
Combined Antagonism of 5-HT2 and NMDA Receptors Reduces the Aggression of Monoamine Oxidase a Knockout Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
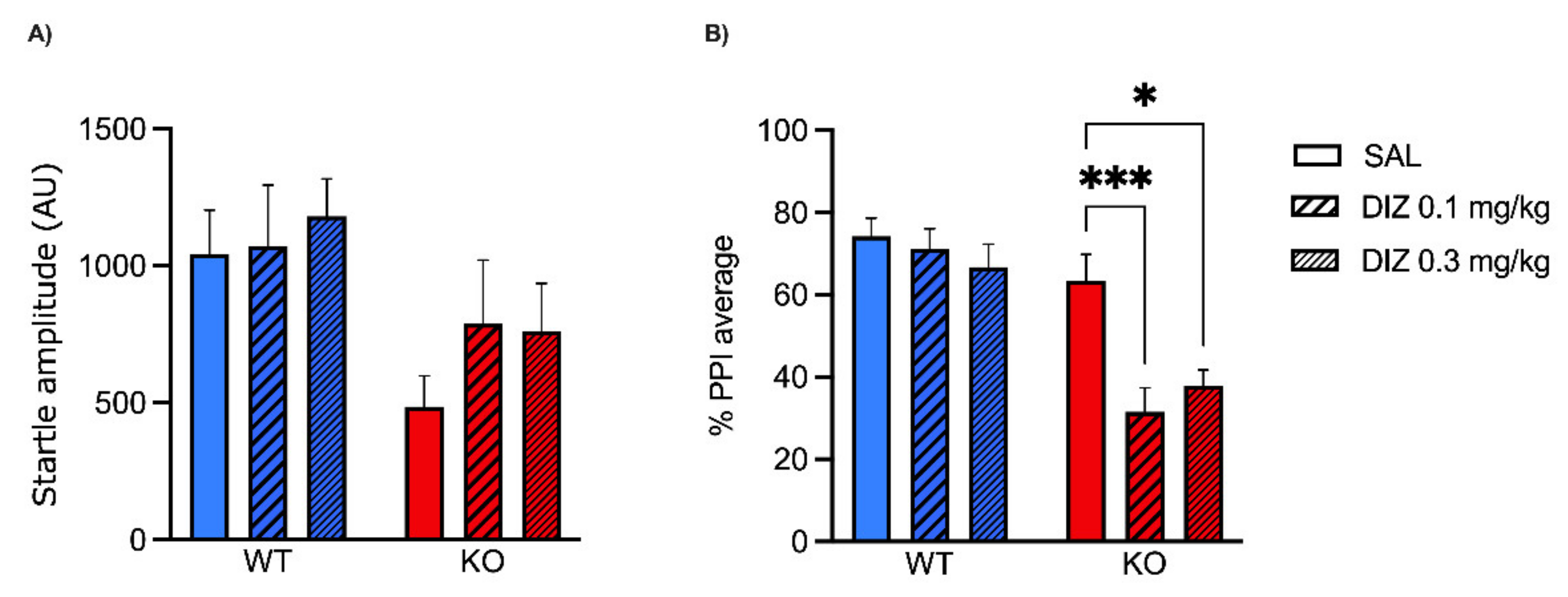
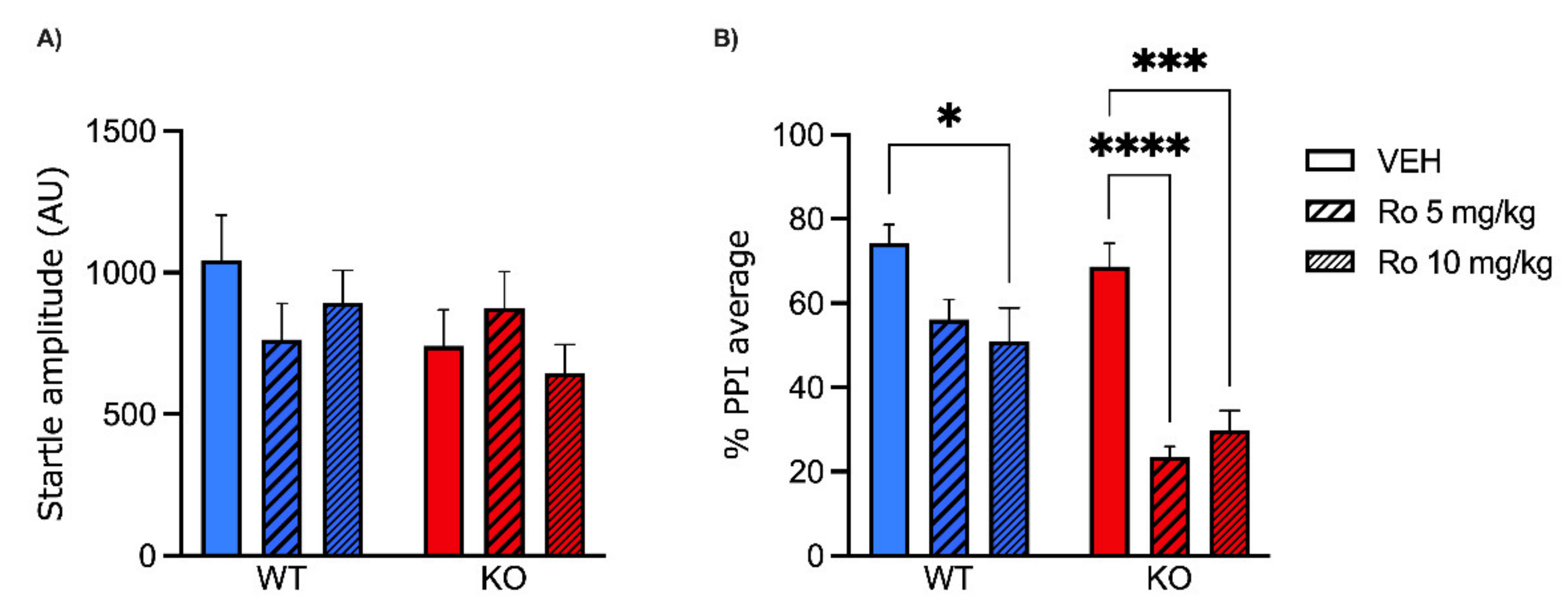
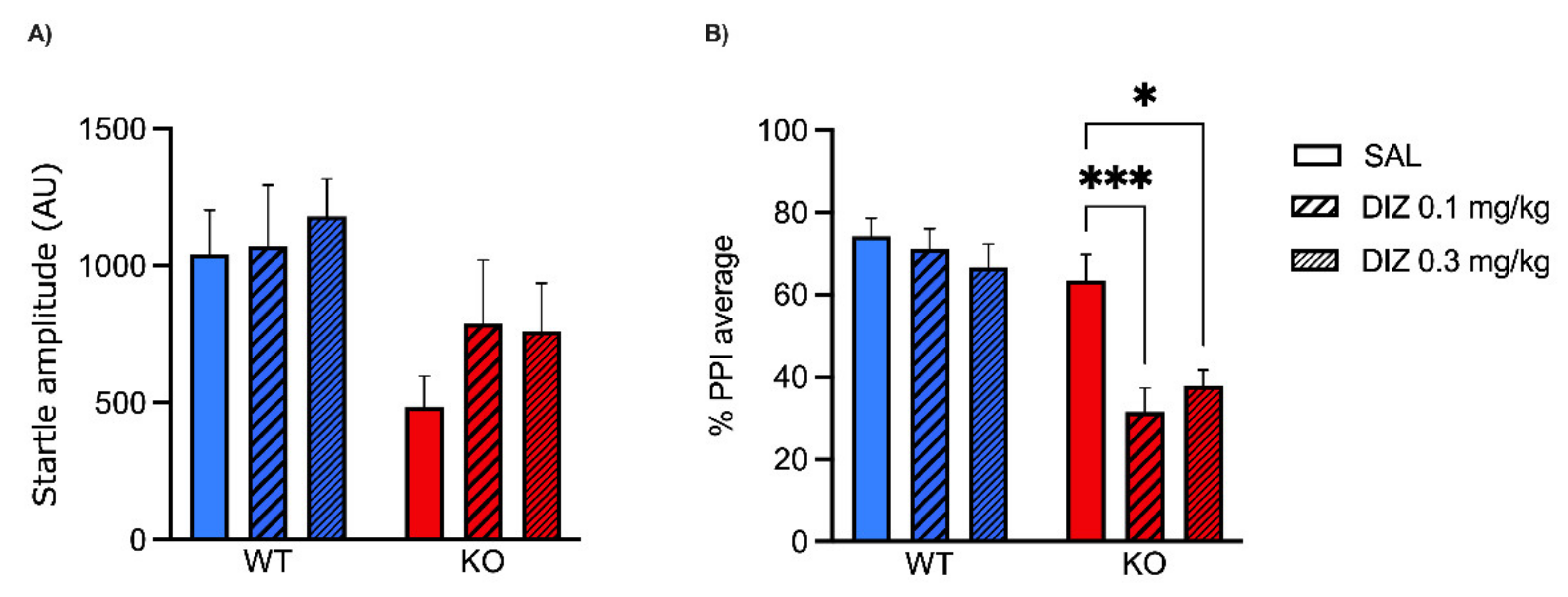
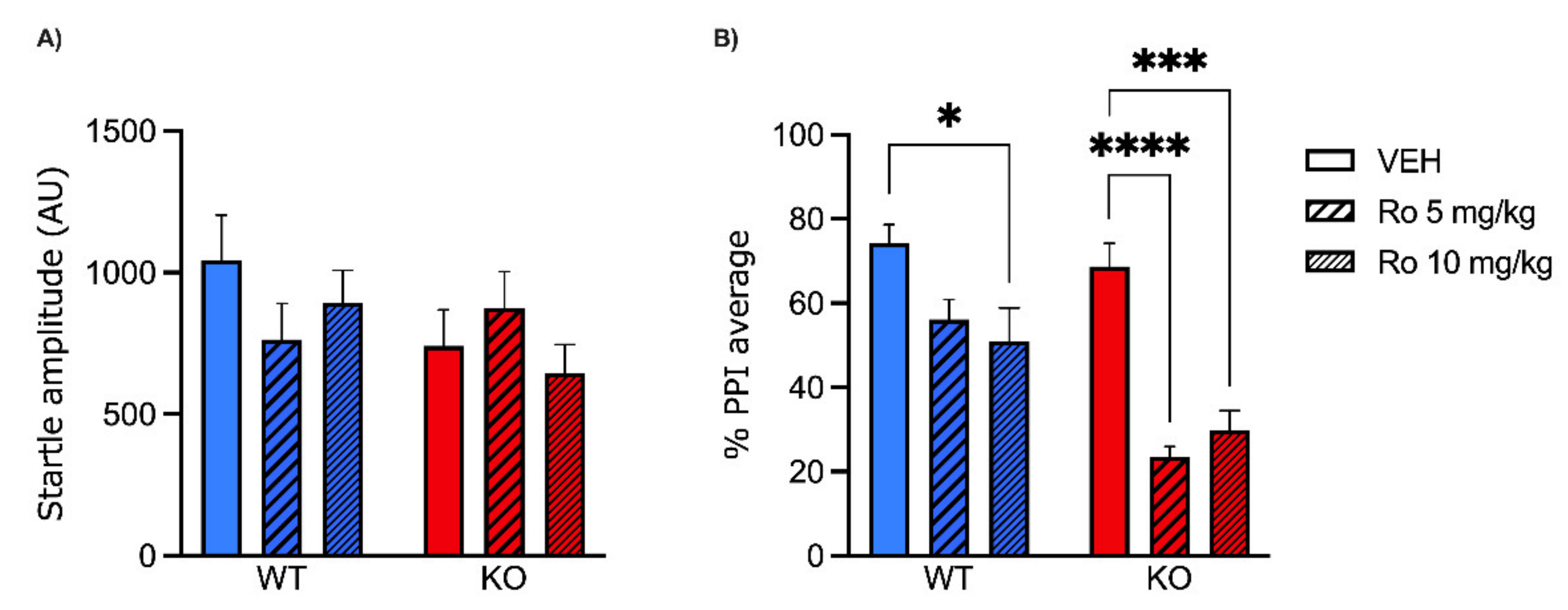
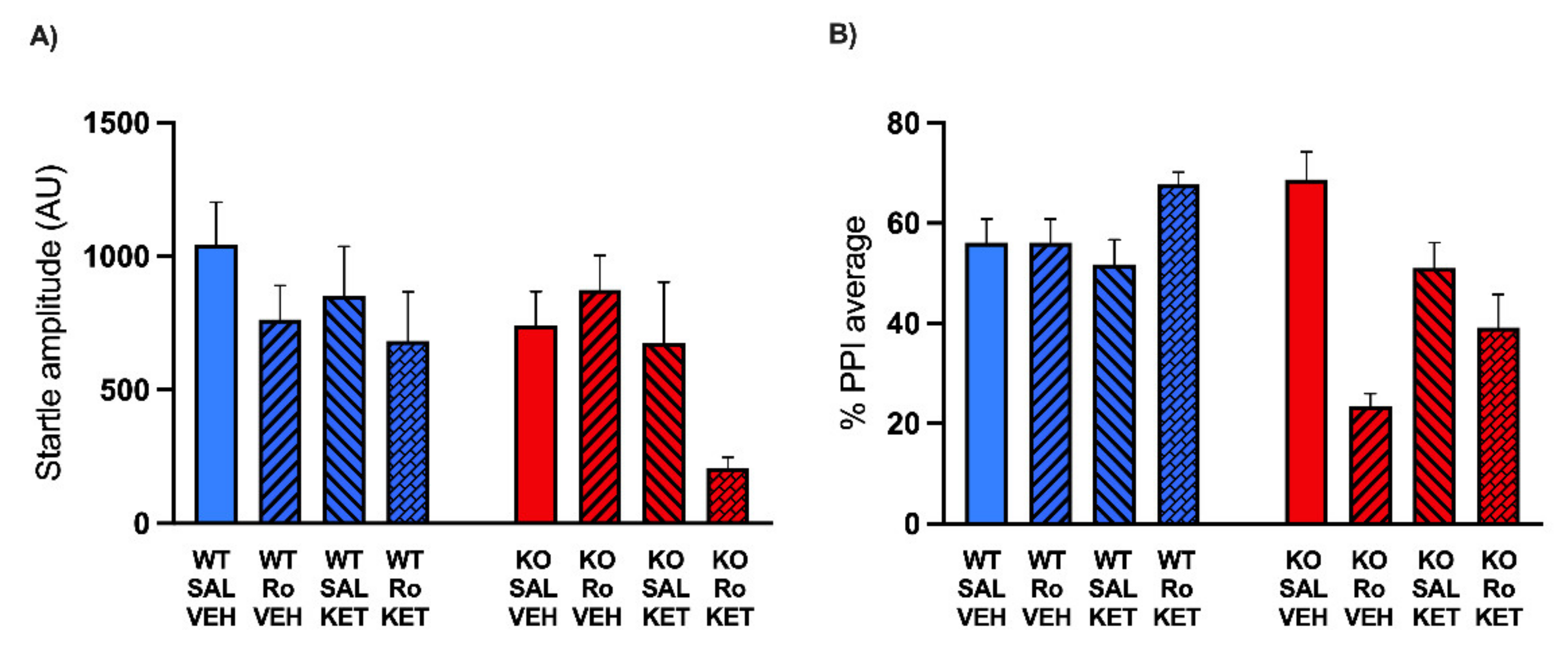
2.1. Low Doses of NMDA Receptor Antagonists DIZ and Ro 25-6981 Impair PPI in MAOA KO, but Not WT Mice
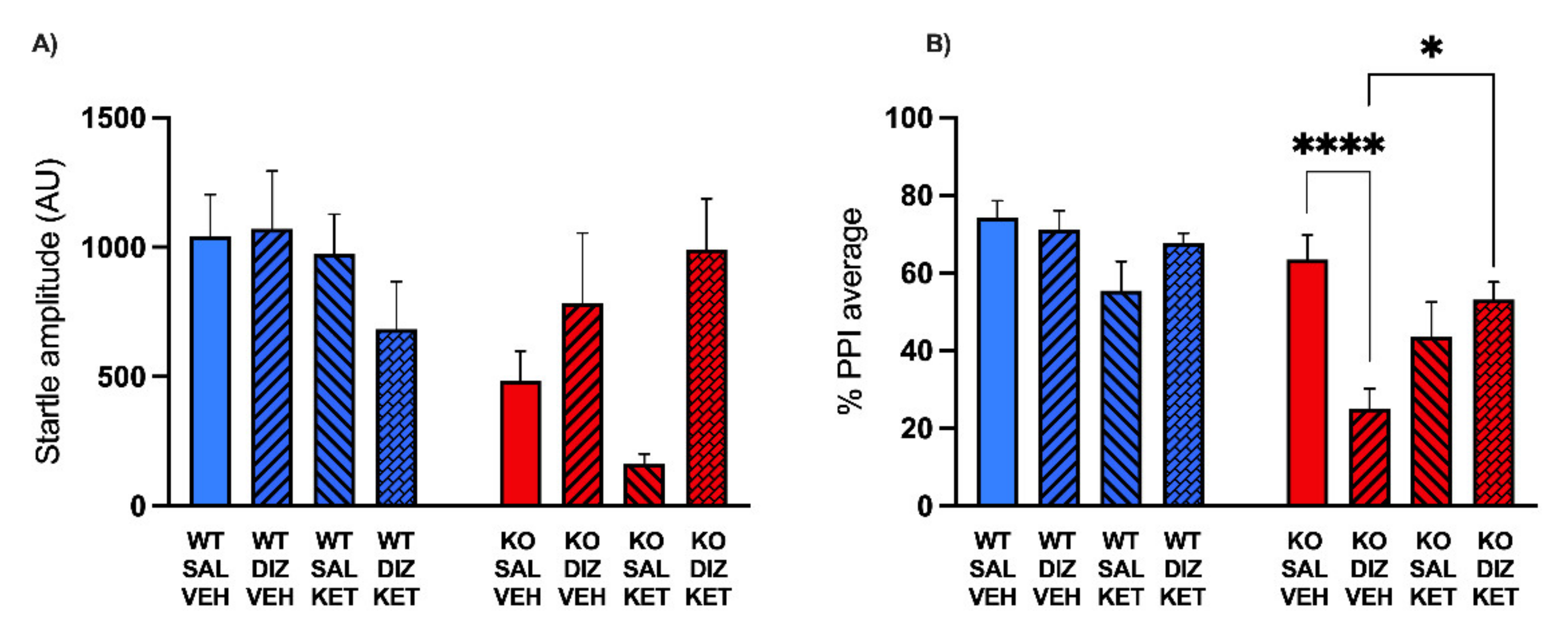
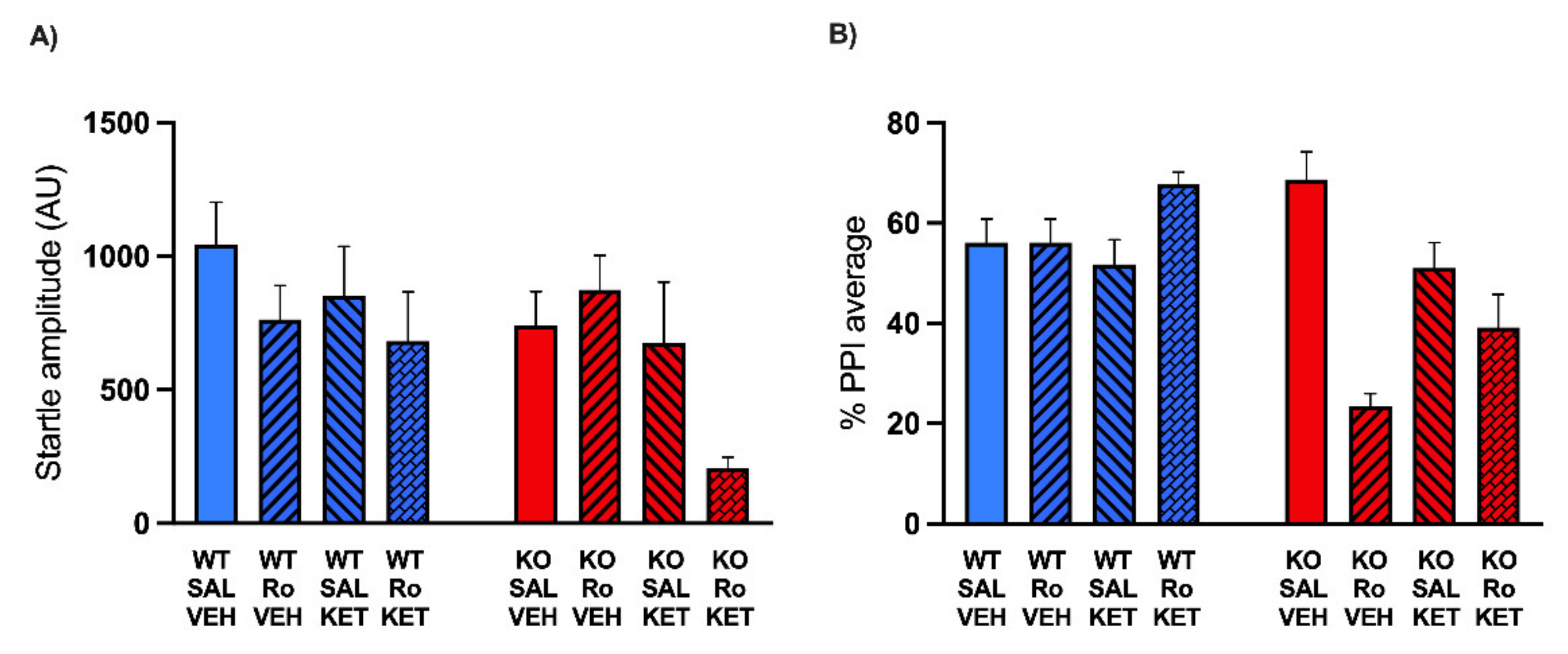
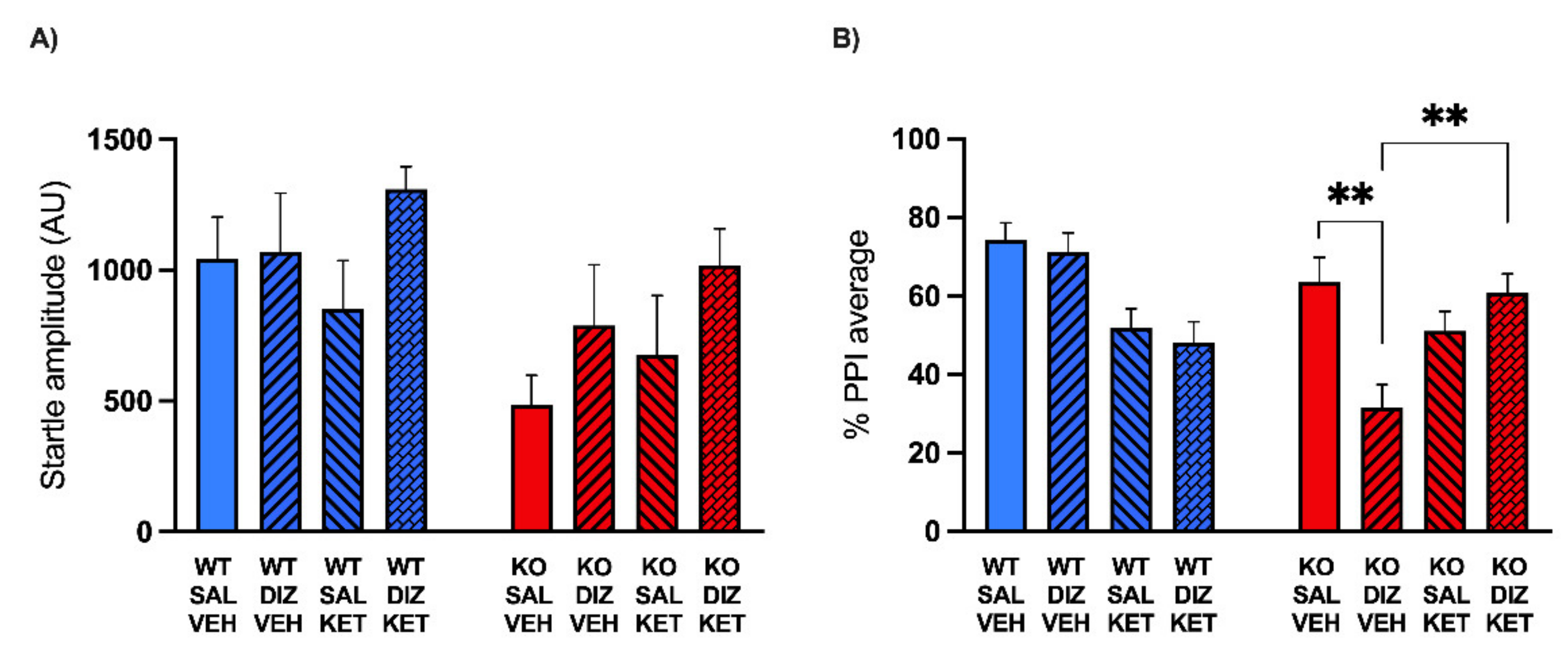
2.2. KET Rescues the PPI Deficits Induced by DIZ, but Not Ro 25-6981, in MAOA KO Mice
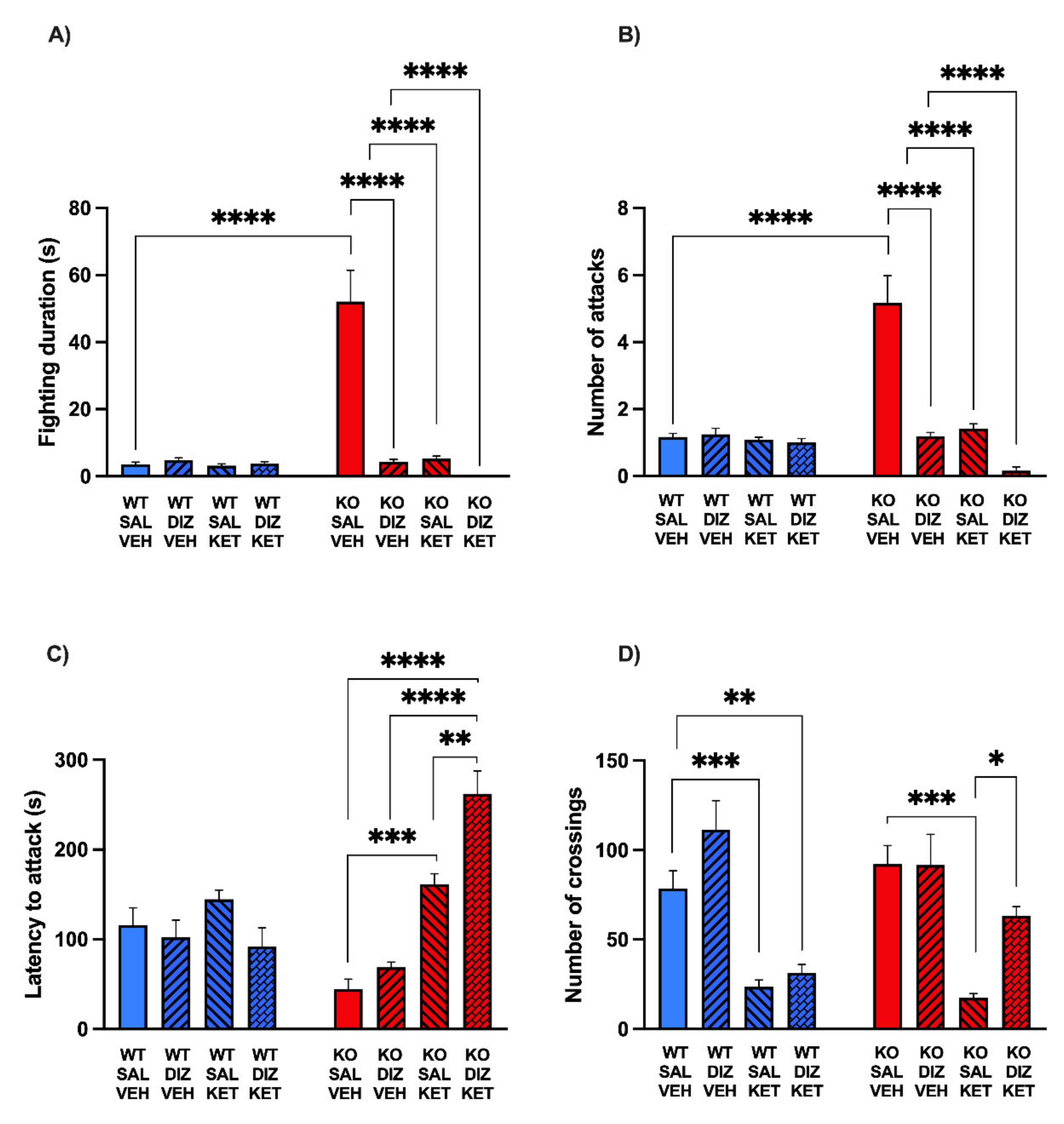
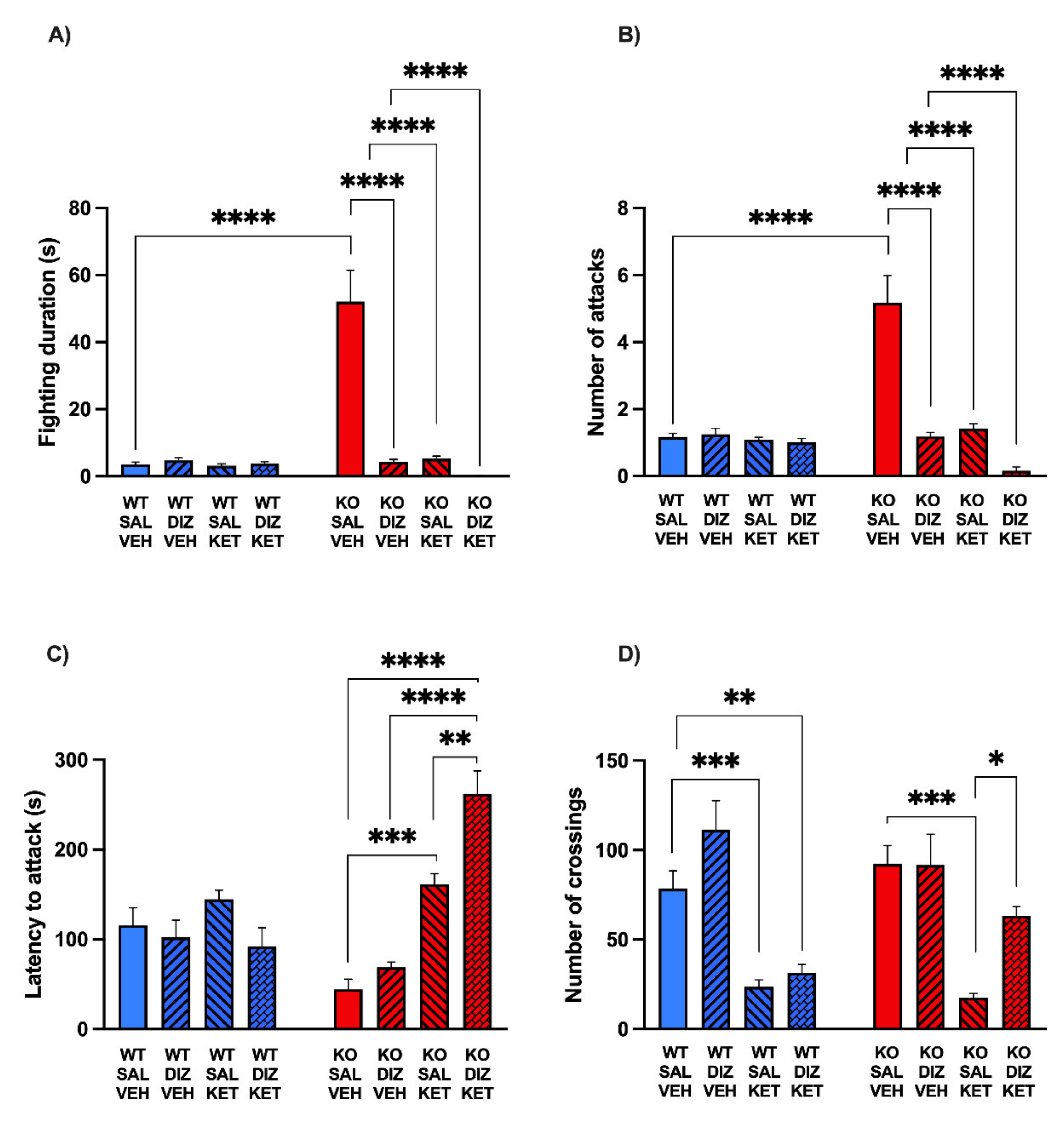
2.3. The Combination of KET and DIZ Reduces the Aggression of MAOA KO Mice without Significant Changes of Locomotor Activity
3. Discussion
4. Materials and Methods
5. Conclusions
5.1. Animal Husbandry
5.2. Drugs
5.3. Surgical and Drug Microinjection Procedures
5.4. Startle Reflex and PPI
5.5. Resident–Intruder Aggression
5.6. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buitelaar, J.K.; Smeets, K.C.; Herpers, P.; Scheepers, F.; Glennon, J.; Rommelse, N.N. Conduct disorders. Eur. Child Adoles. Psy. 2013, 22, S49–S54. [Google Scholar] [CrossRef]
- Kazdin, A.E.; Mazurick, J.L. Dropping out of child psychotherapy: Distinguishing early and late dropouts over the course of treatment. J. Consult. Clin. Psych. 1994, 62, 1069–1074. [Google Scholar] [CrossRef]
- Cherek, D.R.; Tcheremissine, O.V.; Lane, S.D.; Nelson, R.J. Psychopharmacology of human aggression: Laboratory and clinical studies. In Biology of Aggression; Nelson, R.J., Ed.; Oxford University Press: New York, NY, USA, 2006; pp. 424–446. [Google Scholar]
- Harris, A.; Lurigio, A.J. Mental illness and violence: A brief review of research and assessment strategies. Aggress. Violent Behav. 2007, 12, 542–551. [Google Scholar] [CrossRef]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliver. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef] [Green Version]
- Godar, S.C.; Fite, P.J.; McFarlin, K.M.; Bortolato, M. The role of monoamine oxidase A in aggression: Current translational developments and future challenges. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 69, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolla, N.J.; Bortolato, M. The role of monoamine oxidase A in the neurobiology of aggressive, antisocial, and violent behavior: A tale of mice and men. Progr. Neurobiol. 2020, 194, 101875. [Google Scholar] [CrossRef]
- Brunner, H.G.; Nelen, M.; Breakefield, X.O.; Ropers, H.H.; Van Oost, B.A. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 1993, 262, 578–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolato, M.; Floris, G.; Shih, J.C. From aggression to autism: New perspectives on the behavioral sequelae of monoamine oxidase deficiency. J. Neural. Transm. 2018, 125, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Cases, O.; Seif, I.; Grimsby, J.; Gaspar, P.; Chen, K.; Pournin, S.; Müller, U.; Aguet, M.; Babinet, C.; Shih, J.C.; et al. Aggressive behavior and atered amounts of brain serotonin and norephinephrine in mice lacking MAOA. Science 1995, 268, 1763–1766. [Google Scholar] [CrossRef] [Green Version]
- Bortolato, M.; Shih, J.C. Behavioral outcomes of monoamine oxidase deficiency: Preclinical and clinical evidence. Int. Rev. Neurobiol. 2011, 100, 13–42. [Google Scholar] [PubMed] [Green Version]
- Godar, S.C.; Bortolato, M.; Frau, R.; Dousti, M.; Chen, K.; Shih, J.C. Maladaptive defensive behaviours in monoamine oxidase A-deficient mice. Int. J. Neuropsychopharmacol. 2011, 14, 1195–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolato, M.; Godar, S.C.; Melis, M.; Soggiu, A.; Roncada, P.; Casu, A.; Flore, G.; Chen, K.; Frau, R.; Urbani, A.; et al. NMDARs mediate the role of monoamine oxidase A in pathological aggression. J. Neurosci. 2012, 32, 8574–8582. [Google Scholar] [CrossRef] [Green Version]
- Daw, N.W.; Stein, P.S.; Fox, K. The role of NMDA receptors in information processing. Annu. Rev. Neurosci. 1993, 16, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Su, C.-L.; Gean, P.-W. Mechanism underlying NMDA blockade-induced inhibition of aggression in post-weaning socially isolated mice. Neuropharmacology 2018, 143, 95–105. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Van Rhijn, J.R.; Shi, Y.; Bormann, M.; Mossink, B.; Frega, M.; Recaioglu, H.; Hakobjan, M.; Klein Gunnewiek, T.; Schoenmaker, C.; Palmer, E.; et al. Brunner syndrome associated MAOA mutations result in NMDAR hyperfunction and increased network activity in human dopaminergic neurons. Neurobiol. Dis. 2021, 163, 105587. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Nagata, A.; Masuzawa, M.; Miyamoto, E.; Yamada, M.; Nishizawa, N.; Shingu, K. NMDA receptor antagonist neurotoxicity and psychotomimetic activity. Masui Jpn. J. Anesthesiol. 2003, 52, 594–602. [Google Scholar]
- Keith, V.A.; Mansbach, R.S.; Geyer, M.A. Failure of haloperidol to block the effects of phencyclidine and dizocilpine on prepulse inhibition of startle. Biol. Psychiatry 1991, 30, 557–566. [Google Scholar] [CrossRef]
- Chaperon, F.; Müller, W.; Auberson, Y.P.; Tricklebank, M.D.; Neijt, H.C. Substitution for PCP, disruption of prepulse inhibition and hyperactivity induced by N-methyl-D-aspartate receptor antagonists: Preferential involvement of the NR2B rather than NR2A subunit. Behav. Pharmacol. 2003, 14, 477–487. [Google Scholar]
- Varty, G.B.; Higgins, G.A. Examination of drug-induced and isolation-induced disruptions of prepulse inhibition as models to screen antipsychotic drugs. Psychopharmacology 1995, 122, 15–26. [Google Scholar] [CrossRef]
- Weisstaub, N.V.; Zhou, M.; Lira, A.; Lambe, E.; González-Maeso, J.; Hornung, J.P.; Sibille, E.; Underwood, M.; Itohara, S.; Dauer, W.T.; et al. Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science 2006, 313, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Shih, J.C.; Chen, K. MAO-A and -B gene knockout mice exhibit distinctly different behavior. Neurobiology 1999, 7, 235–246. [Google Scholar] [PubMed]
- Bortolato, M.; Aru, G.N.; Fà, M.; Frau, R.; Orrù, M.; Salis, P.; Casti, A.; Luckey, G.C.; Mereu, G.; Gessa, G.L. Activation of D1, but not D2 receptors potentiates dizocilpine-mediated disruption of prepulse inhibition of the startle. Neuropsychopharmacology 2005, 30, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Godar, S.C.; Mosher, L.J.; Scheggi, S.; Devoto, P.; Moench, K.M.; Strathman, H.J.; Jones, C.M.; Frau, R.; Melis, M.; Gambarana, C.; et al. Gene-environment interactions in antisocial behavior are mediated by early-life 5-HT2A receptor activation. Neuropharmacology 2019, 159, 107513. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.L.; Chu, A.; Bahamón, B.; Takahashi, A.; Debold, J.F.; Miczek, K.A. NMDA receptor antagonism: Escalation of aggressive behavior in alcohol-drinking mice. Psychopharmacology 2012, 224, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.L.; Terunuma, M.; Wang, T.L.; Hewage, N.; Bicakci, M.B.; Moss, S.J.; DeBold, J.F.; Miczek, K.A. A Role for Prefrontal Cortical NMDA Receptors in Murine Alcohol-Heightened Aggression. Neuropsychopharmacology 2018, 43, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Zoicas, I.; Kornhuber, J. The Role of the N-Methyl-D-Aspartate Receptors in Social Behavior in Rodents. Int. J. Mol. Sci. 2019, 20, 5599. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.B.; Vertes, R.P. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 2007, 212, 149–179. [Google Scholar] [CrossRef]
- Homayoun, H.; Moghaddam, B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.; von der Malsburg, C.; Singer, W. Dynamic coordination in brain and mind. In Dynamic Coordination in the Brain: From Neurons to Mind; MIT Press: Cambridge, MA, USA, 2010; Volume 5, pp. 1–24. [Google Scholar]
- Nelson, S.B.; Sur, M. NMDA receptors in sensory information processing. Curr. Opin. Neurobiol. 1992, 2, 484–488. [Google Scholar] [CrossRef]
- Wang, H.; Stradtman, G.G., 3rd; Wang, X.J.; Gao, W.J. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc. Natl. Acad. Sci. USA 2008, 105, 16791–16796. [Google Scholar] [CrossRef] [Green Version]
- Sigurdsson, T.; Stark, K.L.; Karayiorgou, M.; Gogos, J.A.; Gordon, J.A. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature 2010, 464, 763–767. [Google Scholar] [CrossRef]
- Mansbach, R.S.; Geyer, M.A. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 1989, 2, 299–308. [Google Scholar] [CrossRef]
- Klamer, D.; Engel, J.A.; Svensson, L. The nitric oxide synthase inhibitor, L-NAME, block phencyclidine-induced disruption of prepulse inhibition in mice. Psychopharmacology 2001, 156, 182–186. [Google Scholar] [CrossRef]
- Linn, G.S.; Negi, S.S.; Gerum, S.V.; Javitt, D.C. Reversal of phencyclidine-induced prepulse inhibition deficits by clozapine in monkeys. Psychopharmacology 2003, 169, 234–239. [Google Scholar] [CrossRef]
- Fejgin, K.; Pålsson, E.; Wass, C.; Svensson, L.; Klamer, D. Nitric oxide signaling in the medial prefrontal cortex is involved in the biochemical and behavioral effects of phencyclidine. Neuropsychopharmacology 2008, 33, 1874–1883. [Google Scholar] [CrossRef] [Green Version]
- Graham, F.K. The more or less startling effects of weak prestimulation. Psychophysiology 1975, 12, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Braff, D.L.; Geyer, M.A. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch. Gen. Psychiatry 1990, 47, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.R.; Geyer, M.A. Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophrenia Bull. 1998, 24, 285–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.J.; Sorensen, S.M.; Kehne, J.H.; Carr, A.A.; Palfreyman, M.G. The role of 5-HT2A receptors in antipsychotic activity. Life Sci. 1995, 56, 2209–2222. [Google Scholar] [CrossRef]
- Seeman, P. Atypical antipsychotics: Mechanism of action. Can. J. Psychiatry 2002, 47, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Isaacson, S.; Mills, R.; Williams, H.; Chi-Burris, K.; Corbett, A.; Dhall, R.; Ballard, C. Pimavanserin for patients with Parkinson’s disease psychosis: A randomised, placebo-controlled phase 3 trial. Lancet 2014, 383, 533–540. [Google Scholar] [CrossRef]
- Varty, G.B.; Higgins, G.A. Reversal of dizocilpine-induced disruption of prepulse inhibition of an acoustic startle response by the 5-HT2 receptor antagonist ketanserin. Eur. J. Pharmacol. 1995, 287, 201–205. [Google Scholar] [CrossRef]
- Geyer, M.; Krebs-Thomson, K.; Varty, G. The Effects of M100907 in Pharmacological and Developmental Animal Models of Prepulse Inhibition Deficits in Schizophrenia. Neuropsychopharmacology 1999, 21, S134–S142. [Google Scholar] [CrossRef]
- Quednow, B.B.; Kometer, M.; Geyer, M.A.; Vollenweider, F.X. Psilocybin-induced deficits in automatic and controlled inhibition are attenuated by ketanserin in healthy human volunteers. Neuropsychopharmacology 2012, 37, 630–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niethammer, M.; Kim, E.; Sheng, M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J. Neurosci. 1996, 16, 2157–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Levy, J.M.; Hou, A.; Winters, C.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Nicoll, R.A.; Reese, T.S. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA 2015, 15, E6983–E6992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Gray, J.A.; Compton-Toth, B.A.; Roth, B.L.; Vos, M.D.; Martinez, A.; Ellis, C.A.; Vallecorsa, T.; Clark, G.J. A direct interaction of PSD-95 with 5-HT2A serotonin receptors regulates receptor trafficking and signal transduction. J. Biol. Chem. 2003, 278, 21901–21908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.I.; Yadav, P.N.; Yao, W.D.; Arbuckle, M.I.; Grant, S.G.; Caron, M.G.; Roth, B.L. PSD-95 is essential for hallucinogen and atypical antipsychotic drug actions at serotonin receptors. J. Neurosci. 2009, 29, 7124–7136. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, P.S.; Ugale, R.R.; Yilmazer-Hanke, D.; Stairs, D.J.; Dravid, S.M. GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br. J. Pharmacol. 2014, 171, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, A.; McClay, J.; Moffitt, T.E.; Mill, J.; Martin, J.; Craig, I.W.; Taylor, A.; Poulton, R. Role of genotype in the cycle of violence in maltreated children. Science 2002, 297, 851–854. [Google Scholar] [CrossRef]
- Alia-Klein, N.; Goldstein, R.Z.; Kriplani, A.; Logan, J.; Tomasi, D.; Williams, B.; Telang, F.; Shumay, E.; Biegon, A.; Craig, I.W.; et al. Brain monoamine oxidase A activity predicts trait aggression. J. Neurosci. 2008, 28, 5099–5104. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.; Bagby, R.M.; Wilson, A.A.; Miler, L.; Clark, M.; Rusjan, P.; Sacher, J.; Houle, S.; Meyer, J.H. Relationship of monoamine oxidase A binding to adaptive and maladaptive personality traits. Psychol. Med. 2011, 41, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Vitalis, T.; Cases, O.; Callebert, J.; Launay, J.M.; Price, D.J.; Seif, I.; Gaspar, P. Effects of monoamine oxidase A inhibition on barrel formation in the mouse somatosensory cortex: Determination of a sensitive developmental period. J. Comp. Neurol. 1998, 393, 169–184. [Google Scholar] [CrossRef]
- Bortolato, M.; Godar, S.C.; Alzghoul, L.; Zhang, J.; Darling, R.D.; Simpson, K.L.; Bini, V.; Chen, K.; Wellman, C.L.; Lin, R.C.; et al. Monoamine oxidase A and A/B knockout mice display autistic-like features. Int. J. Neuropsychopharmacol. 2013, 16, 869–888. [Google Scholar] [CrossRef] [Green Version]
- Sakaue, M.; Ago, Y.; Sowa, C.; Sakamoto, Y.; Nishihara, B.; Koyama, Y.; Baba, A.; Matsuda, T. Modulation by 5-HT2A receptors of aggressive behavior in isolated mice. Jpn. J. Pharmacol. 2002, 89, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Brogden, R.N.; Sorkin, E.M. Ketanserin. Drugs 1990, 40, 903–949. [Google Scholar] [CrossRef]
- Nilsson, M.; Carlsson, A.; Carlsson, M.L. Glycine and D-serine decrease MK-801-induced hyperactivity in mice. J. Neural Transm. 1997, 104, 1195–1205. [Google Scholar] [CrossRef]
- Maj, J.; Rogóz, Z.; Skuza, G. Locomotor hyperactivity induced by MK-801 in rats. Pol. J. Pharmacol. Pharm. 1991, 43, 449–458. [Google Scholar] [PubMed]
- Hargreaves, E.L.; Cain, D.P. MK801-induced hyperactivity: Duration of effects in rats. Pharmacol. Biochem. Behav. 1995, 51, 13–19. [Google Scholar] [CrossRef]
- Rosell, D.R.; Thompson, J.L.; Slifstein, M.; Xu, X.; Frankle, W.G.; New, A.S.; Goodman, M.; Weinstein, S.R.; Laruelle, M.; Abi-Dargham, A.; et al. Increased serotonin 2A receptor availability in the orbitofrontal cortex of physically aggressive personality disordered patients. Biol. Psychiatry 2010, 67, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Higgins, G.A.; Enderlin, M.; Haman, M.; Fletcher, P.J. The 5-HT2A receptor antagonist M100,907 attenuates motor and ’impulsive-type’ behaviours produced by NMDA receptor antagonism. Psychopharmacology 2003, 170, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.S.; Vishnoi, S.; Madan, R.; Bhardwaj, N.; Tabassum, H.; Akram, M.; Parvez, S. Clozapine Improves Behavioral and Biochemical Outcomes in a MK-801-Induced Mouse Model of Schizophrenia. J. Environ. Pathol. Toxicol. Oncol. 2020, 39, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lan, N.C.; Heinzmann, C.; Gal, A.; Klisak, I.; Orth, U.; Lai, E.; Grimsby, J.; Sparkes, R.S.; Mohandas, T.; Shih, J.C. Human monoamine oxidase A and B genes map to Xp 11.23 and are deleted in a patient with Norrie disease. Genomics 1989, 4, 552–559. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 5th ed.; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Frau, R.; Pillolla, G.; Bini, V.; Tambaro, S.; Devoto, P.; Bortolato, M. Inhibition of 5α-reductase attenuates behavioral effects of D1-, but not D2-like receptor agonists in C57BL/6 mice. Psychoneuroendocrinology 2013, 38, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frau, R.; Fanni, S.; Serra, V.; Simola, N.; Godar, S.C.; Traccis, F.; Devoto, P.; Bortolato, M.; Melis, M. Dysfunctional mesocortical dopamine circuit at pre-adolescence is associated to aggressive behavior in MAO-A hypomorphic mice exposed to early life stress. Neuropharmacology 2019, 159, 107517. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frau, R.; Pardu, A.; Godar, S.; Bini, V.; Bortolato, M. Combined Antagonism of 5-HT2 and NMDA Receptors Reduces the Aggression of Monoamine Oxidase a Knockout Mice. Pharmaceuticals 2022, 15, 213. https://doi.org/10.3390/ph15020213
Frau R, Pardu A, Godar S, Bini V, Bortolato M. Combined Antagonism of 5-HT2 and NMDA Receptors Reduces the Aggression of Monoamine Oxidase a Knockout Mice. Pharmaceuticals. 2022; 15(2):213. https://doi.org/10.3390/ph15020213
Chicago/Turabian StyleFrau, Roberto, Alessandra Pardu, Sean Godar, Valentina Bini, and Marco Bortolato. 2022. "Combined Antagonism of 5-HT2 and NMDA Receptors Reduces the Aggression of Monoamine Oxidase a Knockout Mice" Pharmaceuticals 15, no. 2: 213. https://doi.org/10.3390/ph15020213
APA StyleFrau, R., Pardu, A., Godar, S., Bini, V., & Bortolato, M. (2022). Combined Antagonism of 5-HT2 and NMDA Receptors Reduces the Aggression of Monoamine Oxidase a Knockout Mice. Pharmaceuticals, 15(2), 213. https://doi.org/10.3390/ph15020213