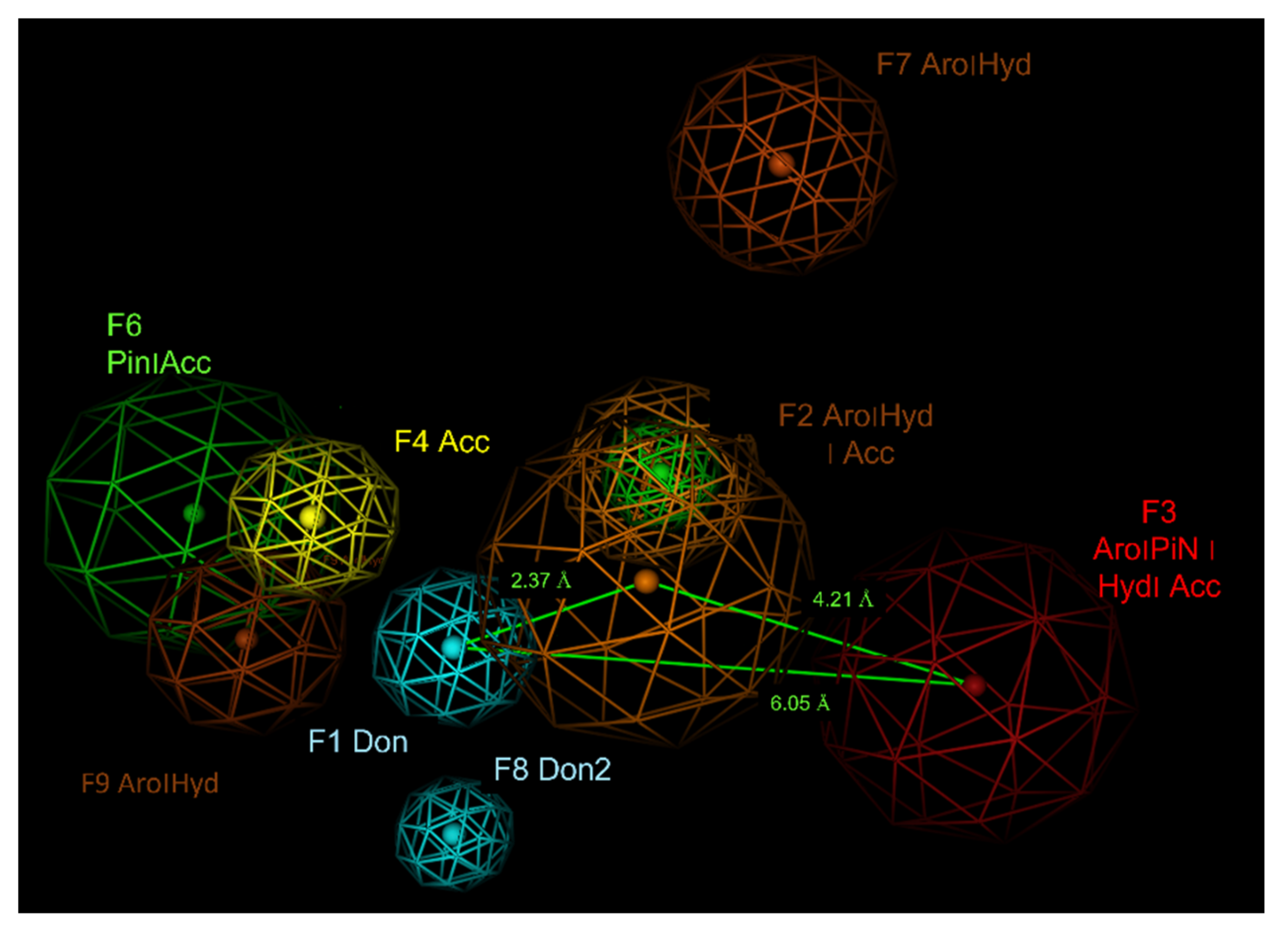
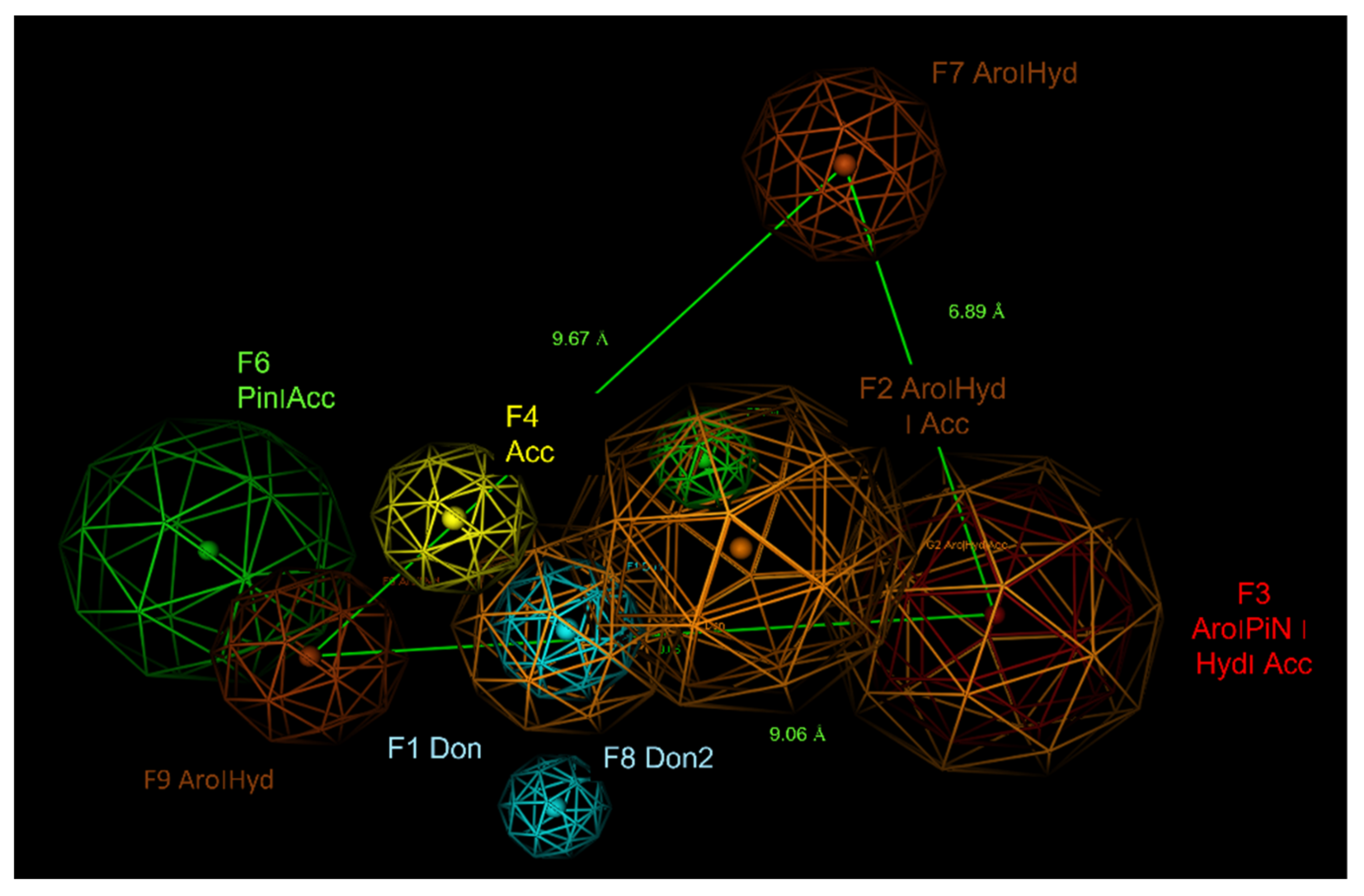
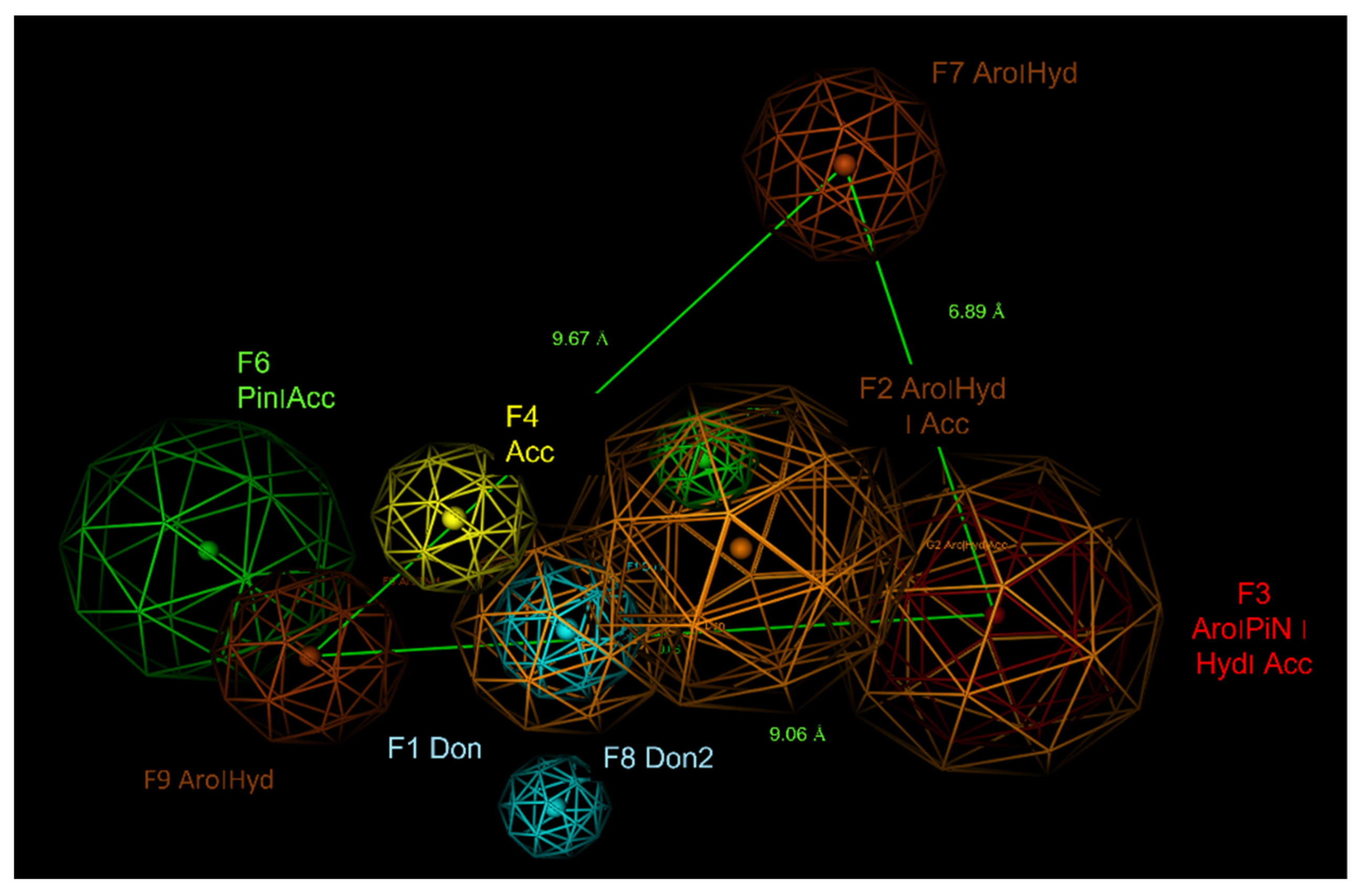
2.6. Pharmacophore Modelling
2.7. In Silico Evaluation of Hybrid Pharmacokinetic Properties
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Experimental Instrumentation
4.1.2. General Procedure for the Synthesis of Structures
4.2. Computational Studies
4.3. Biological Evaluations
4.3.1. Cell Culture
4.3.2. Antibodies
4.3.3. Fluorescence Assay for CFTR Activity
4.3.4. Western Blot Analysis of CFTR Expression Pattern
4.3.5. Short-Circuit Current Recordings
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Kym, P.R.; Wang, X.; Pizzonero, M.; Van der Plas, S.E. Recent Progress in the Discovery and Development of Small-Molecule Modulators of CFTR. Prog. Med. Chem. 2018, 57, 235–276. [Google Scholar] [CrossRef] [PubMed]
- Csanady, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the CFTR anion channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016, 27, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Bobadilla, J.L.; Macek, M., Jr.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations--Correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef]
- Ward, C.L.; Krouse, M.E.; Gruenert, D.C.; Kopito, R.R.; Wine, J.J. Cystic Fibrosis Gene Expression Is Not Correlated with Rectifying Cl- Channels. Proc. Natl. Acad. Sci. USA 1991, 88, 5277–5281. [Google Scholar] [CrossRef] [Green Version]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered Chloride Ion Channel Kinetics Associated with the Delta F508 Cystic Fibrosis Mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the conformational defect ΔF508. Mol. Med. Trends 2012, 18, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.W.; Liu, J.; Li, H.Y.; Sheppard, D.N. Targeting F508del-CFTR to develop new rational therapies for cystic fibrosis. Acta Pharmacol. Sin. 2011, 32, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, B.; Searle, X.; Yeung, C.; Bogdan, A.; Greszler, S.; Singh, A.; Fan, Y.; Swensen, A.M.; Vortherms, T.; et al. Discovery of 4-[(2R,4R)-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic acid (ABBV/GLPG-2222), a potent cystic fibrosis transmembrane conductance regulator (CFTR) corrector for the Treatment of Cystic Fibrosis. J. Med. Chem. 2018, 61, 1436–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jih, K.Y.; Hwang, T.C. VX-770 Potentiates CFTR Function by Promoting Decoupling between the Gating Cycle and ATP Hydrolysis Cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, M.D.; Farinha, C.M. CFTR mutant rescue: A multi-task approach for a better result in the treatment of cystic fibrosis. Curr. Pharm. Des. 2013, 19, 3497–3508. [Google Scholar] [CrossRef] [Green Version]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Proteostasis adaptation for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores DF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature and correctors reveal the rescue mechanism of F508del-CFTR via VX-809 and suggest multiple agents for a complete correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some gating potentiators, including VX-770, diminish DeltaF508- CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra97. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, B.W.; Welsh, M.J. Progress along the path of discovery that leads to the treatment and cure of cystic fibrosis. Am. J. Resp. Critical Care. Med. 2017, 195, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Fajac, I.; Wainwright, C.E. New treatments aimed at the basic defects of cystic fibrosis. Presse Med. 2017, 46, e165–e175. [Google Scholar] [CrossRef]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX16-659-101 Study Group, VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX16-445-001 Study Group, VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Caci, E.; Millo, E.; Armirotti, A.; Damonte, G.; Zegarra-Moran, O.; Galietta, L.J.V. Dual activity of aminoarylthiazoles on the trafficking and gating defects of the cystic fibrosis transmembrane conductance regulator chloride channel caused by cystic fibrosis mutations. J. Biol. Chem. 2011, 286, 15215–15226. [Google Scholar] [CrossRef] [Green Version]
- Pesce, E.; Bellotti, M.; Liessi, N.; Guariento, S.; Damonte, G.; Cichero, E.; Galatini, A.; Salis, A.; Gianotti, A.; Pedemonte, N.; et al. Synthesis and structure-activity relationship of aminoarylthiazole derivatives as correctors of the chloride transport defect in cystic fibrosis. Eur. J. Med. Chem. 2015, 99, 14–35. [Google Scholar] [CrossRef]
- Liessi, N.; Cichero, E.; Pesce, E.; Arkel, M.; Salis, A.; Tomati, V.; Paccagnella, M.; Damonte, G.; Tasso, B.; Galietta, L.J.V.; et al. Synthesis and biological evaluation of novel thiazole- VX-809 hybrid derivatives as F508del correctors by QSAR-based filtering tools. Eur. J. Med. Chem. 2018, 144, 179–200. [Google Scholar] [CrossRef]
- Parodi, A.; Righetti, G.; Pesce, E.; Salis, A.; Tasso, B.; Urbinati, C.; Tomati, V.; Damonte, G.; Rusnati, M.; Pedemonte, N.; et al. Discovery of novel VX-809 hybrid derivatives as F508del-CFTR correctors by molecular modeling, chemical synthesis and biological assays. Eur. J. Med. Chem. 2020, 208, 112833. [Google Scholar] [CrossRef]
- Righetti, G.; Casale, M.; Liessi, N.; Tasso, B.; Salis, A.; Tonelli, M.; Millo, E.; Pedemonte, N.; Fossa, P.; Cichero, E. Molecular Docking and QSAR Studies as Computational Tools Exploring the Rescue Ability of F508del CFTR Correctors. Int. J. Mol. Sci. 2020, 21, 8084. [Google Scholar] [CrossRef]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ursi, P.; Uggeri, M.; Urbinati, C.; Millo, E.; Paiardi, G.; Milanesi, L.; Ford, R.C.; Clews, J.; Meng, X.; Bergese, P.; et al. Exploitation of a novel biosensor based on the full-length human F508de1-CFTR with computational studies, biochemical and biological assays for the characterization of a new Lumacaftor/Tezacaftor analogue. Sens. Actuators B Chem. 2019, 301, 127131. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cai, W.; Zhang, G.; Yang, T.; Liu, Q.; Cheng, Y.; Zhou, L.; Ma, Y.; Cheng, Z.; Lu, S.; et al. Discovery of novel N-(5-(arylcarbonyl)thiazol-2-yl)amides and N-(5-(arylcarbonyl)thiophen-2-yl)amides as potent RORγt inhibitors. Bioorg. Med. Chem. 2014, 22, 692–702. [Google Scholar] [CrossRef]
- Doiron, J.E.; Le, C.A.; Ody, B.K.; Brace, J.B.; Post, S.J.; Thacker, N.L.; Hill, H.M.; Breton, G.W.; Mulder, M.J.; Chang, S.; et al. Evaluation of 1,2,3-Triazoles as Amide Bioisosteres In Cystic Fibrosis Transmembrane Conductance Regulator Modulators VX-770 and VX-809. Chem. Eur. J. 2019, 25, 3662–3674. [Google Scholar] [CrossRef] [PubMed]
- Kym, P.R.; Wang, X.; Searle, X.B.; Liu, B.; Yeung, M.C. Preparation of Substituted Tetrahydropyrans as Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators. US Patent US2016012, 5 May 2016. [Google Scholar]
- Eckford, P.D.; Ramjeesingh, M.; Molinski, S.; Pasyk, S.; Dekkers, J.F.; Li, C.; Ahmadi, S.; Ip, W.; Chung, T.E.; Du, K.; et al. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol. 2014, 21, 666–678. [Google Scholar] [CrossRef] [Green Version]
- Francesconi, V.; Cichero, E.; Kanov, E.V.; Laurini, E.; Pricl, S.; Gainetdinov, R.R.; Tonelli, M. Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human Taar1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals 2020, 13, 391. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Laube, M.; Gassner, C.; Sharma, S.K.; Cunther, R.; Pigorsch, A.; Konig, J.; Kockerling, M.; Wuest, F.; Pietzsch, J.; Kniess, T. Diaryl-Substituted (Dihydro)pyrrolo[3,2,1-hi]indoles, a Class of Potent COX-2 Inhibitors with Tricyclic Core Structure. J. Org. Chem. 2015, 80, 5611–5624. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Böhm, H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichmann, L.; Wang, Y.-T.; Fischer, W.B. Docking assay of small molecule antivirals to p7 of HCV. Comput. Biol. Chem. 2014, 53, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C.; et al. Substantial improvements in large-scale redocking and screening using the novel HYDEscoring function. J. Comput. Aided Mol. Des. 2012, 26, 701–723. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Calautti, A.; Francesconi, V.; Tonelli, M.; Schenone, S.; Fossa, P. Probing In Silico the Benzimidazole Privileged Scaffold for the Development of Drug-like Anti-RSV Agents. Pharmaceuticals 2021, 14, 1307. [Google Scholar] [CrossRef]
- Righetti, G.; Casale, M.; Tonelli, M.; Liessi, N.; Fossa, P.; Pedemonte, N.; Millo, E.; Cichero, E. New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals 2020, 13, 445. [Google Scholar] [CrossRef]
- Scudieri, P.; Caci, E.; Bruno, S.; Ferrera, L.; Schiavon, M.; Sondo, E.; Tomati, V.; Gianotti, A.; Zegarra-Moran, O.; Pedemonte, N.; et al. Association of TMEM16A chloride channel overexpression with airway goblet cell metaplasia. J. Physiol. 2012, 590, 6141–6155. [Google Scholar] [CrossRef]
- Cui, L.; Aleksandrov, L.; Chang, X.B.; Hou, Y.X.; He, L.; Hegedus, T.; Gentzsch, M.; Aleksandrov, A.; Balch, W.E.; Riordan, J.R. Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 2007, 365, 981–994. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | EC50 (µM) |
|---|---|---|
| 12b | 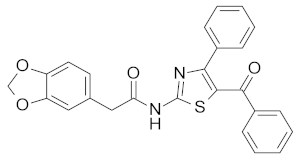 | NA |
| 13b |  | NA |
| 14b | 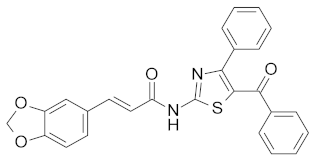 | NA |
| 17 | 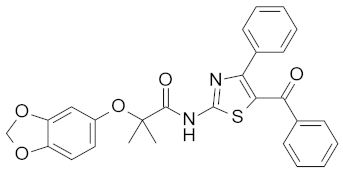 | NA |
| 18 | 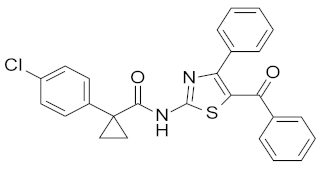 | NA |

| Compound | R | R1 | EC50 (µM) |
|---|---|---|---|
| 9a | 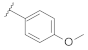 | 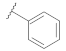 | 0.35 |
| 9b | | | 0.064 |
| 9c | | 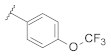 | 0.14 |
| 9d | | 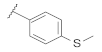 | 0.10 |
| 9e | | 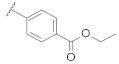 | 0.56 |
| 9f | | 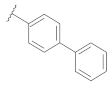 | 0.14 |
| 9g | | 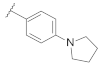 | 0.033 |
| 9h | |  | 0.12 |
| 9i | | | 0.19 |
| 9j | | 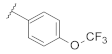 | 0.037 |
| 9k | | | 0.36 |
| 9l | | | 0.49 |
| 9m | | | 0.74 |
| 9n | | | 0.6 |
| 9o | | | 0.21 |
| 9p | 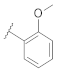 | | 0.22 |
| 9q | | | 0.15 |
| 9r | 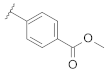 | | 0.12 |
| 9s | | | 0.13 |
| 9t | 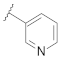 | | 0.13 |
| 9u | 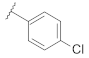 | | 0.64 |
| 9v | | | 0.28 |
| 9w | | | 1.52 |
| 9x | | | 0.37 |
| 9y | | | 0.076 |
| 9z | | | 0.30 |
| ID | Score | Radius (Å) | Expression |
|---|---|---|---|
| F1 | 100% | 1.39 | Don |
| F2 | 100% | 2.21 | Aro|Hyd|Acc |
| F3 | 100% | 2.35 | Aro|PiN|Hyd|Acc |
| F4 | 94% | 1.36 | Acc |
| F5 | 88% | 1.20 | PiN |
| F6 | 84% | 2.22 | PiN|Acc |
| F7 | 81 | 1.19 | Aro|Hyd| |
| F8 | 81 | 1.19 | Don2 |
| F9 | 81 | 1.53 | Aro|Hyd| |
| Compound | cLogP a | MW b | HBA c | HBD d | nRot_bond e | HIA f (%) | Vd g(l/kg) b | %PPB h | LogKaHSA i,d | %F l (oral) e |
|---|---|---|---|---|---|---|---|---|---|---|
| VX-809 | 3.97 | 452.41 | 7 | 2 | 5 | 100 | 0.31 | 98 | 4.26 | 99.6 |
| VX-661 | 2.55 | 520.5 | 8 | 4 | 8 | 100 | 2.3 | 93 | 3.38 | 97.2 |
| VX-445 | 3.91 | 597.65 | 11 | 1 | 9 | 100 | 0.26 | 99 | 5.25 | 99.1 |
| 2a | 3.85 | 468.52 | 6 | 1 | 6 | 100 | 3.1 | 100 | 5.01 | 56 |
| 2b | 4.25 | 515.38 | 7 | 1 | 7 | 100 | 3.1 | 97 | 4.16 | 69.7 |
| 2c | 3.87 | 470.93 | 7 | 1 | 7 | 100 | 3.8 | 98 | 4.32 | 71.8 |
| 2d | 3.87 | 470.93 | 7 | 1 | 7 | 100 | 2.9 | 98 | 4.32 | 71.8 |
| 3b | 4.01 | 457.34 | 5 | 1 | 4 | 100 | 3 | 97 | 4.18 | 78 |
| 3e | 3.03 | 396.44 | 5 | 1 | 4 | 100 | 2.8 | 95 | 4.09 | 92.4 |
| 4d | 3.46 | 490.32 | 5 | 1 | 4 | 100 | 2.9 | 96 | 4.29 | 79.7 |
| 5b | 2.59 | 380.42 | 6 | 2 | 4 | 100 | 2.2 | 95 | 3.8 | 99 |
| 5c | 2.62 | 394.44 | 6 | 2 | 4 | 100 | 2.4 | 95 | 3.82 | 98.6 |
| 6a | 4.17 | 493.32 | 5 | 1 | 4 | 100 | 4.3 | 98 | 4.21 | 84.7 |
| 6c | 2.97 | 416.4 | 6 | 2 | 4 | 100 | 2.7 | 96 | 3.86 | 98.7 |
| 7a | 3.96 | 514.62 | 6 | 1 | 7 | 100 | 4.2 | 100 | 5 | 45.2 |
| 7h | 3.86 | 498.55 | 7 | 1 | 7 | 100 | 3.1 | 100 | 5.02 | 50.8 |
| 7j | 4.58 | 526.6 | 7 | 1 | 9 | 100 | 4.5 | 99 | 5.06 | 36.6 |
| 7m | 5.27 | 544.62 | 6 | 1 | 7 | 100 | 5.1 | 100 | 5.76 | 13.5 |
| 7n | 3.83 | 537.63 | 7 | 1 | 7 | 100 | 4.5 | 100 | 5.1 | 68.1 |
| 7q | 3.86 | 498.55 | 7 | 1 | 7 | 100 | 3.2 | 100 | 5.02 | 50.8 |
| 9a | 3.86 | 498.55 | 7 | 1 | 7 | 100 | 3.1 | 100 | 5.02 | 50.8 |
| 9b | 3.53 | 528.58 | 8 | 1 | 8 | 100 | 2.9 | 99 | 5.04 | 61.3 |
| 9d | 4.99 | 544.64 | 7 | 1 | 8 | 100 | 4.1 | 100 | 4.99 | 36.6 |
| 9g | 3.82 | 567.66 | 8 | 1 | 8 | 100 | 4.5 | 99 | 5.01 | 64.6 |
| 9f | 5.76 | 574.65 | 7 | 1 | 8 | 100 | 5 | 100 | 5.76 | 11 |
| 9j | 4.06 | 582.55 | 8 | 1 | 9 | 100 | 4.3 | 100 | 5.25 | 61.4 |
| 9q | 4.06 | 582.55 | 8 | 1 | 9 | 100 | 4.4 | 100 | 5.25 | 61.4 |
| 9s | 5.26 | 590.71 | 6 | 1 | 8 | 100 | 5.4 | 100 | 5.74 | 7 |
| 9t | 4.72 | 545.61 | 7 | 1 | 7 | 100 | 4.3 | 100 | 5.49 | 32 |
| 9u | 5.63 | 579.07 | 6 | 1 | 7 | 100 | 5.6 | 100 | 5.96 | 4.9 |
| 9v | 4.99 | 544.64 | 7 | 1 | 8 | 100 | 4 | 100 | 4.99 | 36.6 |
| 9x | 4.83 | 533 | 7 | 1 | 7 | 100 | 4.2 | 100 | 5.28 | 25.8 |
| 9y | 4 | 572.65 | 8 | 1 | 9 | 100 | 4.1 | 100 | 5.07 | 44.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parodi, A.; Righetti, G.; Pesce, E.; Salis, A.; Tomati, V.; Pastorino, C.; Tasso, B.; Benvenuti, M.; Damonte, G.; Pedemonte, N.; et al. Journey on VX-809-Based Hybrid Derivatives towards Drug-like F508del-CFTR Correctors: From Molecular Modeling to Chemical Synthesis and Biological Assays. Pharmaceuticals 2022, 15, 274. https://doi.org/10.3390/ph15030274
Parodi A, Righetti G, Pesce E, Salis A, Tomati V, Pastorino C, Tasso B, Benvenuti M, Damonte G, Pedemonte N, et al. Journey on VX-809-Based Hybrid Derivatives towards Drug-like F508del-CFTR Correctors: From Molecular Modeling to Chemical Synthesis and Biological Assays. Pharmaceuticals. 2022; 15(3):274. https://doi.org/10.3390/ph15030274
Chicago/Turabian StyleParodi, Alice, Giada Righetti, Emanuela Pesce, Annalisa Salis, Valeria Tomati, Cristina Pastorino, Bruno Tasso, Mirko Benvenuti, Gianluca Damonte, Nicoletta Pedemonte, and et al. 2022. "Journey on VX-809-Based Hybrid Derivatives towards Drug-like F508del-CFTR Correctors: From Molecular Modeling to Chemical Synthesis and Biological Assays" Pharmaceuticals 15, no. 3: 274. https://doi.org/10.3390/ph15030274
APA StyleParodi, A., Righetti, G., Pesce, E., Salis, A., Tomati, V., Pastorino, C., Tasso, B., Benvenuti, M., Damonte, G., Pedemonte, N., Cichero, E., & Millo, E. (2022). Journey on VX-809-Based Hybrid Derivatives towards Drug-like F508del-CFTR Correctors: From Molecular Modeling to Chemical Synthesis and Biological Assays. Pharmaceuticals, 15(3), 274. https://doi.org/10.3390/ph15030274