
Interrogation of the Structure–Activity Relationship of a Lipophilic Nitroaromatic Prodrug Series Designed for Cancer Gene Therapy Applications

 , , and
, , and
Abstract
:1. Introduction
2. Results
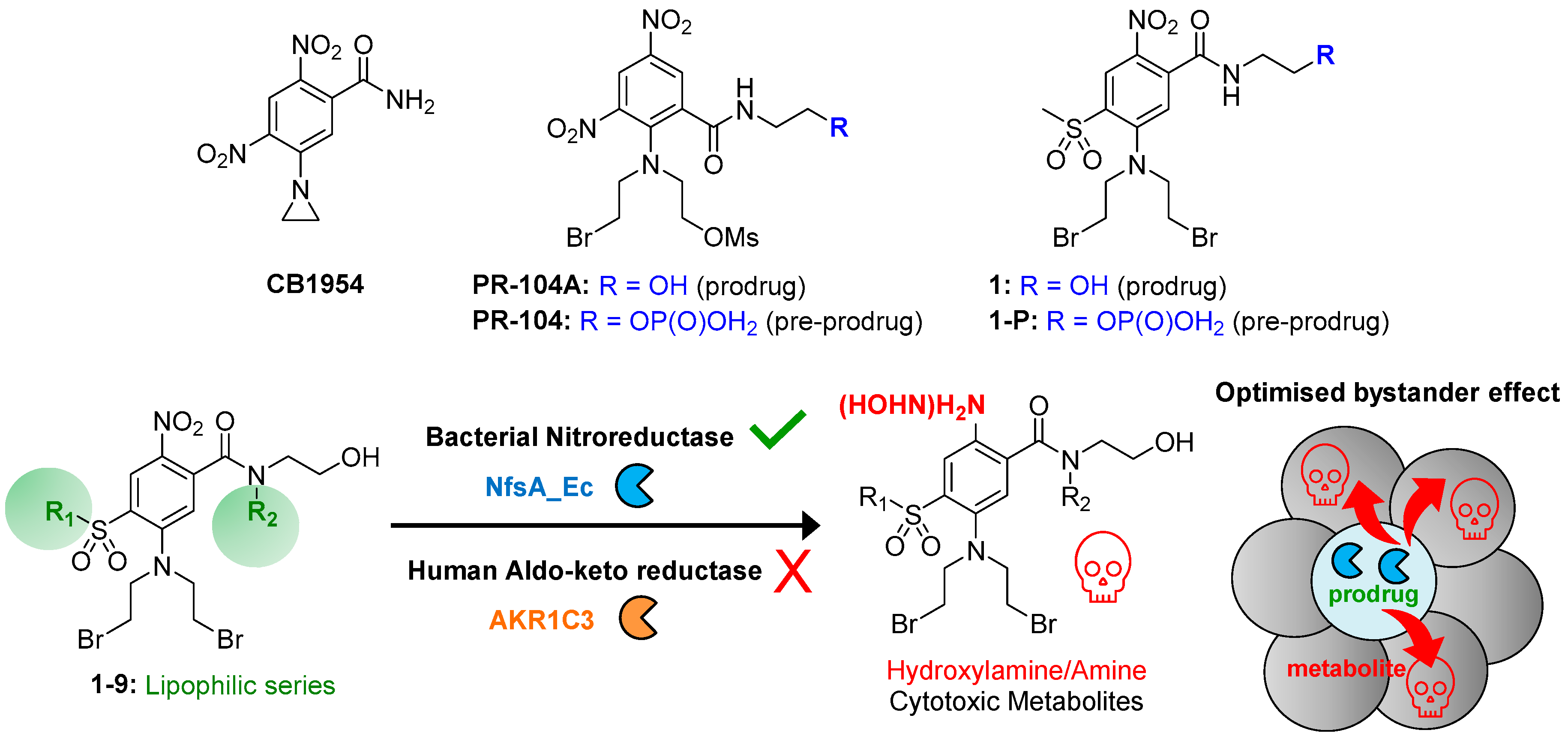
2.1. Drug Design Rationale
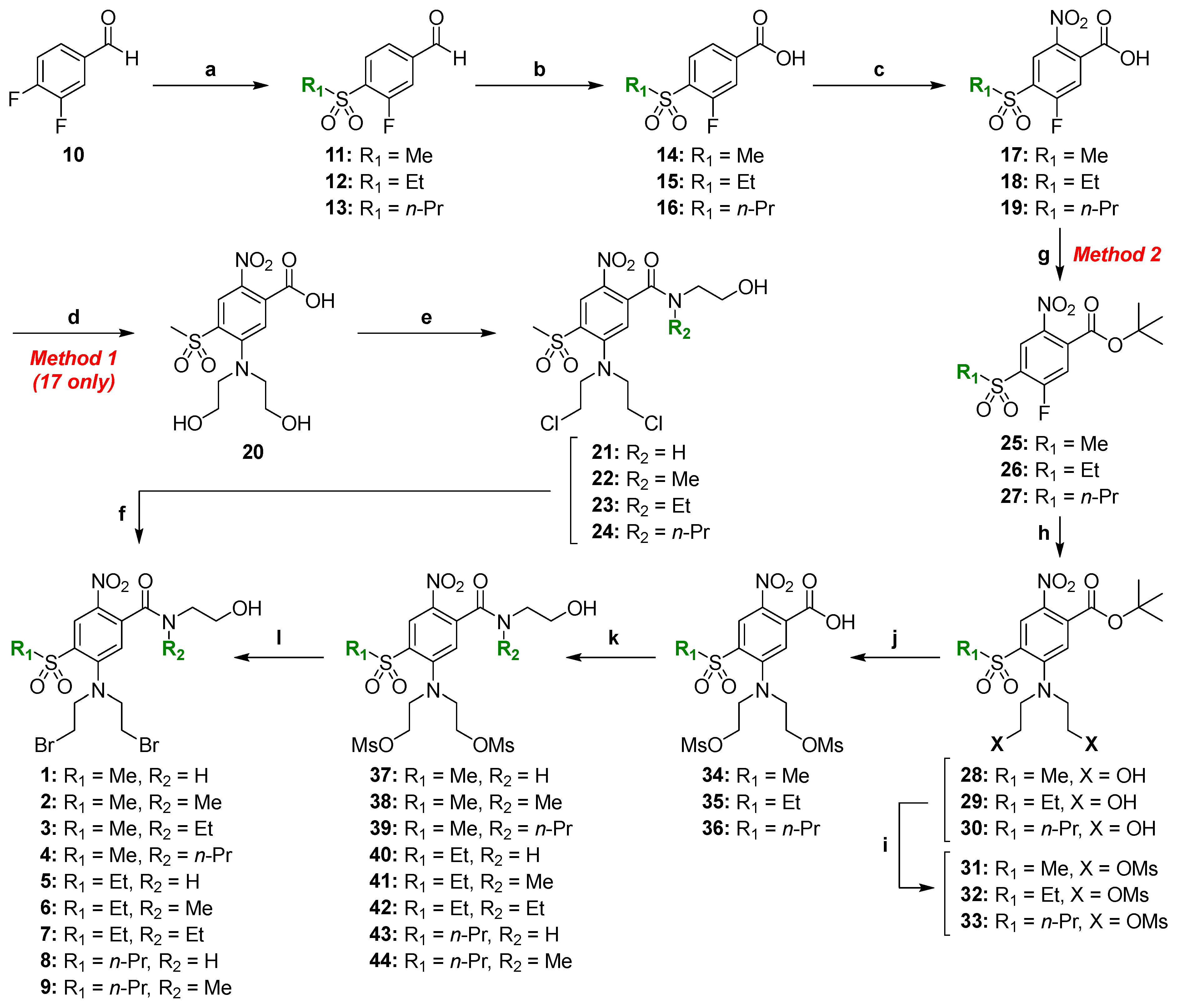
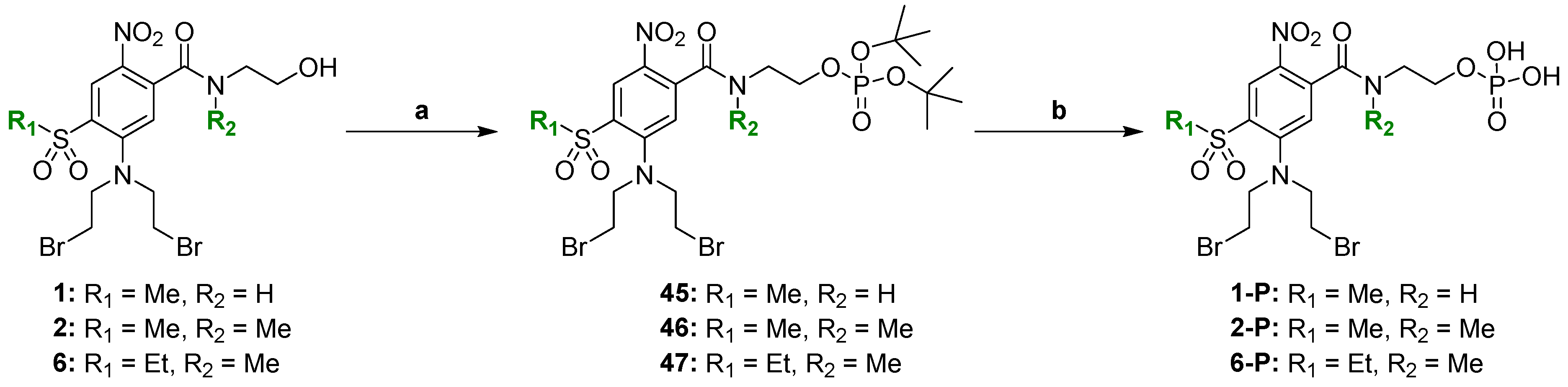
2.2. Chemical Synthesis
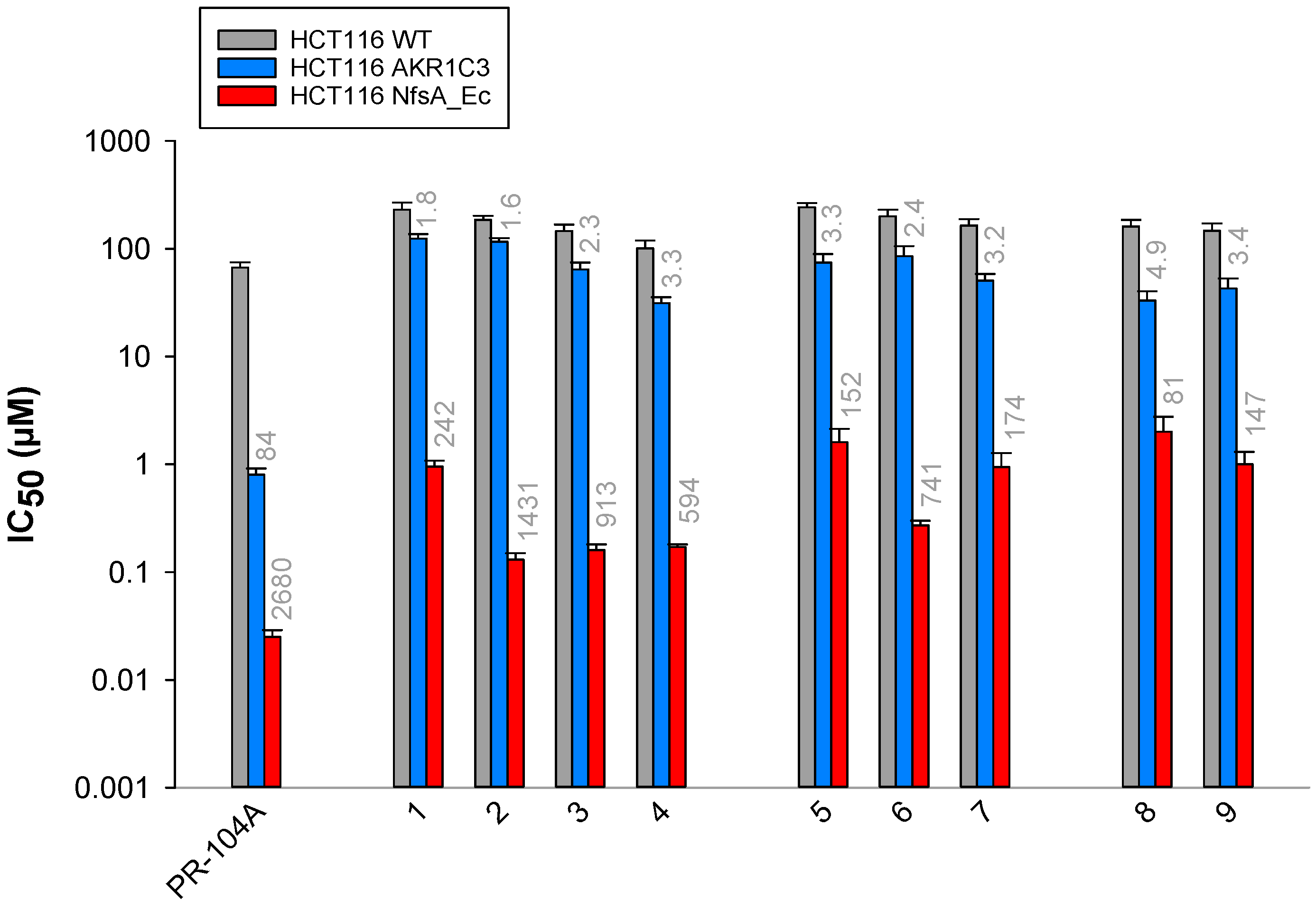
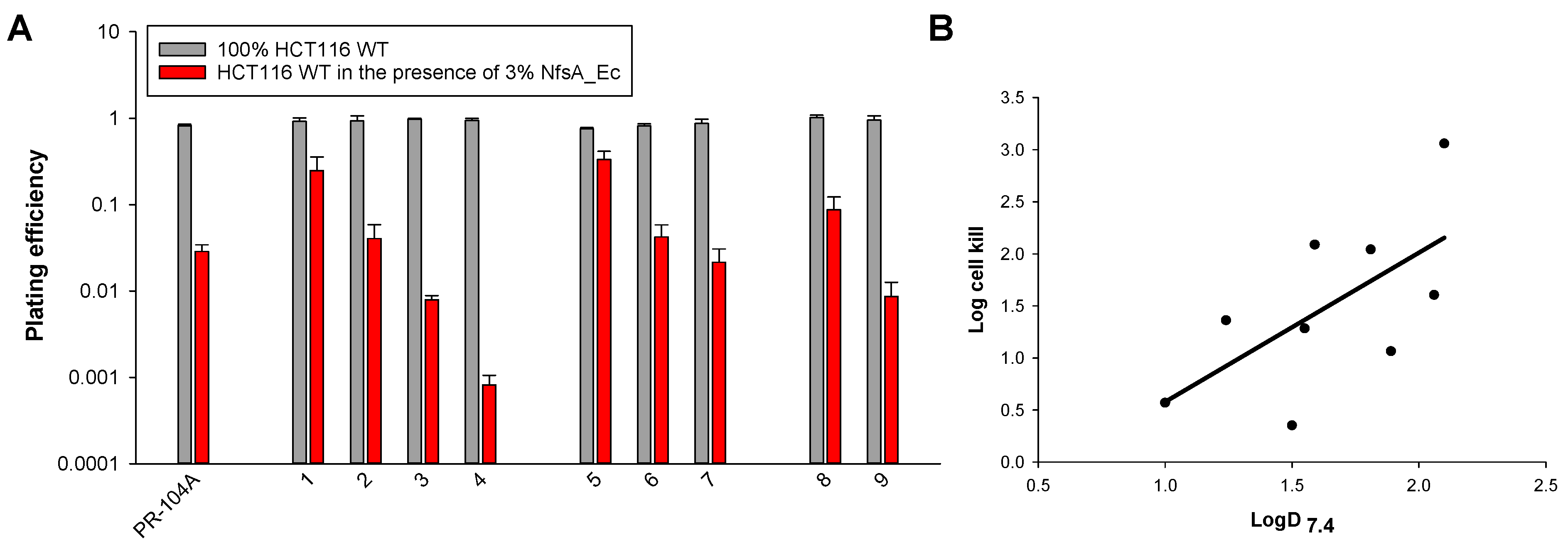
2.3. Anti-Proliferative Activity In Vitro
2.4. Interrogation of the Lipophilic SAR in Three Dimensions
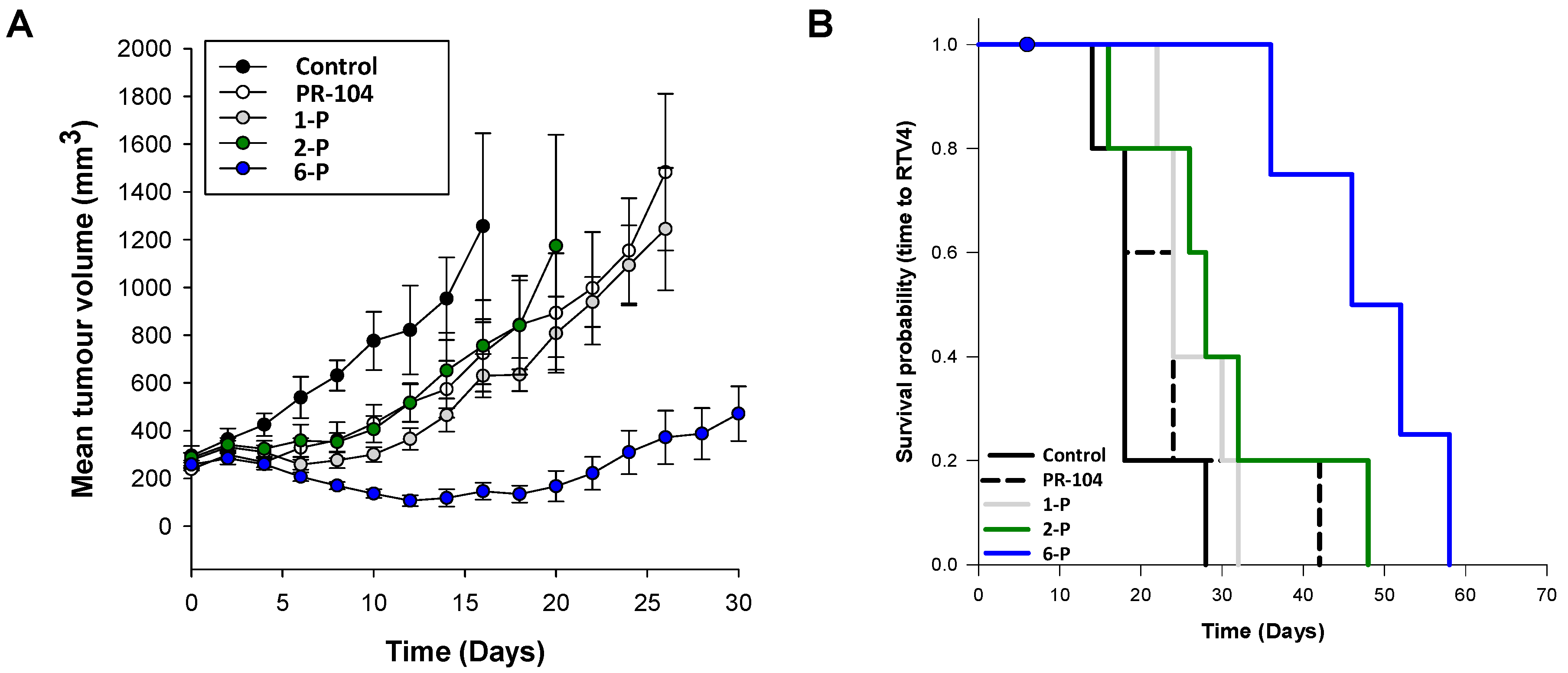
2.5. Activity of the Lead Compounds against NfsA_Ec-Expressing Tumors
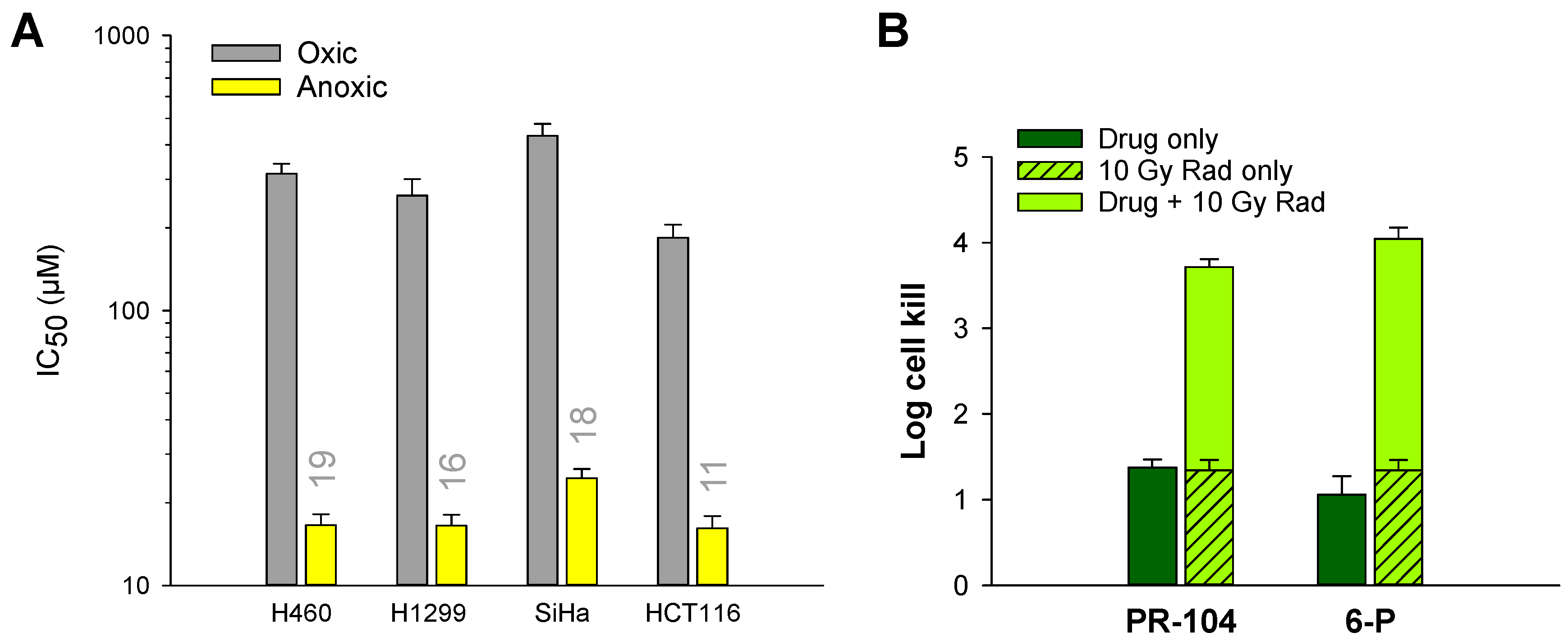
2.6. Hypoxia Selectivity of 6 and 6-P
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.1.1. Synthesis of Compound 2—Method 1
4.1.2. Synthesis of Compound 1—Method 2
4.1.3. Synthesis of Compounds 5 and 6—Method 2
4.1.4. Synthesis of Compound 2-P
4.2. Drug Information and Formulation
4.3. Determination of LogD7.4
4.4. Cell Lines and Candidate Gene Expression
4.5. Anti-Proliferative (IC50) Assays
4.6. Multicellular Layer Assays
4.7. Animal Husbandry
4.8. Tumor Growth Delay Assay
4.9. Tumor Excision Assay
4.10. Ex Vivo Phenotyping of Tumor Xenografts
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McNeish, I.A.; Searle, P.F.; Young, L.S.; Kerr, D.J. Gene directed enzyme prodrug therapy for cancer. Adv. Drug Deliv. Rev. 1997, 26, 173–184. [Google Scholar] [CrossRef]
- Zhang, J.; Kale, V.; Chen, M. Gene-Directed Enzyme Prodrug Therapy. AAPS J. 2015, 17, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, S.M.; Abboud, C.N.; Whartenby, K.A.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The “bystander effect”: Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993, 53, 5274–5283. [Google Scholar] [PubMed]
- Wilson, W.R.; Pullen, S.M.; Hogg, A.; Helsby, N.A.; Hicks, K.O.; Denny, W.A. Quantitation of bystander effects in nitroreductase suicide gene therapy using three-dimensional cell cultures. Cancer Res. 2002, 62, 1425–1432. [Google Scholar]
- Portsmouth, D.; Hlavaty, J.; Renner, M. Suicide genes for cancer therapy. Mol. Aspects Med. 2007, 28, 4–41. [Google Scholar] [CrossRef]
- Bridgewater, J.A.; Knox, R.J.; Pitts, J.D.; Collins, M.K.; Springer, C.J. The bystander effect of the nitroreductase/CB1954 enzyme/prodrug system is due to a cell-permeable metabolite. Hum. Gene Ther. 1997, 8, 709–717. [Google Scholar] [CrossRef]
- Rainov, N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Faulds, D.; Heel, R.C. Ganciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in cytomegalovirus infections. Drugs 1990, 39, 597–638. [Google Scholar] [CrossRef]
- Mesnil, M.; Yamasaki, H. Bystander Effect in Herpes Simplex Virus-Thymidine Kinase/Ganciclovir Cancer Gene Therapy: Role of Gap-junctional Intercellular Communication1. Cancer Res. 2000, 60, 3989–3999. [Google Scholar] [PubMed]
- Williams, E.M.; Little, R.F.; Mowday, A.M.; Rich, M.H.; Chan-Hyams, J.V.; Copp, J.N.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Nitroreductase gene-directed enzyme prodrug therapy: Insights and advances toward clinical utility. Biochem. J. 2015, 471, 131–153. [Google Scholar] [CrossRef]
- Denny, W.A. Nitroreductase-based GDEPT. Curr. Pharm. Des. 2002, 8, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Anlezark, G.M.; Knox, R.J.; Friedlos, F.; Sherwood, R.F.; Melton, R.G. The bioactivation of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954)-II: A comparison of an Escherichia coli nitroreductase and Walker DT diaphorase. Biochem. Pharmacol. 1992, 44, 2297–2301. [Google Scholar] [CrossRef]
- Liu, S.C.; Ahn, G.O.; Kioi, M.; Dorie, M.J.; Patterson, A.V.; Brown, J.M. Optimised Clostridium-directed enzyme prodrug therapy improves the antitumor activity of the novel DNA crosslinking agent PR-104. Cancer Res. 2008, 68, 7995–8003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, A.V.; Ferry, D.M.; Edmunds, S.J.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA crosslinking agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, G.A.; Copp, J.N.; Mowday, A.M.; Guise, C.P.; Syddall, S.P.; Williams, E.M.; Horvat, C.N.; Swe, P.M.; Ashoorzadeh, A.; Denny, W.A.; et al. Creation and screening of a multi-family bacterial oxidoreductase library to discover novel nitroreductases that efficiently activate the bioreductive prodrugs CB1954 and PR-104A. Biochem. Pharmacol. 2013, 85, 1091–1103. [Google Scholar] [CrossRef]
- Copp, J.N.; Mowday, A.M.; Williams, E.M.; Guise, C.P.; Ashoorzadeh, A.; Sharrock, A.V.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Engineering a Multifunctional Nitroreductase for Improved Activation of Prodrugs and PET Probes for Cancer Gene Therapy. Cell Chem. Biol. 2017, 24, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Heap, J.T.; Theys, J.; Ehsaan, M.; Kubiak, A.M.; Dubois, L.; Paesmans, K.; Van Mellaert, L.; Knox, R.; Kuehne, S.A.; Lambin, P.; et al. Spores of Clostridium engineered for clinical efficacy and safety cause regression and cure of tumours in vivo. Oncotarget 2014, 5, 1761–1769. [Google Scholar] [CrossRef] [Green Version]
- Chung-Faye, G.; Palmer, D.; Anderson, D.; Clark, J.; Downes, M.; Baddeley, J.; Hussain, S.; Murray, P.I.; Searle, P.; Seymour, L.; et al. Virus-directed, Enzyme Prodrug Therapy with Nitroimidazole Reductase: A Phase I and Pharmacokinetic Study of its Prodrug, CB1954. Clin. Cancer. Res. 2001, 7, 2662–2668. [Google Scholar]
- Tang, M.H.; Helsby, N.A.; Wilson, W.R.; Tingle, M.D. Aerobic 2- and 4-nitroreduction of CB 1954 by human liver. Toxicology 2005, 216, 129–139. [Google Scholar] [CrossRef]
- McKeage, M.J.; Gu, Y.; Wilson, W.R.; Hill, A.; Amies, K.; Melink, T.J.; Jameson, M.B. A phase I trial of PR-104, a pre-prodrug of the bioreductive prodrug PR-104A, given weekly to solid tumour patients. BMC Cancer 2011, 11, 432. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.; Choy, S.F.; Hicks, K.O.; Melink, T.J.; Holford, N.H.G.; Wilson, W.R. A combined pharmacokinetic model for the hypoxia-targeted prodrug PR-104A in humans, dogs, rats and mice predicts species differences in clearance and toxicity. Cancer Chemother. Pharmacol. 2011, 67, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Abbattista, M.R.; Ashoorzadeh, A.; Guise, C.P.; Mowday, A.M.; Mittra, R.; Silva, S.; Hicks, K.O.; Bull, M.R.; Jackson-Patel, V.; Lin, X.; et al. Restoring Tumour Selectivity of the Bioreductive Prodrug PR-104 by Developing an Analogue Resistant to Aerobic Metabolism by Human Aldo-Keto Reductase 1C3. Pharmaceuticals 2021, 14, 1231. [Google Scholar] [CrossRef] [PubMed]
- Van Der Wiel, A.M.A.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.; Lieuwes, N.; Biemans, R.; Lin, X.; et al. Selectively Targeting Tumor Hypoxia with the Hypoxia-Activated Prodrug CP-506. Mol. Cancer Ther. 2021, 20, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Toenjes, S.T.; Gustafson, J.L. Atropisomerism in medicinal chemistry: Challenges and opportunities. Future Med. Chem. 2018, 10, 409–422. [Google Scholar] [CrossRef]
- Betson, M.S.; Clayden, J.; Helliwell, M.; Johnson, P.; Lai, L.W.; Pink, J.H.; Stimson, C.C.; Vassiliou, N.; Westlund, N.; Yasin, S.A.; et al. Conformational preference in aromatic amides bearing chiral ortho substituents: Its origin and application to relayed stereocontrol. Org. Biomol. Chem. 2006, 4, 424–443. [Google Scholar] [CrossRef]
- Pros, G.J.; Bloomfield, A.J. Why Do N-Alkylated Anilides Bend Over? the Factors Dictating the Divergent Conformational Preferences of 2° and 3° N-Aryl Amides. J. Phys. Chem. A 2019, 123, 7609–7618. [Google Scholar] [CrossRef]
- Bain, A.D. Annual Reports on NMR Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2008; Volume 63. [Google Scholar]
- Greenberg, A.; Breneman, C.M.; Liebmann, J. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Ribet, J.P.; Pena, R.; Maurel, J.L.; Belin, C.; Tillard, M.; Vacher, B.; Bonnaud, B.; Colpaert, F. Conformational analysis and crystal structure of {[1-(3-chloro-4- fluorobenzoyl)-4-fluoropiperidin-4yl]methyl}[(5-methylpyridin-2-yl)methyl]amine, fumaric acid salt. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 62, 353–363. [Google Scholar] [CrossRef]
- Finkelstein, H. Darstellung organischer Jodide aus den entsprechenden Bromiden und Chloriden. Ber. Dtsch. Chem. Ges. 1910, 43, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.B.; March, J. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th ed.; Wiley-Interscience: New York, NY, USA, 2007; ISBN 978-0-471-72091-1. [Google Scholar]
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 978-0-470-05348-5. [Google Scholar]
- Mowday, A.M.; Copp, J.N.; Syddall, S.P.; Dubois, L.J.; Wang, J.; Lieuwes, N.G.; Biemans, R.; Ashoorzadeh, A.; Abbattista, M.R.; Williams, E.M.; et al. E. coli nitroreductase NfsA is a reporter gene for non-invasive PET imaging in cancer gene therapy applications. Theranostics 2020, 10, 10548–10562. [Google Scholar] [CrossRef]
- Atwell, G.J.; Yang, S.; Pruijn, F.B.; Pullen, S.M.; Hogg, A.; Patterson, A.V.; Wilson, W.R.; Denny, W.A. Synthesis and Structure-Activity Relationships for 2,4-Dinitrobenzamide-5 mustards as Prodrugs for the Escherichia coli nfsB Nitroreductase in Gene Therapy. J. Med. Chem. 2007, 50, 1197–1212. [Google Scholar] [CrossRef]
- Freeman-Cook, K.D.; Hoffman, R.L.; Johnson, T.W. Lipophilic efficiency: The most important efficiency metric in medicinal chemistry. Future Med. Chem 2013, 5, 113–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komoto, J.; Yamada, T.; Watanabe, K.; Takusagawa, F. Crystal structure of human prostaglandin F synthase (AKR1C3). Biochemistry 2004, 43, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.L.; Ride, J.P.; Bunce, C.M.; Desmond, J.C.; Cummings, S.M.; White, S.A. Crystal structures of prostaglandin D(2) 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res. 2004, 64, 1802–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, D.C.; Macann, A.; Wilson, W.R. Therapeutic targeting of the hypoxic tumour microenvironment. Nat. Rev. Clin. Oncol. 2021, 18, 751–772. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Chang, W.H.; Lai, A.G. The hypoxic tumour microenvironment: A safe haven for immunosuppressive cells and a therapeutic barrier to overcome. Cancer Lett. 2020, 487, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Kratzke, R.A. Oncolytic virus therapy for cancer: The first wave of translational clinical trials. Transl. Res. 2013, 161, 355–364. [Google Scholar] [CrossRef]
- Pipiya, T.; Sauthoff, H.; Huang, Y.Q.; Chang, B.; Cheng, J.; Heitner, S.; Chen, S.; Rom, W.N.; Hay, J.G. Hypoxia reduces adenoviral replication in cancer cells by downregulation of viral protein expression. Gene Ther. 2005, 12, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Atwell, G.J.; Denny, W.A. Synthesis of asymmetric halomesylate mustards with aziridineethanol/alkali metal halides: Application to an improved synthesis of the hypoxia prodrug PR-104. Tetrahedron 2007, 63, 5470–5476. [Google Scholar] [CrossRef]
- Atwell, G.J.; Denny, W.A. Synthesis of 3H- and 2H4-labelled versions of the hypoxia-activated pre-prodrug 2-[(2-bromoethyl)-2,4-dinitro-6-[[[2-(phosphonooxy)ethyl]amino]carbonyl]anilino]ethyl methanesulfonate (PR-104). J. Label. Comp. Radiopharm. 2007, 50, 7–12. [Google Scholar] [CrossRef]
- Shah, J.C.; Chen, J.R.; Chow, D. Preformulation study of etoposide: Identification of physicochemical characteristics responsible for the low and erratic bioavailability of etoposide. Pharm. Res. 1989, 6, 408–412. [Google Scholar] [CrossRef]
- Rivory, L.P.; Avent, K.M.; Pond, S.M. Effects of lipophilicity and protein binding on the hepatocellular uptake and hepatic disposition of two anthracyclines, doxorubicin and iododoxorubicin. Cancer Chemother. Pharmacol. 1996, 38, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Siim, B.G.; Hicks, K.O.; Pullen, S.M.; van Zijl, P.L.; Denny, W.A.; Wilson, W.R. Comparison of aromatic and tertiary amine N-oxides of acridine DNA intercalators as bioreductive drugs. Cytotoxicity, DNA binding, cellular uptake, and metabolism. Biochem. Pharmacol. 2000, 60, 969–978. [Google Scholar] [CrossRef]
- Guise, C.P.; Abbattista, M.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helsby, N.A.; Atwell, G.J.; Yang, S.; Palmer, B.D.; Anderson, R.F.; Pullen, S.M.; Ferry, D.M.; Hogg, A.; Wilson, W.R.; Denny, W.A. Aziridinyldinitrobenzamides: Synthesis and structure-activity relationships for activation by E. coli nitroreductase. J. Med. Chem. 2004, 47, 3295–3307. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
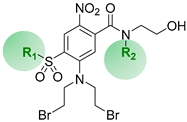 | |||
| Cmpd | R1 | R2 | LogD7.4 a |
| 1 | Me | H | 1.0 |
| 2 | Me | Me | 1.2 |
| 3 | Me | Et | 1.6 |
| 4 | Me | n-Pr | 2.1 |
| 5 | Et | H | 1.5 |
| 6 | Et | Me | 1.6 |
| 7 | Et | Et | 1.9 b |
| 8 | n-Pr | H | 1.8 b |
| 9 | n-Pr | Me | 2.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashoorzadeh, A.; Mowday, A.M.; Guise, C.P.; Silva, S.; Bull, M.R.; Abbattista, M.R.; Copp, J.N.; Williams, E.M.; Ackerley, D.F.; Patterson, A.V.; et al. Interrogation of the Structure–Activity Relationship of a Lipophilic Nitroaromatic Prodrug Series Designed for Cancer Gene Therapy Applications. Pharmaceuticals 2022, 15, 185. https://doi.org/10.3390/ph15020185
Ashoorzadeh A, Mowday AM, Guise CP, Silva S, Bull MR, Abbattista MR, Copp JN, Williams EM, Ackerley DF, Patterson AV, et al. Interrogation of the Structure–Activity Relationship of a Lipophilic Nitroaromatic Prodrug Series Designed for Cancer Gene Therapy Applications. Pharmaceuticals. 2022; 15(2):185. https://doi.org/10.3390/ph15020185
Chicago/Turabian StyleAshoorzadeh, Amir, Alexandra M. Mowday, Christopher P. Guise, Shevan Silva, Matthew R. Bull, Maria R. Abbattista, Janine N. Copp, Elsie M. Williams, David F. Ackerley, Adam V. Patterson, and et al. 2022. "Interrogation of the Structure–Activity Relationship of a Lipophilic Nitroaromatic Prodrug Series Designed for Cancer Gene Therapy Applications" Pharmaceuticals 15, no. 2: 185. https://doi.org/10.3390/ph15020185
APA StyleAshoorzadeh, A., Mowday, A. M., Guise, C. P., Silva, S., Bull, M. R., Abbattista, M. R., Copp, J. N., Williams, E. M., Ackerley, D. F., Patterson, A. V., & Smaill, J. B. (2022). Interrogation of the Structure–Activity Relationship of a Lipophilic Nitroaromatic Prodrug Series Designed for Cancer Gene Therapy Applications. Pharmaceuticals, 15(2), 185. https://doi.org/10.3390/ph15020185