
Nitroaromatic Hypoxia-Activated Prodrugs for Cancer Therapy


Abstract
:1. Introduction: The Problem of Tumour Hypoxia
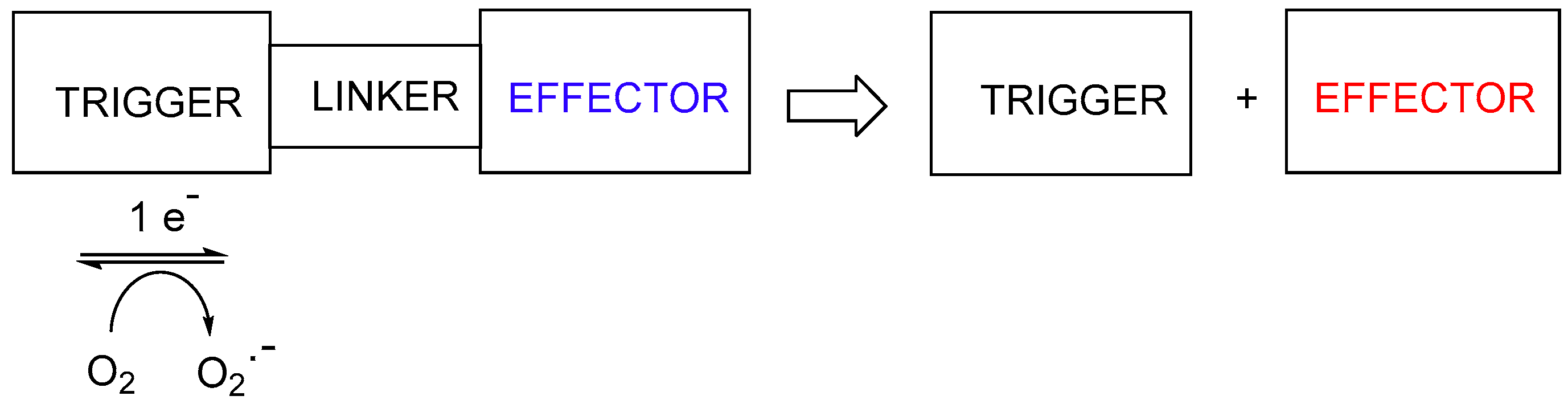
2. Prodrugs Activated by Electron Redistribution
3. Prodrugs Activated by Fragmentation
3.1. 4-Nitrobenzyl Triggers
3.2. Nitrophenyl Triggers
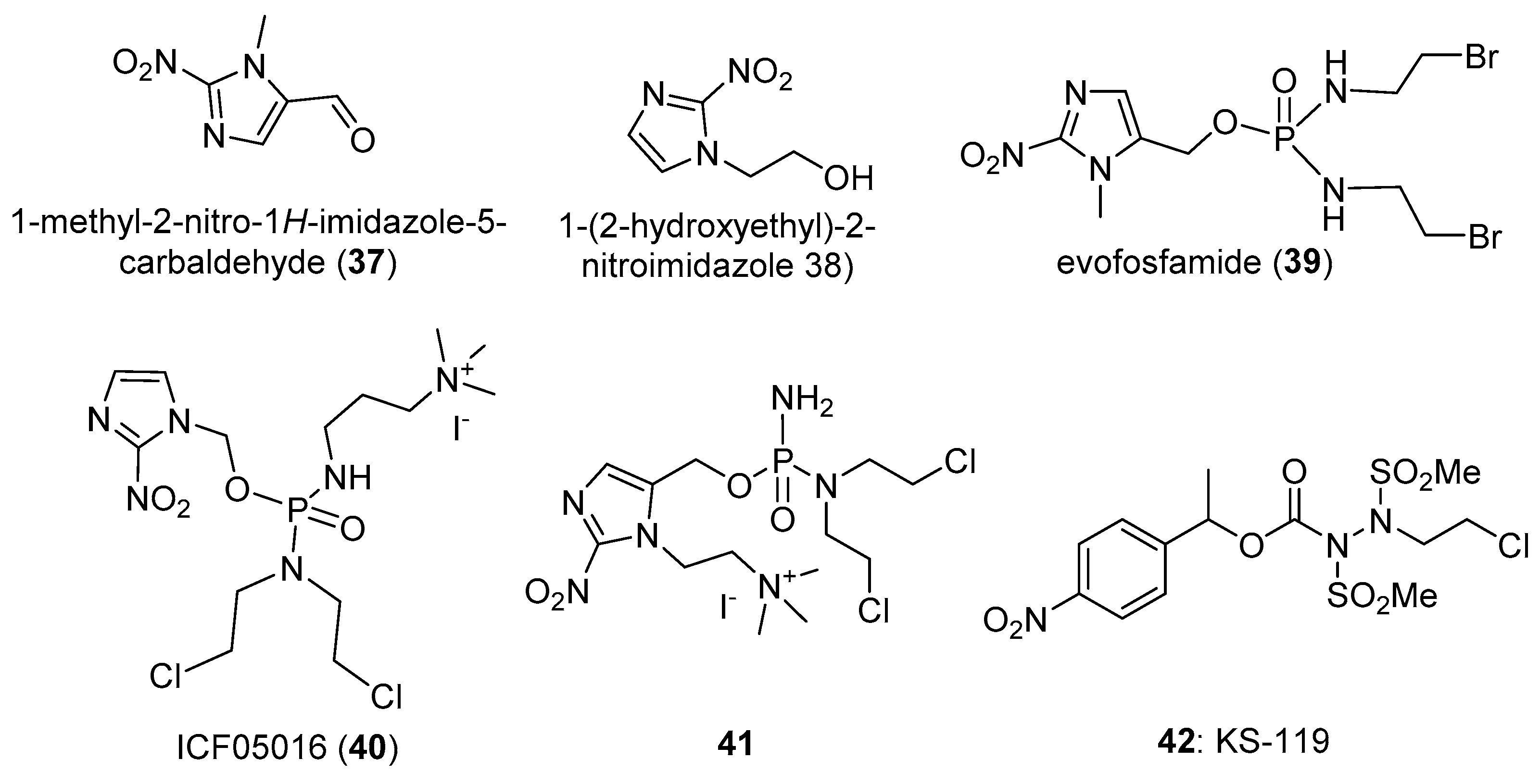
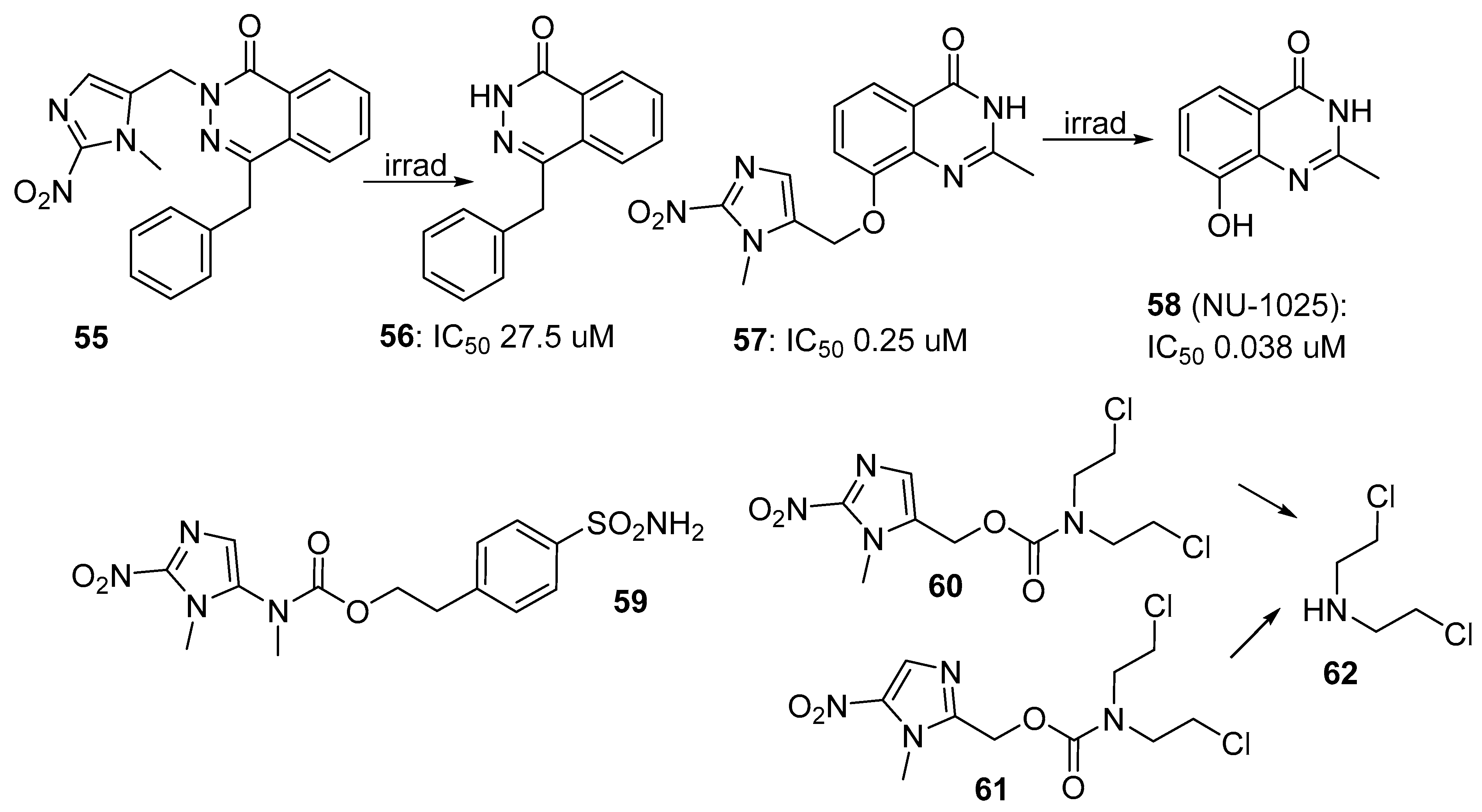
3.3. 2-Nitroimidazole Triggers
3.4. 4-Nitroimidazole Triggers
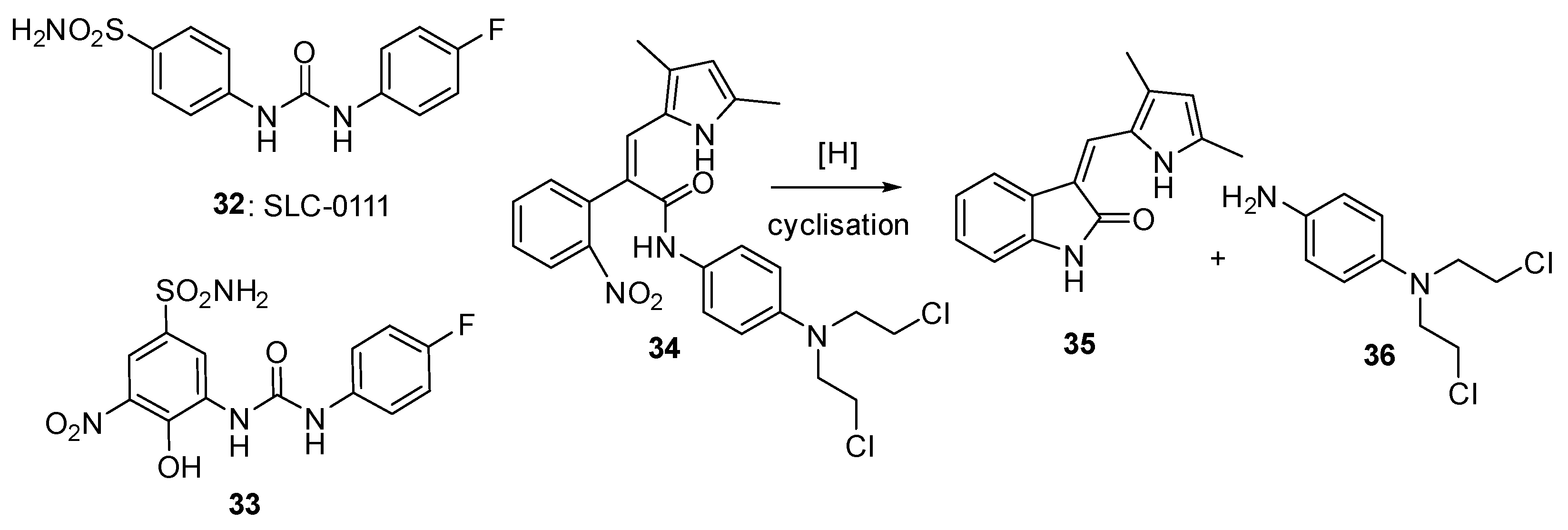
3.5. Other Nitroheterocyclic Triggers
3.6. Nitrobenzo[e]indole Prodrugs
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hockel, M.; Vaupel, P. Biological consequences of tumor hypoxia. Sem. Oncol. 2001, 28 (Suppl. S8), 36–41. [Google Scholar] [CrossRef]
- Rockwell, S.; Moulder, J.E. Hypoxic fractions of human tumors xenografted into mice: A review. Int. J. Rad. Oncol. Biol. Phys. 1990, 19, 197–202. [Google Scholar] [CrossRef]
- Brown, J.M.; Giaccia, A.J. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2013, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Lee, H.-Y.; Liou, J.P. Nitro-group-containing drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Some reactions and properties of nitro radical-anions important in biology and medicine. Environ. Health Perspect. 1985, 64, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Wilson, W.R. Considerations for the design of nitrophenyl mustards as agents with selective toxicity for hypoxic tumor cells. J. Med. Chem. 1986, 29, 879–887. [Google Scholar] [CrossRef]
- Knox, R.J.; Burke, P.J.; Chen, S.; Kerr, D.J. CB 1954: From the Walker tumor to NQO2 and VDEPT. Curr. Pharm. Des. 2003, 9, 2091–2104. [Google Scholar] [CrossRef]
- Palmer, B.D.; Wilson, W.R.; Pullen, S.M.; Denny, W.A. Hypoxia-selective antitumor agents. 3. Relationships between structure and cytotoxicity against cultured tumor cells for substituted N.N-bis(2-chloroethyl)anilines. J. Med. Chem. 1990, 33, 112–121. [Google Scholar] [CrossRef]
- Hoy, C.A.; Thompson, I.H.; Mooney, C.L.; Salazar, E.P. Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 1985, 45, 1737–1743. [Google Scholar]
- Palmer, B.D.; Wilson, W.R.; Cliffe, S.; Denny, W.A. Hypoxia-selective antitumor agents. 5. Synthesis of water-soluble nitroaniline mustards with selective cytotoxicity for hypoxic mammalian cells. J. Med. Chem. 1992, 35, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.D.; Wilson, W.R.; Anderson, R.F.; Boyd, M.; Denny, W.A. Hypoxia-selective antitumor agents. 14. Synthesis and hypoxic cell cytotoxicity of regioisomers of the hypoxia-selective cytotoxin 5-[N,N-(2-chloroethyl)amino]-2,4-dinitrobenzamide. J. Med. Chem. 1996, 39, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Atwell, G.J.; Denny, W.A. Synthesis of asymmetric halomesylate mustards with aziridineethanol/alkali metal halides: Application to an improved synthesis of the hypoxia prodrug PR-104. Tetrahedron 2007, 63, 5470–5476. [Google Scholar] [CrossRef]
- Patterson, A.V.; Ferry, D.M.; Edmunds, S.J.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.R.; Bogle, G.; Wang, J.; Patel, K.; Pruijn, F.B.; Wilson, W.R.; Hicks, K.O. Agent-based modeling using three-dimensional cell cultures. Front. Pharmacol. 2018, 9, 1013. [Google Scholar] [CrossRef]
- Stornetta, A.; Deng, K.-C.; Kieren, C.; Danielli, S.; Liyanage, H.D.S.; Sturla, S.J.; Wilson, W.R.; Gu, Y. Drug-DNA adducts as biomarkers for metabolic activation of the nitro-aromatic nitrogen mustard prodrug PR-104A. Biochem. Pharmacol. 2018, 154, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Guise, C.P.; Abbattista, M.R.; Tipparaju, S.R.; Lambie, N.K.; Su, J.; Li, D.; Wilson, W.R.; Dachs, G.U.; Patterson, A.V. Diflavin oxidoreductases activate the bioreductive prodrug PR-104A under hypoxia. Mol. Pharmacol. 2012, 81, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Thall, P.F.; Yi, C.A.; Borthakur, G.; Coveler, A.; Bueso-Ramos, C.; Benito, J.; Konoplev, S.; Gu, Y.; Ravandi, F.; et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 2015, 100, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Foehrenbacher, A.; Patel, K.; Abbattista, M.R.; Guise, C.P.; Secomb, T.W.; Wilson, W.R.; Hicks, K.O. The role of bystander effects in the antitumor activity of the hypoxia-activated prodrug PR-104. Front. Oncol. 2013, 3, 263. [Google Scholar] [CrossRef] [Green Version]
- Guise, C.P.; Abbattista, M.R.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Van der Wiel, A.M.A.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.M.; Lieuwes, N.G.; Biemans, R.; Lin, X.; et al. Selectively targeting tumor hypoxia with the hypoxia-activated prodrug CP-506. Mol. Cancer Ther. 2021, 20, 2372. [Google Scholar] [CrossRef] [PubMed]
- Phase 1/2 Clinical Trial of CP-506 (HAP) in Monotherapy or with Carboplatin or ICI. ClinicalTrials.gov. NCT04954599; 6 July 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04954599 (accessed on 31 January 2022).
- Salter-Blanc, A.J.; Bylaska, E.J.; Johnston, H.J.; Tratnyek, P.G. Predicting reduction rates of energetic nitroaromatic compounds using calculated one-electron reduction potentials. Environ. Sci. Technol. 2015, 49, 3778–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Lai, X.; Li, J.; Yu, R.; Zhuang, Z.; Sun, G.; Cui, X.; Zhang, N.; Zhao, L.; Upadhyaya, P.U.; et al. NBGNU: A hypoxia-activated tripartite combi-nitrosourea prodrug overcoming AGT-mediated chemoresistance. Future Med. Chem. 2019, 11, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Al-Hilal, T.A.; Hossain, M.A.; Alobaida, A.; Alam, F.; Keshavarz, A.; Nozik-Grayck, E.; Stenmark, K.R.; German, N.A.; Ahsan, F.J. Design, synthesis and biological evaluations of a long-acting, hypoxia-activated prodrug of fasudil, a ROCK inhibitor, to reduce its systemic side-effects. J. Control. Release 2021, 334, 237–247. [Google Scholar] [CrossRef]
- Liu, W.; Liu, H.; Peng, X.; Zhou, G.; Liu, D.; Li, S.; Zhang, J.; Wang, S. Hypoxia-activated anticancer prodrug for bioimaging, tracking drug release, and anticancer application. Bioconjug. Chem. 2018, 29, 3332–3343. [Google Scholar] [CrossRef]
- Yeoh, Y.Q.; Horsley, J.R.; Polyak, S.W.; Abell, A.D. ANTIBAC 16. A hypoxia-activated antibacterial prodrug. Bioorg. Med. Chem. Lett. 2020, 30, 127140. [Google Scholar] [CrossRef]
- Ghosh, C.; Manjunath, G.B.; Akkapeddi, P.; Venkateswarlu, Y.; Hoque, J.; Uppu, D.S.S.M.; Mohini, M.; Konai, M.M.; Haldar, J. Molecular antibacterial peptoid mimics: The simpler the better! J. Med. Chem. 2014, 57, 1428–1436. [Google Scholar] [CrossRef]
- Luo, X.; Li, A.; Chi, X.; Lin, Y.; Liu, X.; Zhang, L.; Su, X.; Yin, Z.; Lin, H.; Gao, J. Hypoxia-activated prodrug enabling synchronous chemotherapy and HIF-1α downregulation for tumor treatment. Bioconj. Chem. 2021, 32, 983–990. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, M.-C.; Luo, M.-Z.; Penketh, P.G.; Baumann, R.P.; Shyam, K.; Sartorelli, A.C. 4-Nitrobenzyloxycarbonyl derivatives of O6-benzylguanine as hypoxia-activated prodrug inhibitors of O6-alkylguanine-DNA alkyltransferase (AGT), which produces resistance to agents targeting the O-6 position of DNA guanine. J. Med. Chem. 2011, 54, 7720–7728. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Seow, H.A.; Baumann, R.P.; Ishiguro, K.; Penketh, P.G.; Shyam, K.; Sartorelli, C. Design of a hypoxia-activated prodrug inhibitor of O6-alkylguanine-DNA alkyltransferase. Bioorg. Med. Chem. Lett. 2012, 22, 6242–6247. [Google Scholar] [CrossRef] [Green Version]
- Baumann, R.P.; Penketh, P.G.; Ishiguro, K.; Shyam, K.; Zhu, Y.L.; Sartorelli, A.C. Reductive activation of the prodrug 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4- nitrophenyl)ethoxy]carbonyl]hydrazine (KS119) selectively occurs in oxygen-deficient cells and overcomes O6-alkylguanine-DNA alkyltransferase mediated KS119 tumor cell resistance. Biochem. Pharmacol. 2010, 79, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penketh, P.G.; Baumann, R.P.; Shyam, K.; Williamson, H.S.; Ishiguro, K.; Zhu, R.; Eriksson, E.S.E.; Eriksson, L.A.; Sartorelli, A.C. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4- nitrophenyl)ethoxy]carbonyl]hydrazine (KS119): A cytotoxic prodrug with two stable conformations differing in biological and physical properties Chem Biol. Drug Des. 2011, 78, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Liu, Y.; Akintujoye, O.M.; Shyam, K.; Grove, T.A.; Sartorelli, A.C.; Rockwell, S. 3. Preliminary studies with a new hypoxia-selective cytotoxin, KS119W, in vitro and in vivo. Radiat. Res. 2012, 178, 126–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, K.P.; Penketh, P.G.; Shyam, K.; Sartorelli, A.C. Differential inhibition of cellular glutathione reductase activity by isocyanates generated from the antitumor prodrugs Cloretazine and BCNU. Biochem. Pharmacol. 2005, 69, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.F.; da Silva Filho, A.J.; Vasconcellos, M.L.A.A.; Luis de Santana, O. One-electron reduction potentials: Calibration of theoretical protocols for Morita–Baylis–Hillman nitroaromatic compounds in aprotic media. Molecules 2018, 23, 2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nocentinii, A.; Trallori, E.; Singh, S.; Lomelino, C.L.; Bartolucci, G.; Di Cesare, M.; Lorenzo, G.C.; McKenna, R.; Gratteri, P.; Supuran, C.T. 4-hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms IX and XII showing hypoxia-enhanced antiproliferative profiles. J. Med. Chem. 2018, 61, 10860–10874. [Google Scholar] [CrossRef] [PubMed]
- Sansom, G.N.; Kirk, N.S.; Guise, C.P.; Anderson, R.F.; Smaill, J.B.; Patterson, A.V.; Kelso, M.J. Prototyping kinase inhibitor-cytotoxin anticancer mutual prodrugs activated by tumour hypoxia: A chemical proof of concept study. Bioorg. Med. Chem. Lett. 2019, 29, 1215–1219. [Google Scholar] [CrossRef]
- O’Connor, L.J.; Cazares-Korner, C.; Saha, J.; Evans, C.N.G.; Stratford, M.R.L.; Hammond, E.M.; Conway, S.J. Efficient synthesis of 2-nitroimidazole derivatives and the bioreductive clinical candidate Evofosfamide (TH-302). Org. Chem. Front. 2015, 2, 1026–1029. [Google Scholar] [CrossRef]
- Takakusagi, I.; Kishimoto, S.; Naz, S.; Matsumoto, S.; Saito, K.; Hart, C.P.; Mitchell, J.B.; Krishna, M.C. Radiotherapy synergizes with the hypoxia-activated prodrug Evofosfamide: In vitro and in vivo studies. Antioxid. Redox Signal. 2018, 28, 131–140. [Google Scholar] [CrossRef]
- Hong, C.R.; Wilson, W.R.; Hicks, K.O. An intratumor pharmacokinetic/pharmacodynamic model for the hypoxia-activated prodrug Evofosfamide (HongTH-302): Monotherapy activity is not dependent on a bystander effect. Neoplasia 2019, 21, 159–171. [Google Scholar] [CrossRef]
- Kishimoto, S.; Brender, J.R.; Chandramouli, G.V.R.; Saida, Y.; Yamamoto, K.; Mitchell, J.B.; Krishna, M.C. Hypoxia-activated prodrug Evofosfamide treatment in pancreatic ductal adenocarcinoma xenografts alters the tumor redox status to potentiate radiotherapy. Antioxid. Redox Signal. 2021, 35, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.J.; Floyd, J.; Fichtel, L.; Michalek, J.; Kanakia, K.P.; Huang, S.; Reardon, D.; Wen, P.Y.; Lee, E.Q. Phase 2 trial of hypoxia activated evofosfamide (TH302) for treatment of recurrent bevacizumab-refractory glioblastoma. Sci. Rep. 2021, 11, 2306. [Google Scholar] [CrossRef] [PubMed]
- Grande, E.; Rodriguez-Antona, C.; Lopez, C.; Alonso-Gordoa, T.; Benavent, M.; Capdevila, J.; Teule, A.; Custodio, A.; Sevilla, I.; Hernando, J.; et al. Sunitinib and Evofosfamide (TH -302) in systemic treatment-naive patients with grade 1/2 metastatic pancreatic neuroendocrine tumors: The GETNE-1408 Trial. Oncologist 2021, 26, 941–949. [Google Scholar] [CrossRef]
- Tran, N.H.; Foster, N.R.; Mahipal, A.; Byrne, T.; Hubbard, J.; Silva, A.; Mody, K.; Alberts, S.; Borad, M.J. Phase IB study of sorafenib and evofosfamide in patients with advanced hepatocellular and renal cell carcinomas (NCCTG N1135, Alliance). Investig. New Drugs 2021, 39, 1072–1080. [Google Scholar] [CrossRef]
- Ghedira, D.; Voissiere, A.; Peyrode, C.; Kraiem, J.; Gerard, Y.; Maubert, E.; Vivier, M.; Miot-Noirault, E.; Chezal, J.-M.; Farhat, F.; et al. Structure-activity relationship study of hypoxia-activated prodrugs for proteoglycan-targeted chemotherapy in chondrosarcoma. Eur. J. Med. Chem. 2018, 158, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Gerard, Y.; Voissiere, A.; Peyrode, C.; Galmier, M.-J.; Maubert, E.; Ghedira, D.; Tarrit, S.; Gaumet, V.; Canitrot, D.; Miot-Noirault, E.; et al. Design, synthesis and evaluation of targeted hypoxia-activated prodrugs applied to chondrosarcoma chemotherapy. Bioorg. Chem. 2020, 98, 103747. [Google Scholar] [CrossRef] [PubMed]
- Penketh, P.; Williamson, H.; Shyam, K. Physicochemical considerations of tumor selective drug delivery and activity confinement with particular reference to 1,2-bis(sulfonyl)-1-alkylhydrazines delivery. Curr. Drug Deliv. 2020, 17, 362–374. [Google Scholar] [CrossRef]
- Karnthaler-Benbakka, C.; Groza, D.; Koblmueller, B.; Terenzi, A.; Holste, K.; Haider, M.; Baier, D.; Berger, W.; Heffeter, P.; Kowol, C.R.; et al. Targeting a targeted drug: An approach toward hypoxia-activatable tyrosine kinase inhibitor prodrugs. ChemMedChem 2016, 11, 2410–2421. [Google Scholar] [CrossRef]
- Calder, E.D.D.; Skwarska, A.; Sneddon, D.; Folkes, L.K.; Mistry, I.N.; Conway, S.J.; Hammond, E.M. Hypoxia-activated pro-drugs of the KDAC inhibitor vorinostat (SAHA). Tetrahedron 2020, 76, 131170. [Google Scholar] [CrossRef]
- Skwarska, A.; Calder, E.D.D.; Sneddon, D.; Bolland, H.; Odyniec, M.L.; Mistry, I.N.; Martin, J.; Folkes, L.K.; Conway, S.J.; Hammond, E.M. Development and pre-clinical testing of a novel hypoxia-activated KDAC inhibitor. Cell Chem. Biol. 2021, 28, 1258–1270.e13. [Google Scholar] [CrossRef]
- Ikeda, Y.; Hisano, H.; Nishikawa, Y.; Nagasaki, Y. Targeting and treatment of tumor hypoxia by newly designed prodrug possessing high permeability in solid tumors. Mol. Pharm. 2016, 13, 2283–2289. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Zhang, Q.; Lu, W. Synthesis and biological evaluation of hypoxia-activated prodrugs of SN-38. Eur. J. Med. Chem. 2017, 132, 135–141. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, C.; Yu, J.; Wang, L.; Lu, W. A novel multifunctional 2-nitroimidazole-based bioreductive linker and its application in hypoxia-activated prodrugs. Bioorg. Chem. 2020, 101, 103975. [Google Scholar] [CrossRef]
- Dragovich, P.S.; Broccatelli, F.; Chen, J.; Fan, P.; Le, H.; Mao, W.; Pillow, T.H.; Polson, A.G.; Wai, J.; Xu, Z.; et al. Design, synthesis, and biological evaluation of pyrrolobenzodiazepine-containing hypoxia-activated prodrugs. Bioorg. Med. Chem. Lett. 2017, 27, 5300–5304. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Jackson, R.K.; Dickson, B.D.; Cheng, G.J.; Lipert, B.; Liew, L.P.; Gu, Y.; Hunter, F.W.; Hay, M.P.; Wilson, W.R. Hypoxia-selective radiosensitisation by SN38023, a bioreductive prodrug of DNA-dependent protein kinase inhibitor IC87361. Biochem. Pharmacol. 2019, 169, 113641. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, E.T.; Geng, L.; Tan, J.; Chen, H.; Shir, Y.; Edwards, E.; Halbrook, J.; Kesicki, E.A.; Kashishian, A.; Hallahan, D.E. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. 2005, 65, 4987–4992. [Google Scholar] [CrossRef] [Green Version]
- Bielec, B.; Schueffl, H.; Terenzi, A.; Berger, W.; Heffeter, P.; Keppler, B.K.; Kowol, C.R. Development and biological investigations of hypoxia-sensitive prodrugs of the tyrosine kinase inhibitor crizotinib Bioorg. Chem. 2020, 99, 103778. [Google Scholar] [CrossRef]
- Dickson, B.D.; Wong, W.W.; Wilson, W.R.; Hay, M.P. Studies towards hypoxia-activated prodrugs of PARP inhibitors. Molecules 2019, 24, 1559. [Google Scholar] [CrossRef] [Green Version]
- Anduran, E.; Aspatwar, A.; Nanda-Kumar, P.; Suylen, D.; Bua, S.; Nocentini, A.; Parkkila, S.; Supuran, C.T.; Dubois, L.; Lambin, P.; et al. Hypoxia-activated prodrug derivatives of carbonic anhydrase inhibitors in benzenesulfonamide series: Synthesis and biological evaluation. Molecules 2020, 25, 2347. [Google Scholar] [CrossRef]
- Hay, M.P.; Wilson, W.R.; Denny, W.A. Design and synthesis of imidazolylmethyl carbamate prodrugs of alkylating agents. Tetrahedron 2000, 56, 645–657. [Google Scholar] [CrossRef]
- Hay, M.P.; Anderson, R.F.; Ferry, D.M.; Wilson, W.R.; Denny, W.A. Synthesis and evaluation of nitroheterocyclic carbamate prodrugs for use with nitroreductase-mediated GDEPT. J. Med. Chem. 2003, 46, 5533–5545. [Google Scholar] [CrossRef] [PubMed]
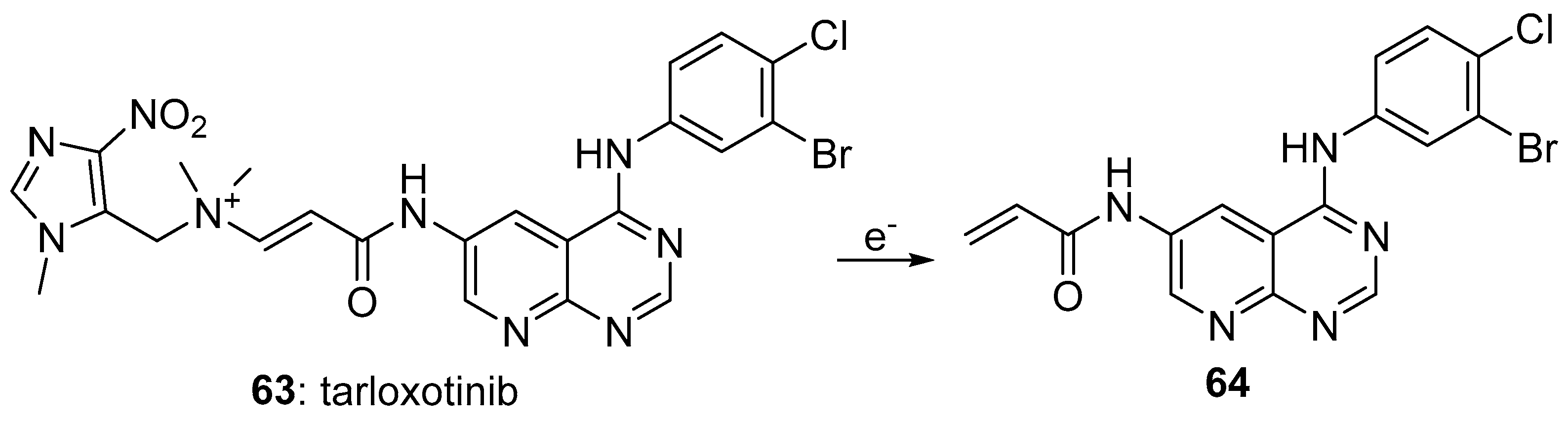
- Estrada-Bernal, A.; Le, A.T.; Doak, A.E.; Tirunagaru, V.G.; Silva, S.; Bull, M.R.; Smaill, J.B.; Patterson, A.V.; Kim, C.; Liu, S.V.; et al. Tarloxotinib is a hypoxia-activated pan-HER kinase inhibitor active against a broad range of HER-family oncogenes. Clin. Cancer Res. 2021, 27, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.; Asselin, M.-C.; Reymen, B.; Jackson, A.; Lambin, P.; West, C.M.L.; O’Connor, J.P.B.; Faivre-Finn, C. Targeting hypoxia to improve non-small cell lung cancer outcome. J. Natl. Cancer Inst. 2018, 110, 14–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, T.; Suda, K.; Nishimo, M.; Fujino, T.; Ohara, S.; Hamada, A.; Soh, J.; Tirunagaru, V.; Vellanki, A.; Doebele, R.C.; et al. Activity and mechanism of acquired resistance to tarloxotinib in HER2 mutant lung cancer: An in vitro study. Trans. Lung Cancer Res. 2021, 10, 3659–3670. [Google Scholar] [CrossRef] [PubMed]
- Study of Tarloxotinib in Patients with NSCLC (EGFR Exon 20i Insertion, HER2-Activating Mutations) and Other Solid Tumors with NRG1/ERBB Gene Fusions. ClinicalTrials.gov. NCT03805841; 16 January 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03805841 (accessed on 31 January 2022).
- Winn, B.A.; Shi, Z.; Carlson, G.J.; Wang, Y.; Nguyen, B.L.; Kelly, E.M.; Ross, R.D.; Hamel, E.; Chaplin, D.J.; Trawick, M.L.; et al. Bioreductively activatable prodrug conjugates of phenstatin designed to target tumor hypoxia. Bioorg. Med. Chem. Lett. 2017, 27, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.; Naylor, M.A.; Everett, S.A.; Stratford, M.R.L.; Lewis, G.; Hill, S.; Patel, K.B.; Wardman, P.; Davis, P.D. Synthesis and biological properties of bioreductively targeted nitrothienyl prodrugs of combretastatin A-4. Mol Cancer Ther. 2006, 5, 2886–2894. [Google Scholar] [CrossRef] [Green Version]
- Winn, B.A.; Laxman, D.; Kuch, B.; MacDonough, M.T.; Strecker, T.E.; Wang, Y.; Shi, Z.; Gerberich, J.I.; Mondal, D.; Ramirez, A.J.; et al. Bioreductively activatable prodrug conjugates of combretastatin A-1 and combretastatin A-4 as anticancer agents targeted toward tumor-associated hypoxia. J. Nat. Prod. 2020, 83, 937–954. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, H.; Wu, J.; Gao, F.; Zhang, C.; Hu, X.; Liu, Q.; Wei, Y.; Zhuang, J.; Huang, X. A novel model system for understanding anticancer activity of hypoxia-activated prodrugs. Mol. Pharm. 2020, 17, 2072–2082. [Google Scholar] [CrossRef]
- Tercel, M.; Lee, A.E.; Hogg, A.; Anderson, R.F.; Lee, H.H.; Siim, B.G.; Denny, W.A.; Wilson, W.R. Hypoxia-selective antitumor agents. 16. Nitroarylmethyl quaternary salts as bioreductive prodrugs of the alkylating agent mechlorethamine. J. Med. Chem. 2001, 44, 3511–3522. [Google Scholar] [CrossRef]
- Atwell, G.J.; Wilson, W.R.; Denny, W.A. Synthesis and cytotoxicity of amino analogues of the potent DNA alkylating agent seco-CBI-TMI. Bioorg. Med. Chem. Lett. 1997, 7, 1493–1496. [Google Scholar] [CrossRef]
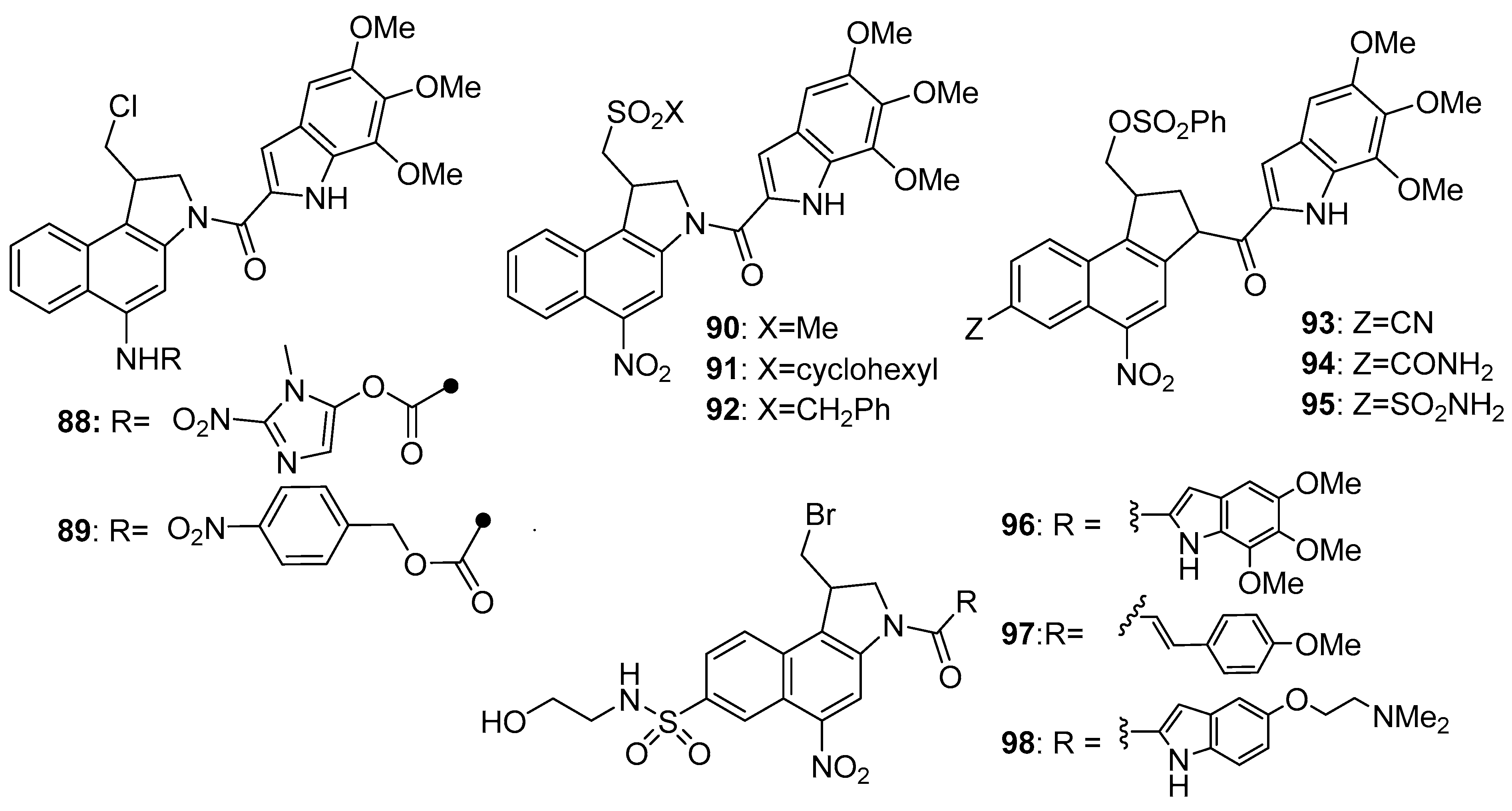
- Wilson, W.R.; Stribbling, S.M.; Pruijn, F.B.; Syddall, S.P.; Patterson, A.V.; Liyanage, H.D.S.; Smith, E.; Botting, K.J.; Tercel, M. Nitro-chloromethylbenzindolines: Hypoxia-activated prodrugs of potent adenine N3 DNA minor groove alkylators. Mol. Cancer Ther. 2009, 8, 2903–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwell, G.J.; Tercel, M.; Boyd, M.; Wilson, W.R.; Denny, W.A. Synthesis and cytotoxicity of 5-amino-1-(chloromethyl)-3-[(5,6,7-trimethoxyindol-2-yl)carbonyl]-1,2-dihydro-3H-benz[e]indole (amino-seco-CBI-TMI) and related 5-(alkylamino) analogs: New DNA minor groove alkylating agents. J. Org. Chem. 1998, 63, 9414–9420. [Google Scholar] [CrossRef]
- Heinrich, D.M.; Youte, J.-J.; Denny, W.A.; Tercel, M. A new enantioselective approach to the core structure of hypoxia selective prodrugs related to the duocarmycins. Tetrahedron Lett. 2011, 52, 7000–7003. [Google Scholar] [CrossRef]
- Tercel, M.; Atwell, G.J.; Yang, S.; Stevenson, R.J.; Botting, K.J.; Boyd, M.; Smith, E.; Anderson, R.F.; Denny, W.A.; Wilson, W.R.; et al. Substituent effects on hypoxic selectivity of nitro seco-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (nitroCBI) prodrugs of DNA minor groove alkylating agents. J. Med. Chem. 2009, 52, 7258–7272. [Google Scholar] [CrossRef]
- Tercel, M.; Yang, S.; Atwell, G.J.; Smith, E.; Gu, Y.; Anderson, R.F.; Denny, W.A.; Wilson, W.R.; Pruijn, F.B. Hypoxic selectivity and solubility—Investigating the properties of A-ring substituted nitro seco-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-ones (nitroCBIs) as hypoxia-activated prodrugs for antitumor therapy. Bioorg. Med. Chem. 2010, 18, 4997–5006. [Google Scholar] [CrossRef]
- Tercel, M.; Atwell, G.J.; Yang, S.; Ashoorzadeh, A.; Stevenson, R.J.; Botting, K.J.; Gu, Y.; Mehta, S.Y.; Denny, W.A.; Wilson, W.R.; et al. Selective treatment of hypoxic tumor cells in vivo: Phosphate pre-prodrugs of nitro analogs of the duocarmycins. Angew. Chem. Int. Ed. 2011, 50, 2606–2609. [Google Scholar] [CrossRef] [PubMed]
- Tercel, M.; Lee, H.H.; Mehta, S.Y.; Youte, J.-J.; Tendoung, S.Y.; Bai, S.; Liyanage, H.D.S.; Pruijn, F.B. Influence of a basic side chain on the properties of hypoxia-selective nitro analogues of the duocarmycins: Demonstration of substantial anticancer activity in combination with irradiation or chemotherapy. J. Med. Chem. 2017, 60, 5834–5856. [Google Scholar] [CrossRef]
- Lee, H.H.; Dickson, B.D.; Stevenson, R.J.; Yang, S.; Tercel, M. Optimized synthesis of a nitroCBI hypoxia-activated prodrug with substantial anticancer activity. Tetrahedron 2019, 75, 3001–3007. [Google Scholar] [CrossRef]
- Hong, C.R.; Mehta, S.Y.; Liyanage, H.D.S.; McManaway, S.P.; Lee, H.H.; Jaiswal, J.K.; Bogle, G.; Tercel, M.; Pruijn, F.B.; Wilson, W.R.; et al. Spatially-resolved pharmacokinetic/pharmacodynamic modelling of bystander effects of a nitrochloromethylbenzindoline hypoxia-activated prodrug. Cancer Chemother. Pharmacol. 2021, 88, 673–687. [Google Scholar] [CrossRef]
- Hay, M.P.; Sykes, B.M.; Denny, W.A.; Wilson, W.R. A 2-nitroimidazole carbamate prodrug of 5-amino-1-(chloromethyl)-3-[(5,6,7-trimethoxyindol-2-yl)carbonyl]-1,2-dihydro-3H-benz[e]indole (amino-seco-CBI-TMI) for use with ADEPT and GDEPT. Bioorg. Med. Chem. Lett. 1999, 15, 2237–2242. [Google Scholar] [CrossRef]
- Hay, M.P.; Atwell, G.J.; Wilson, W.R.; Pullen, S.M.; Denny, W.A. Structure-activity relationships for 4-nitrobenzylcarbamates of 5-aminobenz[e]indoline minor groove alkylating agents as prodrugs for gene therapy in conjunction with E. coli nitroreductase. J. Med. Chem. 2003, 46, 2456–2466. [Google Scholar] [CrossRef]
- Ashoorzadeh, A.; Atwell, G.J.; Pruijn, F.B.; Wilson, W.R.; Tercel, M.; Denny, W.A.; Stevenson, R.J. The effect of sulfonate leaving groups on the hypoxia-selective toxicity of nitro analogs of the duocarmycins. Bioorg. Med. Chem. 2011, 19, 4851–4860. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.J.; Denny, W.A.; Tercel, M.; Pruijn, F.B.; Ashoorzadeh, A. Nitro seco analogs of the duocarmycins containing sulfonate leaving groups as hypoxia-activated prodrugs for cancer therapy. J. Med. Chem. 2012, 55, 2780–2802. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.J.; Denny, W.A.; Ashoorzadeh, A.; Pruijn, F.B.; van Leeuwen, F.; Tercel, M. The effect of a bromide leaving group on the properties of nitro analogs of the duocarmycins as hypoxia-activated prodrugs and phosphate pre-prodrugs for antitumor therapy. Bioorg. Med. Chem. 2011, 19, 5989–5998. [Google Scholar] [CrossRef] [PubMed]
- Su, M.X.; Zhang, L.-L.; Huang, Z.-J.; Shi, J.-J.; Lu, J.-L. Investigational hypoxia-activated prodrugs: Making sense of future development. Curr. Drug Targets 2019, 20, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Anduran, E.; Dubois, L.J.; Lambin, P.; Winum, J.-Y. Hypoxia-activated prodrug derivatives of anti-cancer drugs: A patent review 2006–2021. Exp. Opin. Ther. Pat. 2021, 31, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Spiegelberg, L.; Houben, R.; Niemans, R.; Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P.; et al. Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
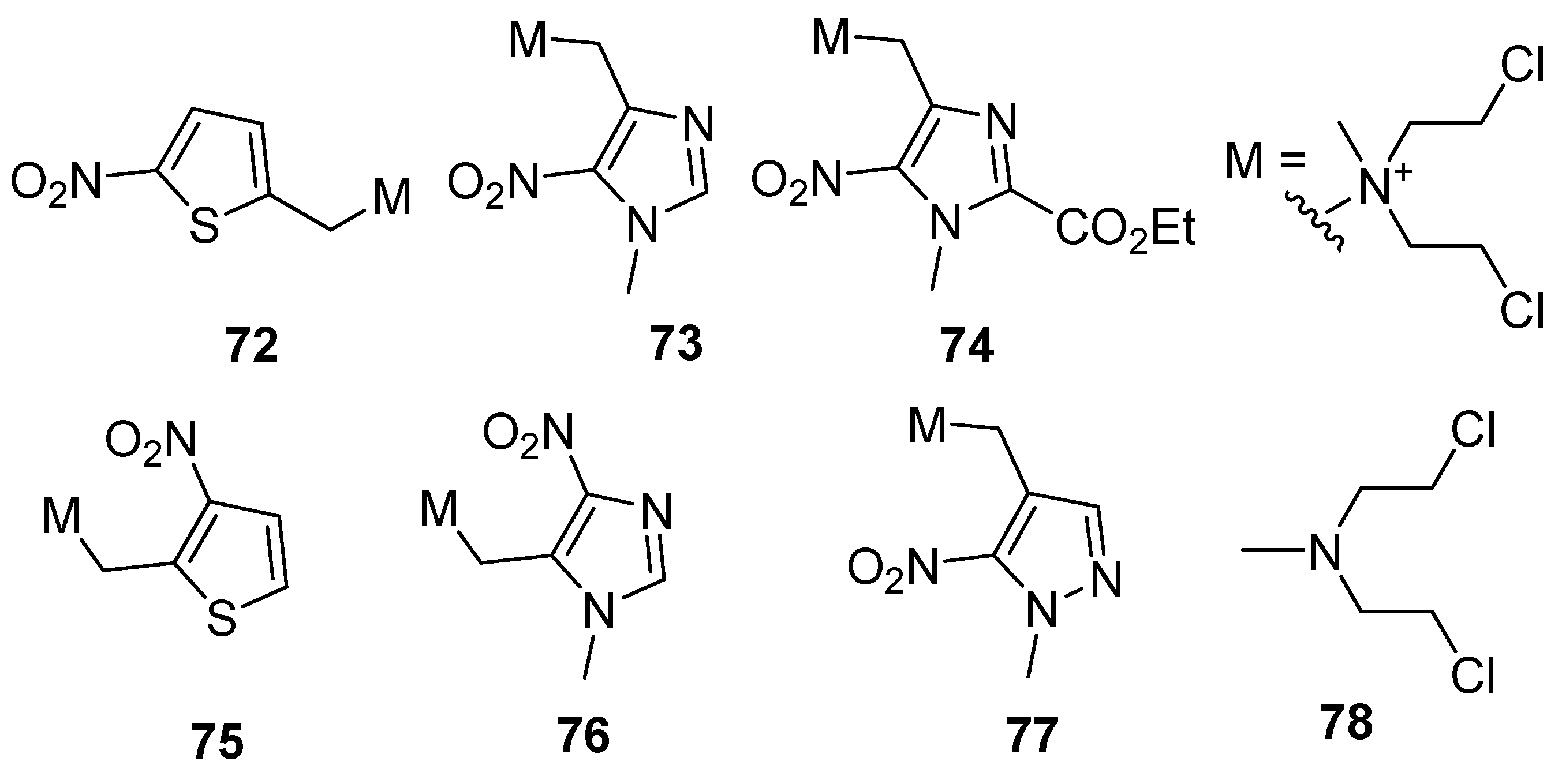
| Cmpd | E1 (mV) | C10 Air (µM) | C10 N2 (µM) | HR |
|---|---|---|---|---|
| 72 | −277 | 22 | 1.3 | 16 |
| 73 | −287 | 3450 | 2.2 | 2500 |
| 74 | −344 | 30 | 4.5 | 14 |
| 75 | −361 | 30 | 6 | 6 |
| 76 | −397 | 57 | 1.2 | 46 |
| 77 | −500 | 245 | 18 | 16 |
| 78 | 1.0 | 0.85 | 1.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denny, W.A. Nitroaromatic Hypoxia-Activated Prodrugs for Cancer Therapy. Pharmaceuticals 2022, 15, 187. https://doi.org/10.3390/ph15020187
Denny WA. Nitroaromatic Hypoxia-Activated Prodrugs for Cancer Therapy. Pharmaceuticals. 2022; 15(2):187. https://doi.org/10.3390/ph15020187
Chicago/Turabian StyleDenny, William A. 2022. "Nitroaromatic Hypoxia-Activated Prodrugs for Cancer Therapy" Pharmaceuticals 15, no. 2: 187. https://doi.org/10.3390/ph15020187
APA StyleDenny, W. A. (2022). Nitroaromatic Hypoxia-Activated Prodrugs for Cancer Therapy. Pharmaceuticals, 15(2), 187. https://doi.org/10.3390/ph15020187