
Cyclooxygenase-2 (COX-2) as a Target of Anticancer Agents: A Review of Novel Synthesized Scaffolds Having Anticancer and COX-2 Inhibitory Potentialities

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Drug Molecules with Dual Anticancer and COX-2 Inhibitory Activity
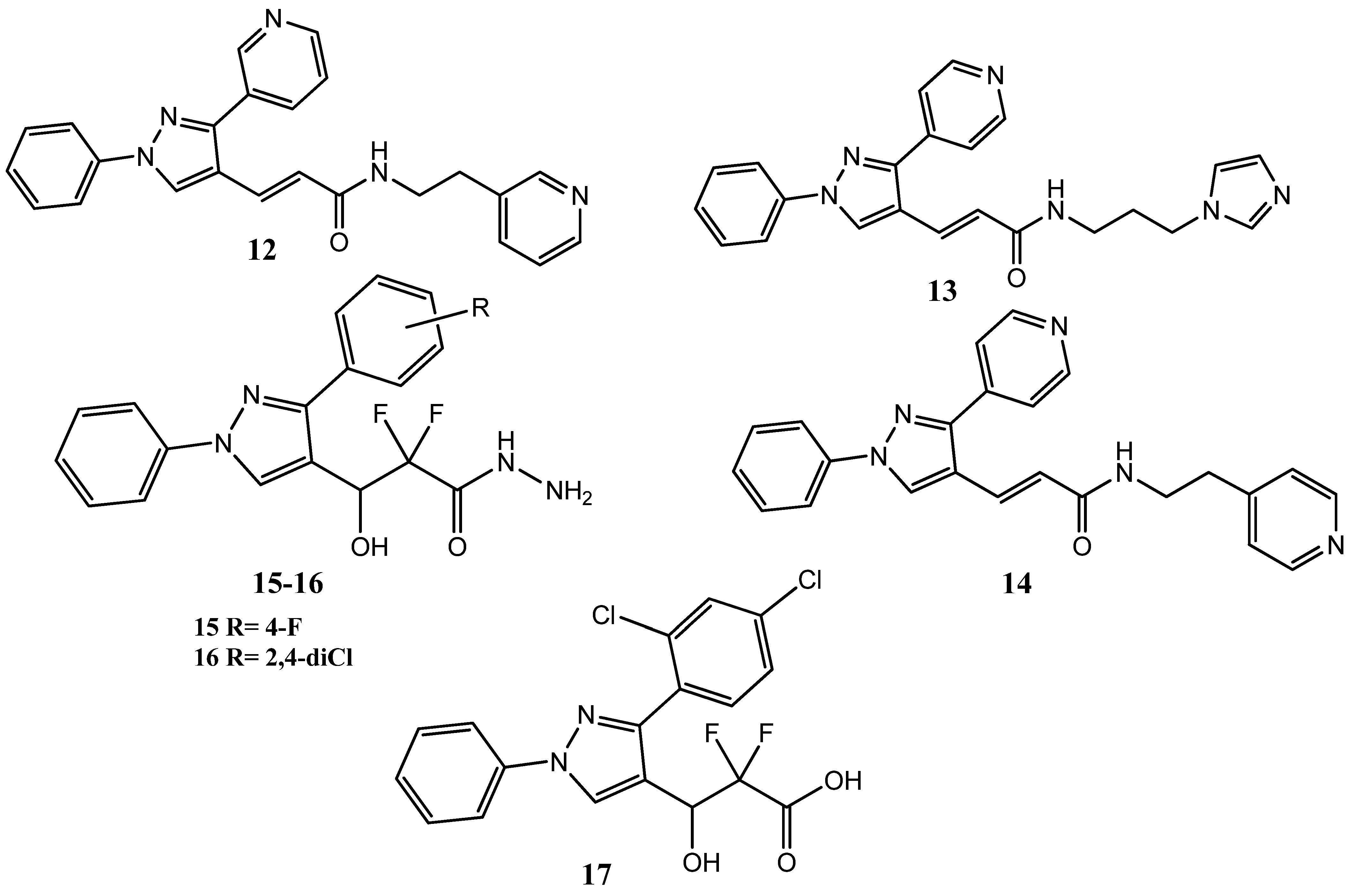
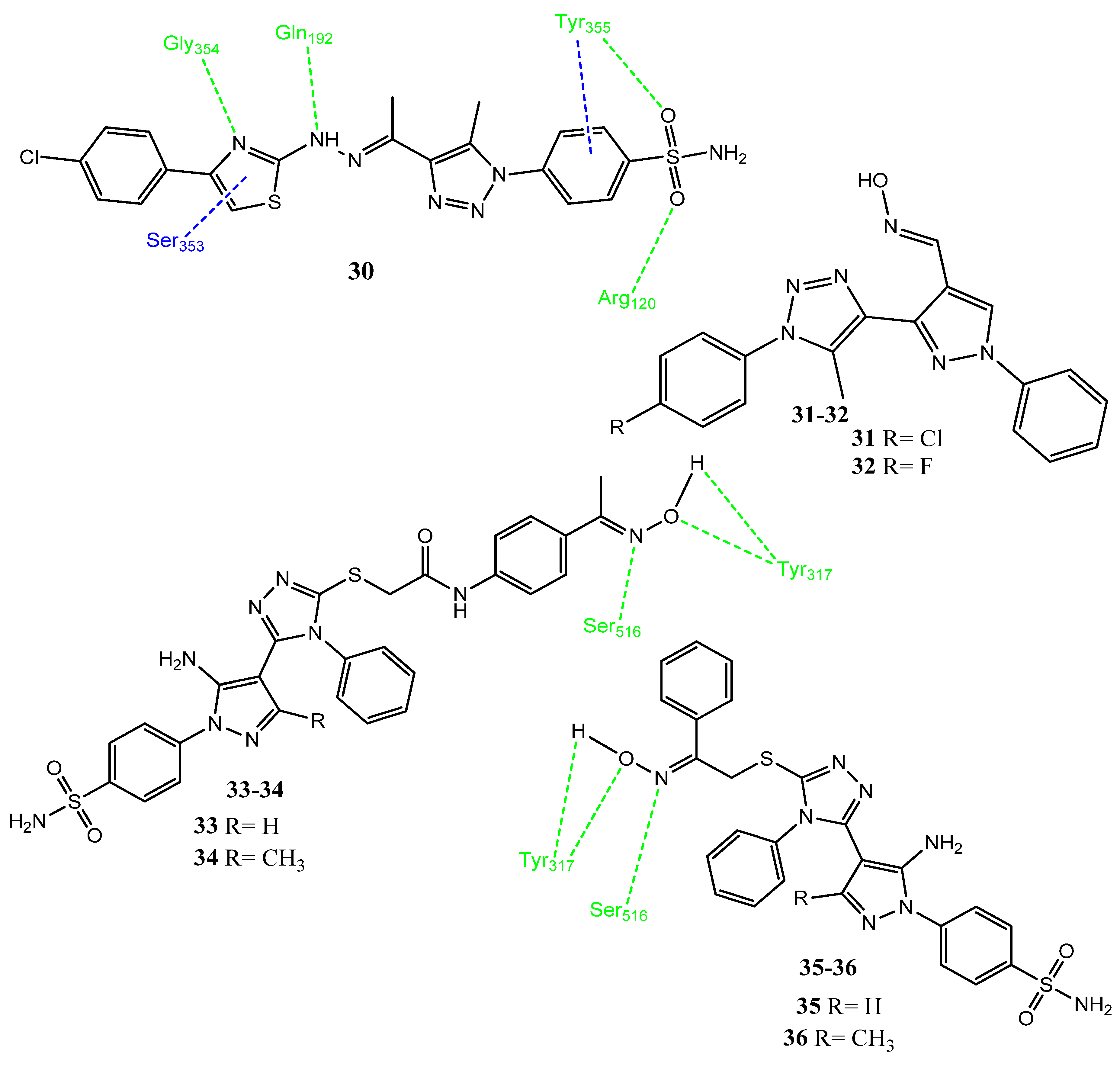
2.1. Pyrazole Derivatives
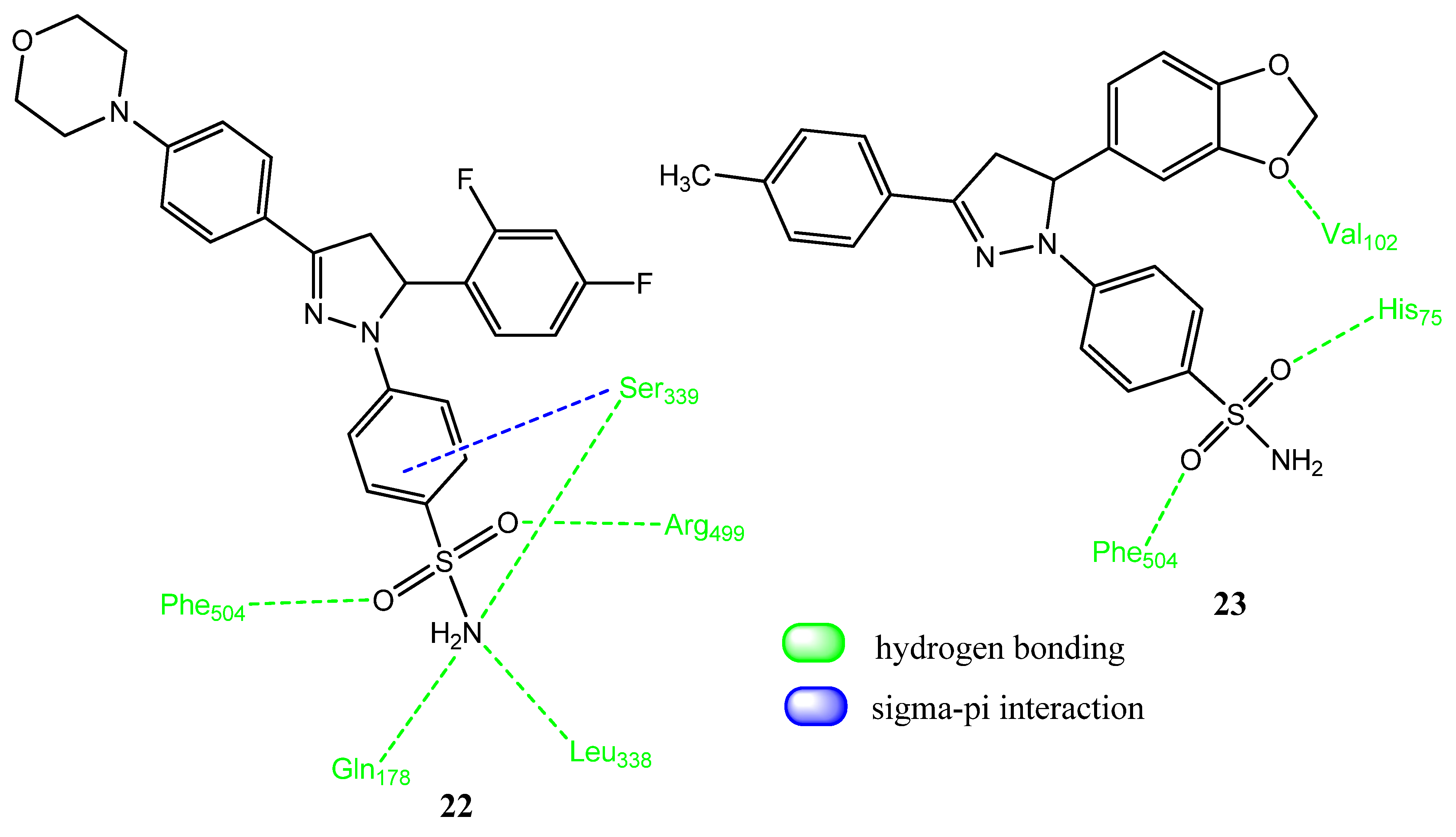
2.2. Pyrazoline Derivatives
2.3. Pyrazole- and Pyrazolone-Based Hybrid Molecules
2.4. Benzimidazole/Benzoxazole-Based Hybrid Molecules
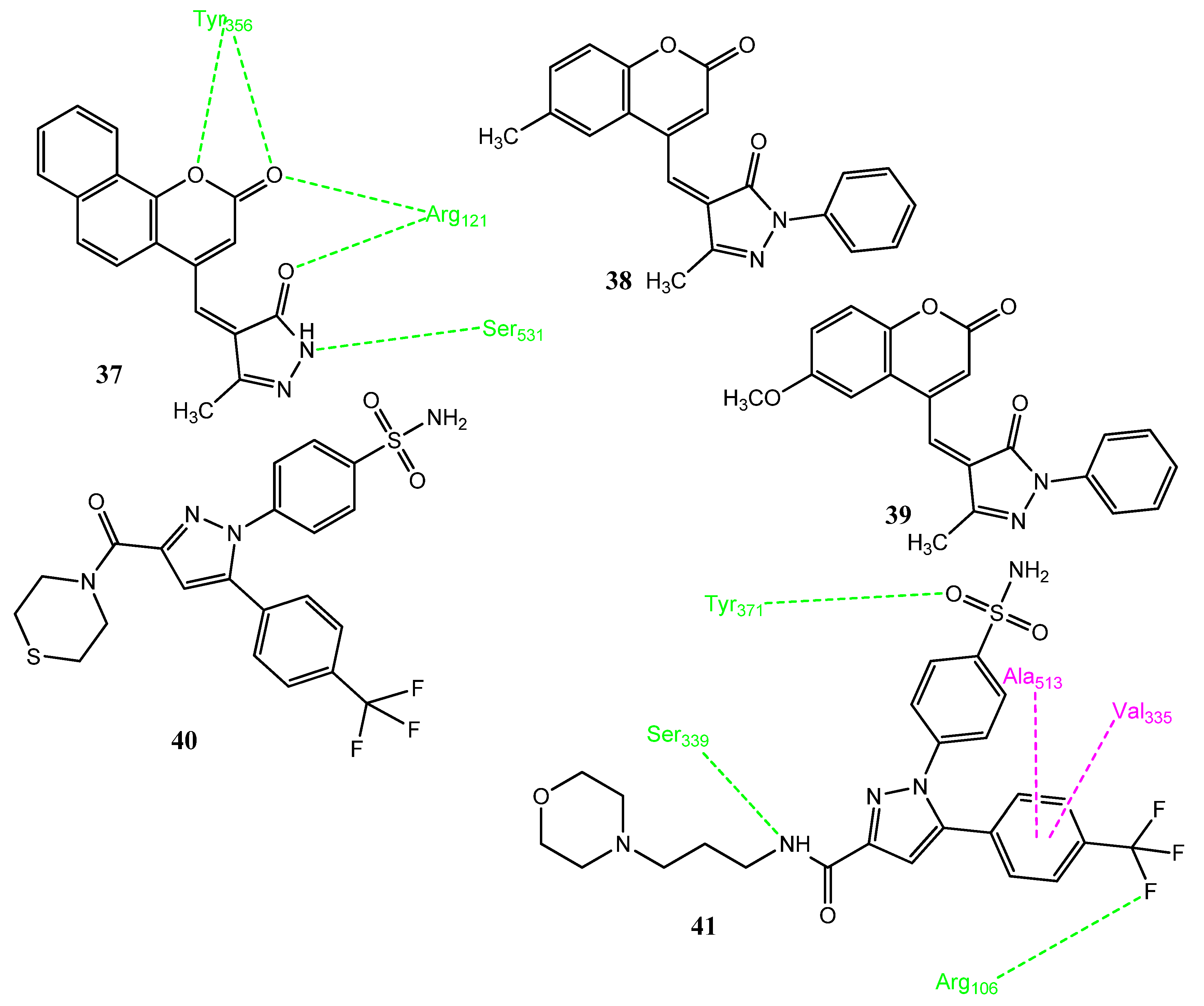
2.5. Natural Product-Based Hybrid Molecules
2.6. Derivatives of Natural Compounds
2.7. Quinoline Derivatives
2.8. Quinazolinone Derivatives
2.9. Indole- and Indanone-Based Molecules
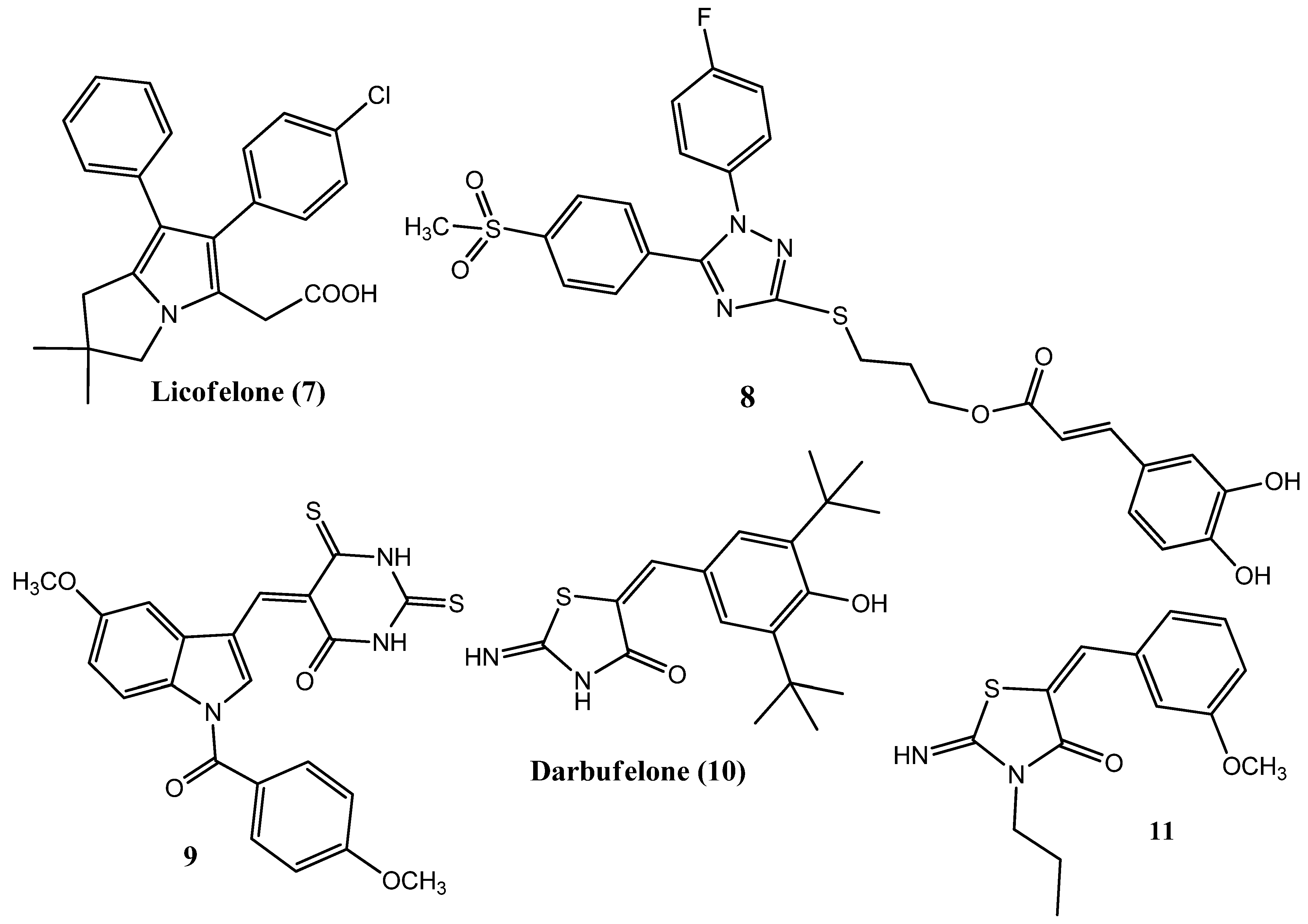
2.10. Pyrrolizines as Anticancer and COX-2 Inhibitors
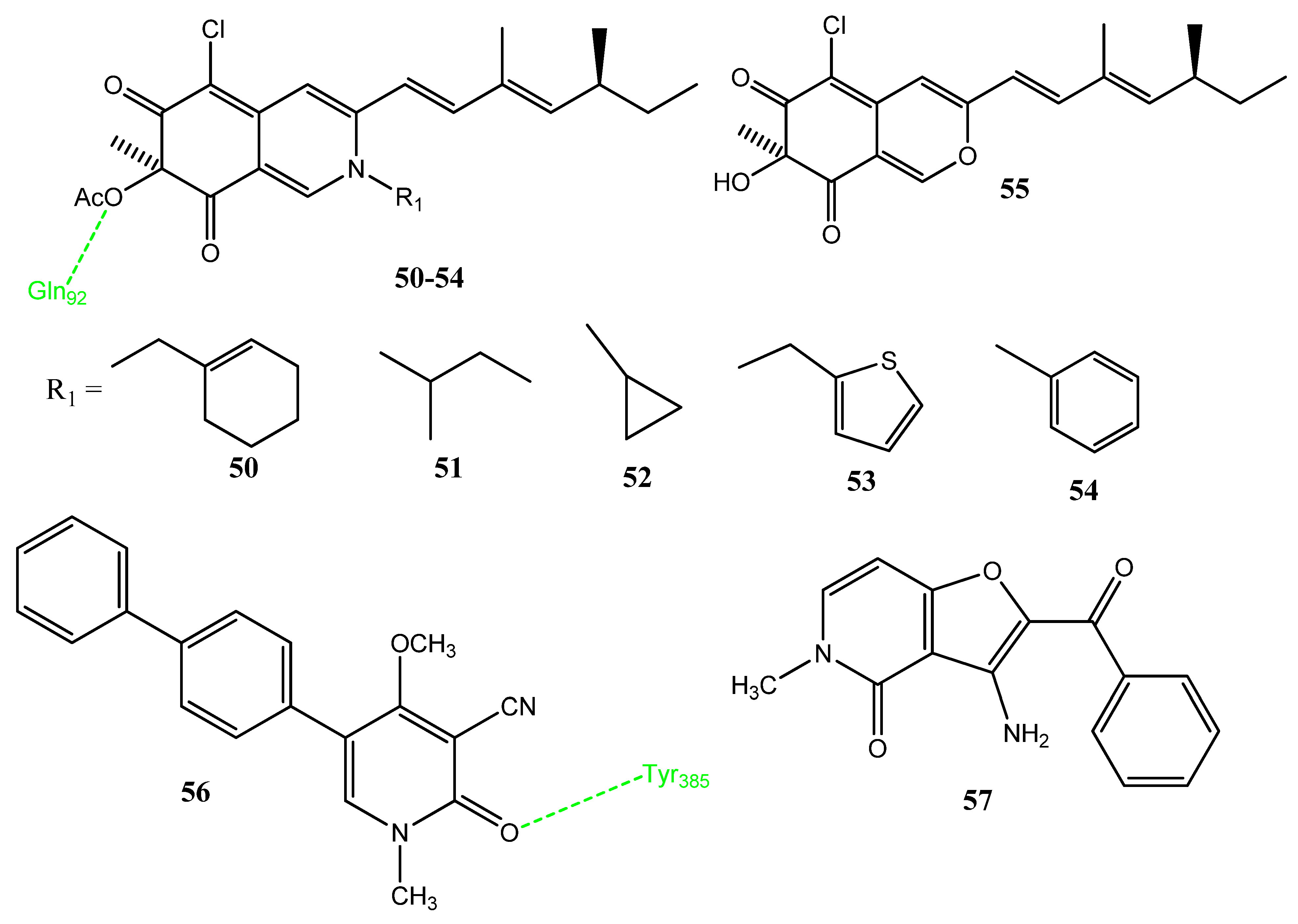
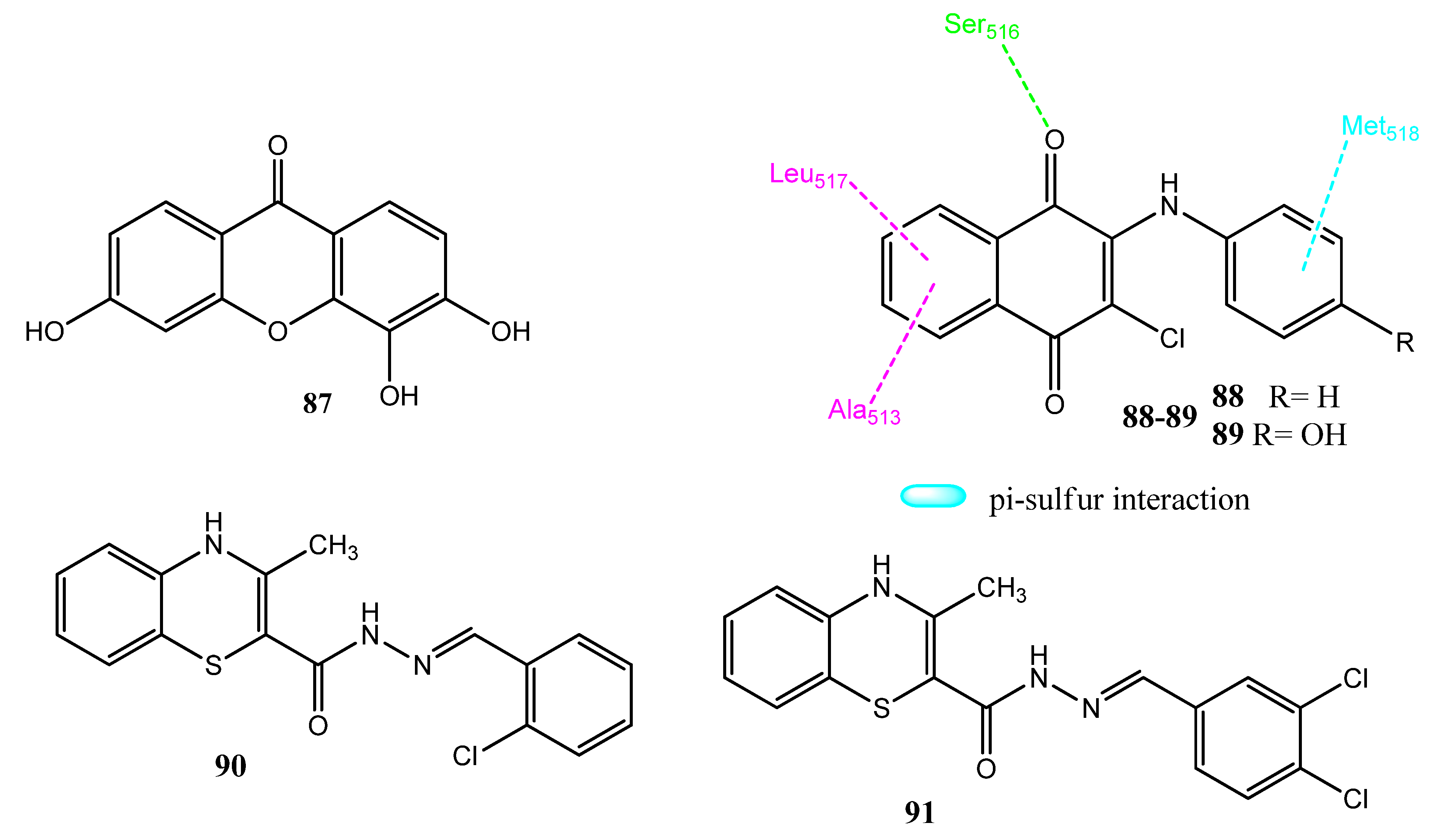
2.11. Xanthone, Naphthoquinone, and Benzothiazine Derivatives
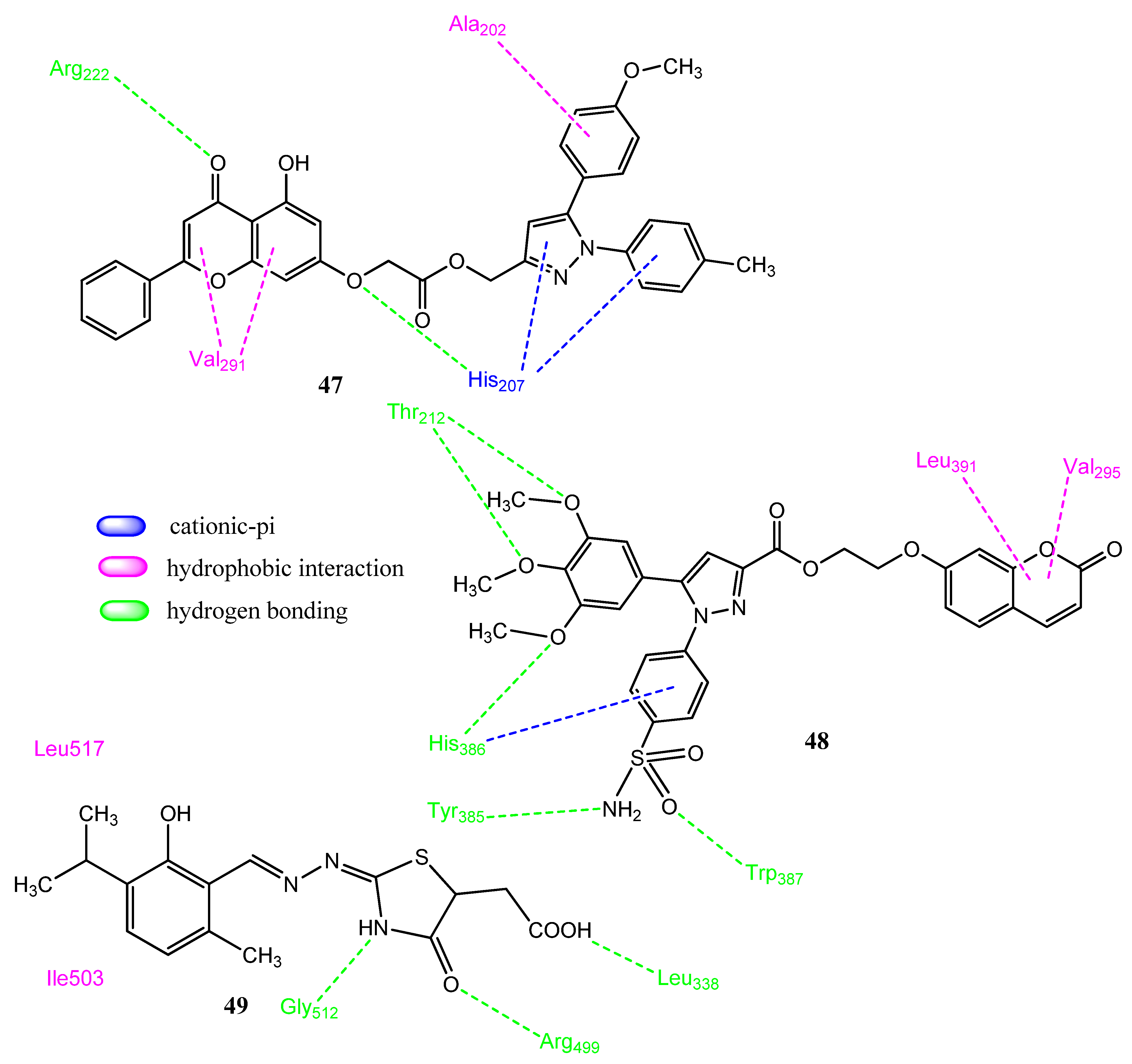
2.12. Miscellaneous Fused Heterocyclic Rings
2.13. Heterocyclic Compounds as Anticancer and COX-2 Inhibitors
2.13.1. Imidazolone and Triazole Hybrid Derivatives
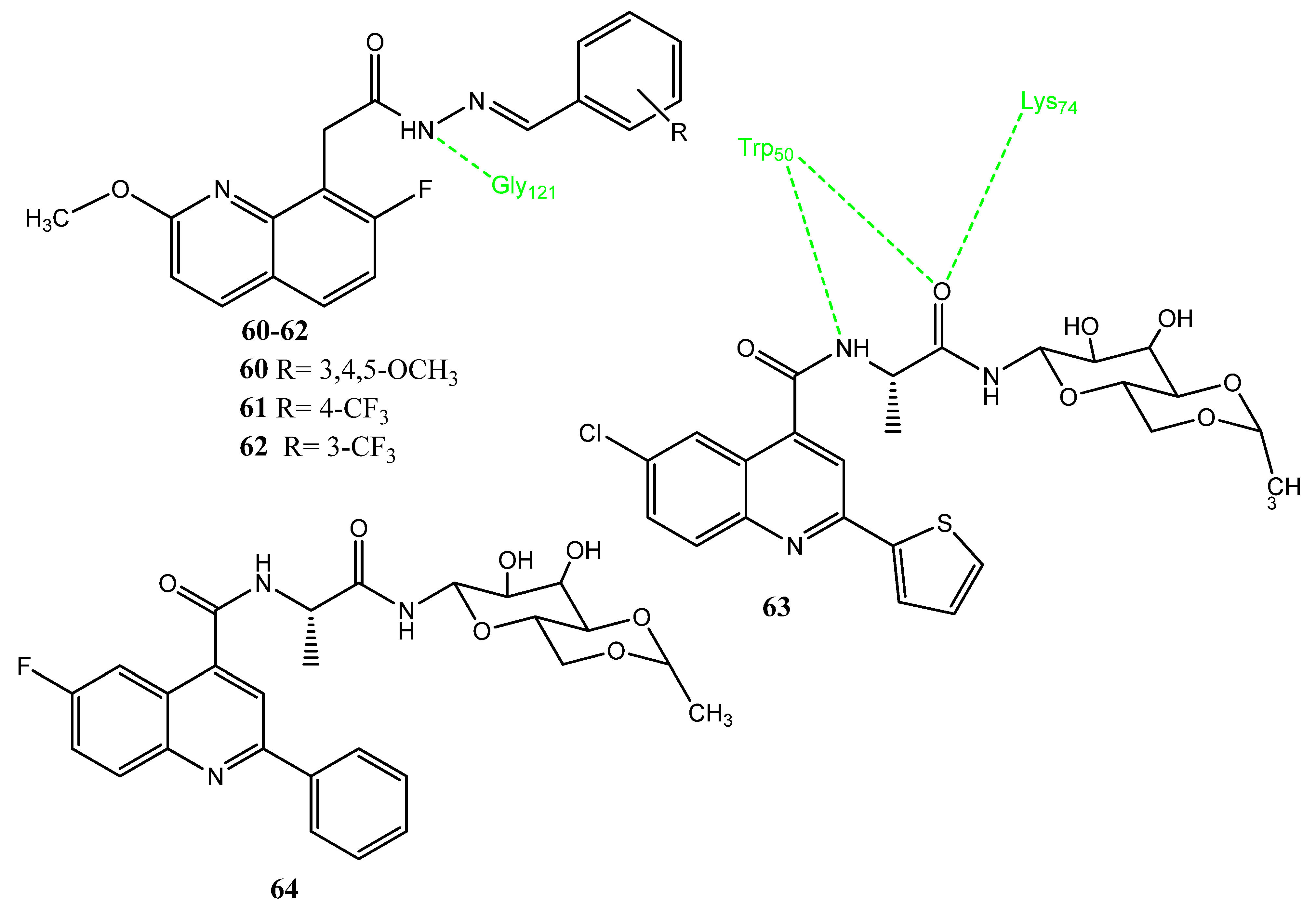
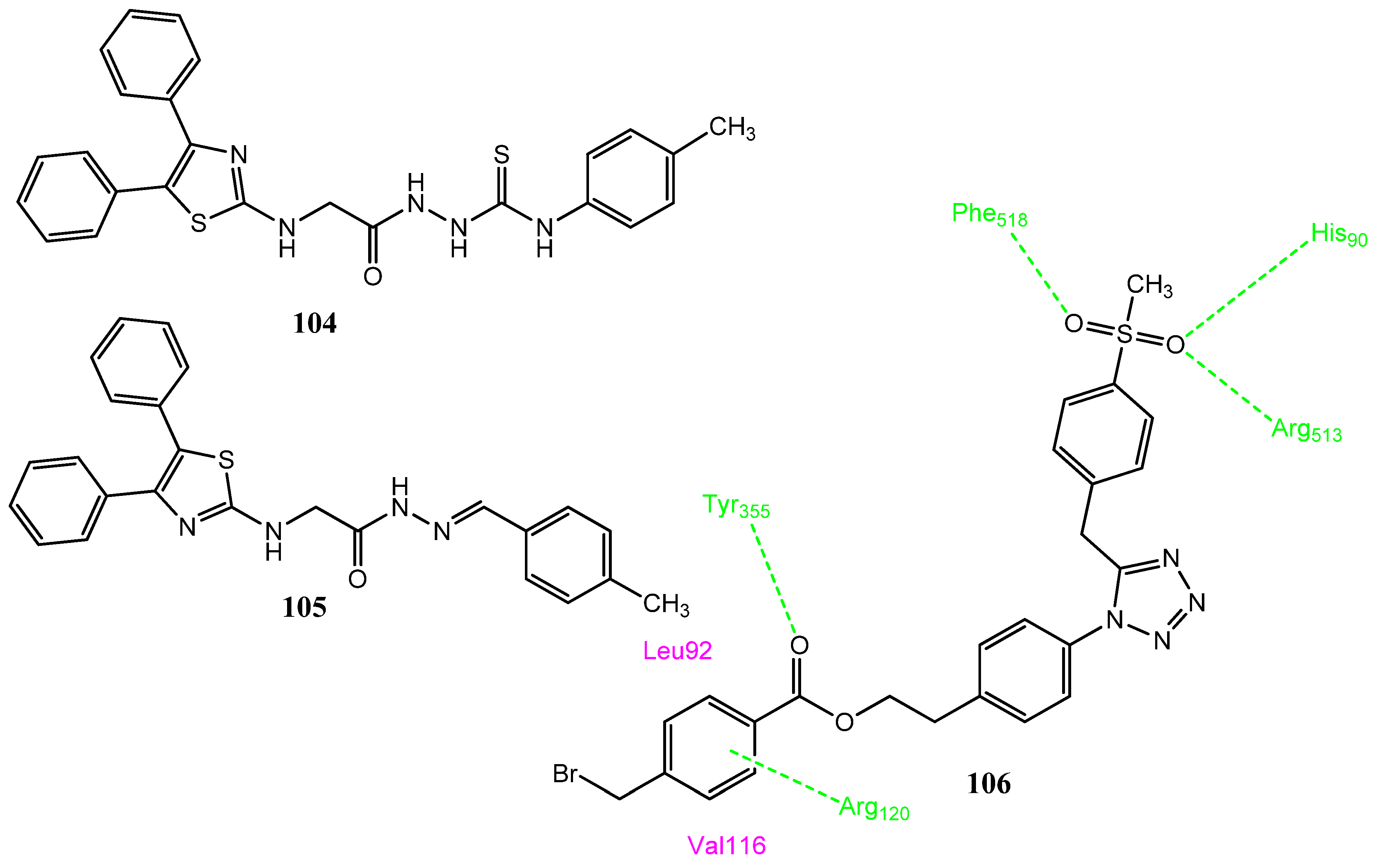
2.13.2. Thiazole Derivatives as COX-2 Inhibitors
2.13.3. Tetrazole Derivatives
2.13.4. Oxadiazole and Pyrimidine Derivatives
2.13.5. Thiadiazole and Cyclopentole Derivatives
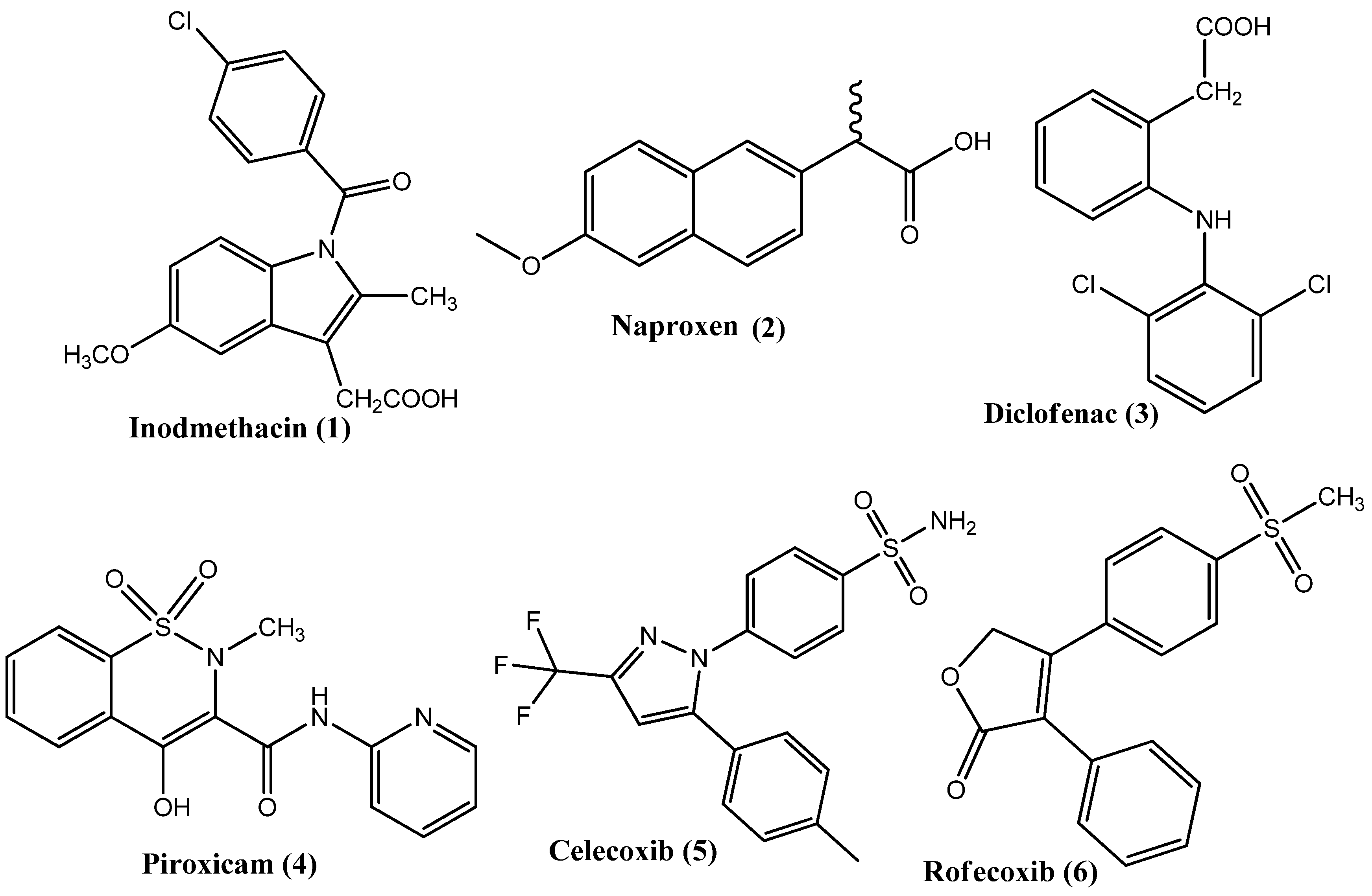
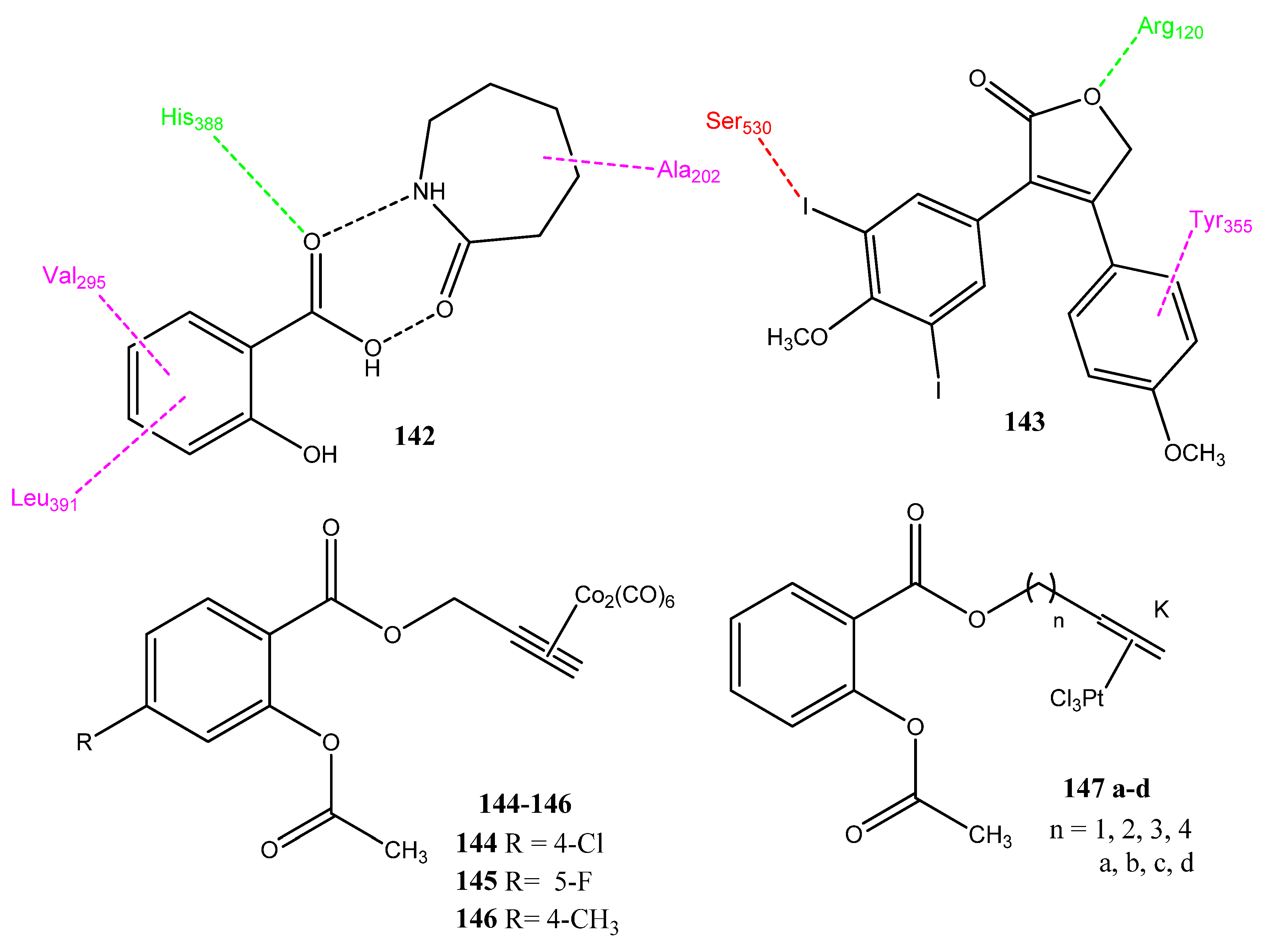
2.14. Derivatives of NSAIDs as Anticancer and COX-2 Inhibitors
2.15. Metal Complexes as Anticancer Agents and COX-2 Inhibitors
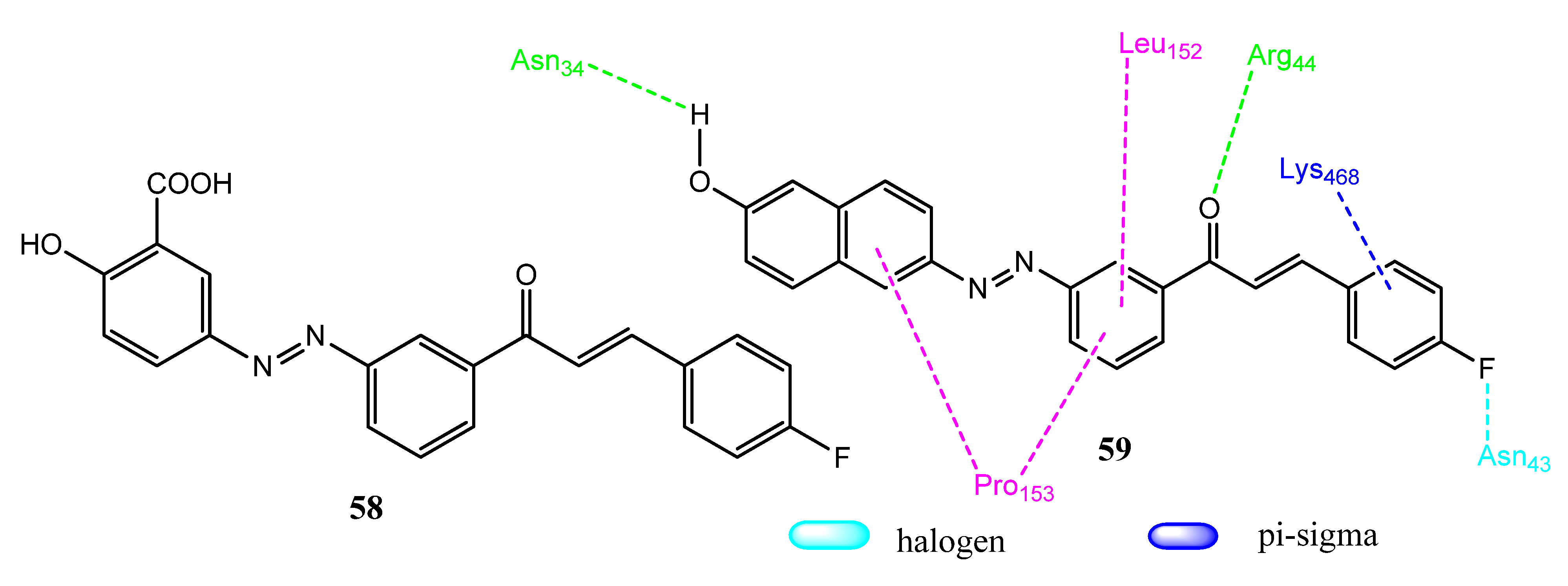
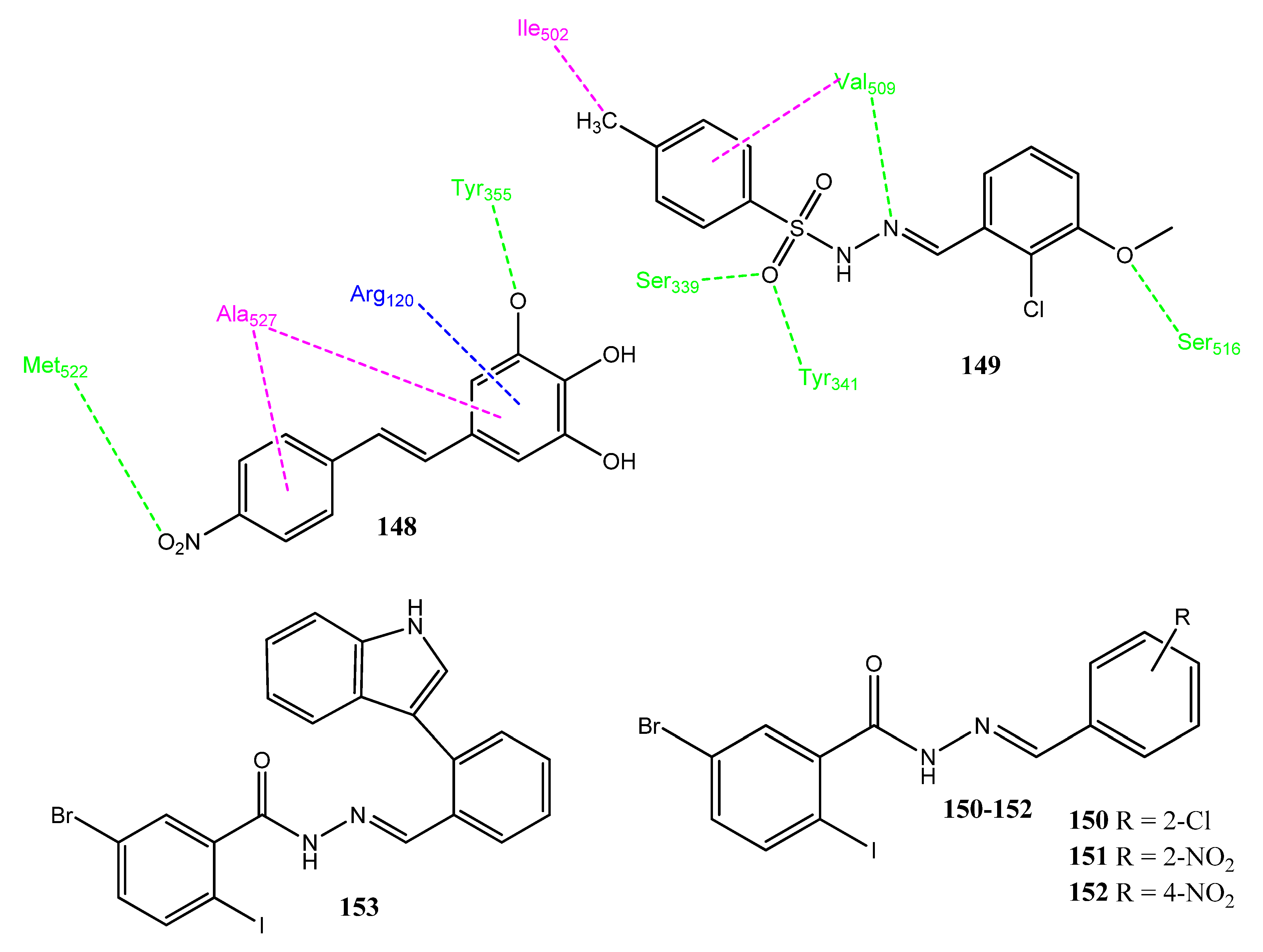
2.16. Stilbene Derivatives
2.17. Hydrazone Derivatives
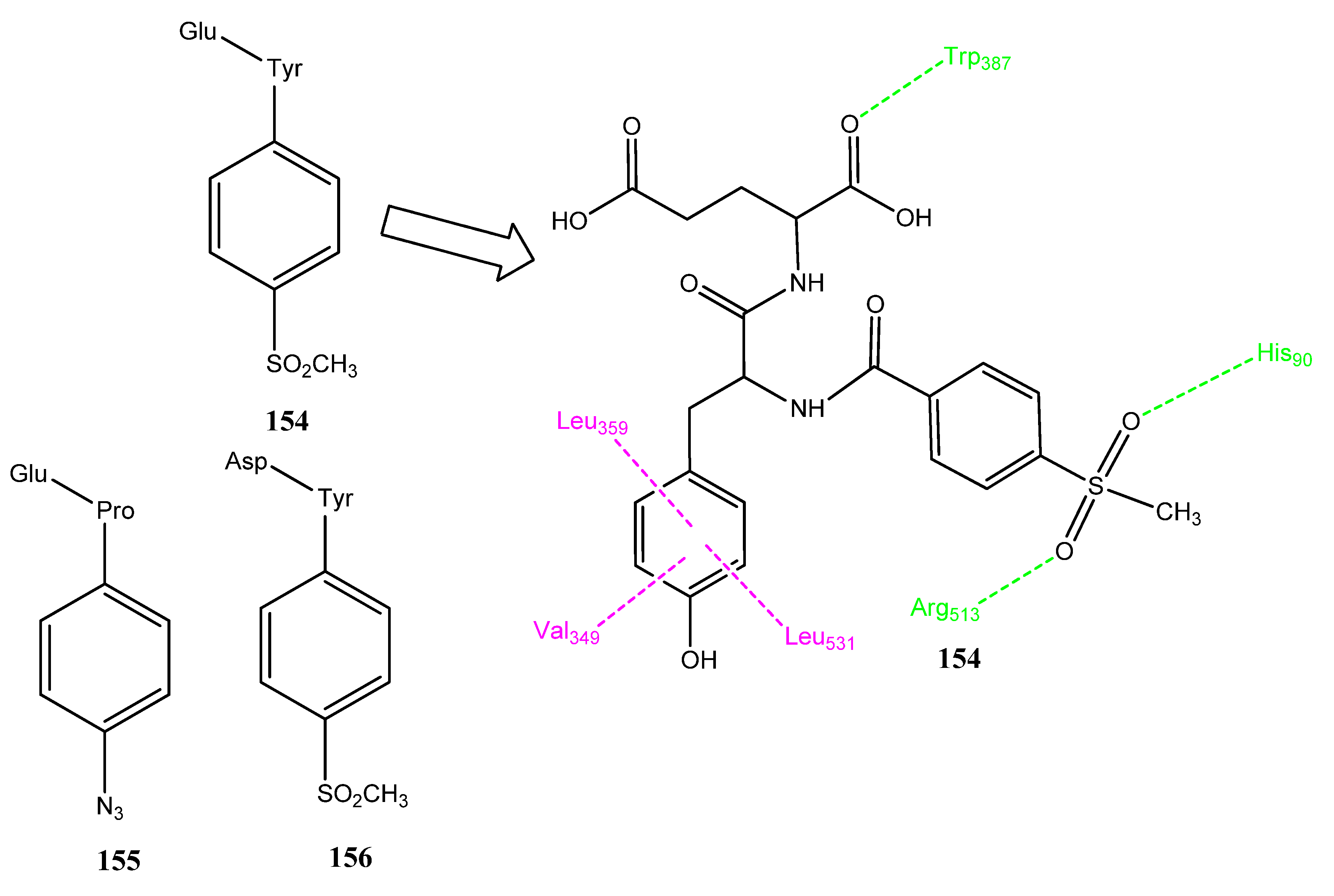
2.18. Peptides as COX-2 Inhibitors
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Trans. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazieh, A.R.; Da’ar, O.B.; Alkaiyat, M.; Zaatreh, Y.A.; Saad, A.A.; Bustami, R.; Alrujaib, M.; Alkattan, K. Cancer Incidence Trends from 1999 to 2015 and Contributions of Various Cancer Types to the Overall Burden: Projections to 2030 and Extrapolation of Economic Burden in Saudi Arabia. Cancer Manag. Res. 2019, 11, 9665–9674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, D.C.; Wilson, L.F. The fractions of cancer attributable to modifiable factors: A global review. Cancer Epidemiol. 2016, 44, 203–221. [Google Scholar] [CrossRef]
- Stanković, T.; Dinić, J.; Podolski-Renić, A.; Musso, L.; Burić, S.S.; Dallavalle, S.; Pešić, M. Dual inhibitors as a new challenge for cancer multidrug resistance treatment. Curr. Med. Chem. 2019, 26, 6074–6106. [Google Scholar] [CrossRef]
- Sano, S.; Chan, K.S.; Carbajal, S.; Clifford, J.; Peavey, M.; Kiguchi, K.; Itami, S.; Nickoloff, B.J.; DiGiovanni, J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 2005, 11, 43–49. [Google Scholar] [CrossRef]
- Mantovani, A. Inflaming metastasis. Nature 2009, 457, 36–37. [Google Scholar] [CrossRef]
- Achiwa, H.; Yatabe, Y.; Hida, T.; Kuroishi, T.; Kozaki, K.-i.; Nakamura, S.; Ogawa, M.; Sugiura, T.; Mitsudomi, T.; Takahashir, T. Prognostic significance of elevated cyclooxygenase 2 expression in primary, resected lung adenocarcinomas. Clin. Cancer Res. 1999, 5, 1001–1005. [Google Scholar]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A role in cancer stem cell survival and repopulation of cancer cells during therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [Green Version]
- Steinmeyer, J. Pharmacological basis for the therapy of pain and inflammation with nonsteroidal anti-inflammatory drugs. Arthritis Res. Ther. 2000, 2, 379–385. [Google Scholar] [CrossRef]
- Brune, K.; Patrignani, P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J. Pain Res. 2015, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; DuBois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Iadarola, M.; Yang, H.-Y.T.; Dionne, R.A. Expression of COX-1 and COX-2 in a clinical model of acute inflammation. J. Pain 2007, 8, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Wu, W.; Shin, V.; Bruce, I.; Wong, B.; Cho, C. Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis 2005, 26, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Greene, E.R.; Huang, S.; Serhan, C.N.; Panigrahy, D. Regulation of inflammation in cancer by eicosanoids. Prostagland. Other Lipid Mediat. 2011, 96, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Cai, X.; Chennamaneni, S.; Yi, X.; Liu, L.; Pink, J.J.; Dowlati, A.; Xu, Y.; Zhou, A.; Su, B. From COX-2 inhibitor nimesulide to potent anti-cancer agent: Synthesis, in vitro, in vivo and pharmacokinetic evaluation. Eur. J. Med. Chem. 2012, 47, 432–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, F.H.; Adsule, S.; Li, Y.; Padhye, S. Back to the future: COX-2 inhibitors for chemoprevention and cancer therapy. Mini Rev. Med. Chem. 2007, 7, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.A.M.; Angeli, A.; El-Azab, A.S.; Hammouda, M.E.A.; El-Sherbeny, M.A.; Supuran, C.T. Synthesis and anti-inflammatory activity of sulfonamides and carboxylates incorporating trimellitimides: Dual cyclooxygenase/carbonic anhydrase inhibitory actions. Bioorg. Chem. 2019, 84, 260–268. [Google Scholar] [CrossRef]
- Vosooghi, M.; Amini, M. The discovery and development of cyclooxygenase-2 inhibitors as potential anticancer therapies. Expert Opin. Drug Discov. 2014, 9, 255–267. [Google Scholar] [CrossRef]
- Kang, S.N.; Hong, S.-S.; Lee, M.-K.; Lim, S.-J. Dual function of tributyrin emulsion: Solubilization and enhancement of anticancer effect of celecoxib. Int. J. Pharm. 2012, 428, 76–81. [Google Scholar] [CrossRef]
- Xu, H.-B.; Shen, F.-M.; Lv, Q.-Z. Celecoxib enhanced the cytotoxic effect of cisplatin in drug-resistant human gastric cancer cells by inhibition of cyclooxygenase-2. Eur. J. Pharmacol. 2015, 769, 1–7. [Google Scholar] [CrossRef]
- Tavolari, S.; Munarini, A.; Storci, G.; Laufer, S.; Chieco, P.; Guarnieri, T. The decrease of cell membrane fluidity by the non-steroidal anti-inflammatory drug Licofelone inhibits epidermal growth factor receptor signalling and triggers apoptosis in HCA-7 colon cancer cells. Cancer Lett. 2012, 321, 187–194. [Google Scholar] [CrossRef]
- Cai, H.; Huang, X.; Xu, S.; Shen, H.; Zhang, P.; Huang, Y.; Jiang, J.; Sun, Y.; Jiang, B.; Wu, X. Discovery of novel hybrids of diaryl-1, 2, 4-triazoles and caffeic acid as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase for cancer therapy. Eur. J. Med. Chem. 2016, 108, 89–103. [Google Scholar] [CrossRef]
- Penthala, N.R.; Ponugoti, P.R.; Kasam, V.; Crooks, P.A. 5-((1-Aroyl-1H-indol-3-yl) methylene)-2-thioxodihydropyrimidine-4, 6 (1H, 5H)-diones as potential anticancer agents with anti-inflammatory properties. Bioorg. Med. Chem. Lett. 2013, 23, 1442–1446. [Google Scholar] [CrossRef] [Green Version]
- Che, X.-H.; Chen, C.-L.; Ye, X.-L.; Weng, G.-B.; Guo, X.-Z.; Yu, W.-Y.; Tao, J.; Chen, Y.-C.; Chen, X. Dual inhibition of COX-2/5-LOX blocks colon cancer proliferation, migration and invasion in vitro. Oncol. Rep. 2016, 35, 1680–1688. [Google Scholar] [CrossRef] [Green Version]
- Ottanà, R.; Carotti, S.; Maccari, R.; Landini, I.; Chiricosta, G.; Caciagli, B.; Vigorita, M.G.; Mini, E. In vitro antiproliferative activity against human colon cancer cell lines of representative 4-thiazolidinones. Part I. Bioorg. Med. Chem. Lett. 2005, 15, 3930–3933. [Google Scholar] [CrossRef]
- Ando, K.; Kawamura, K. Sulfamoylheteroaryl Pyrazole Compounds as Anti-Inflammatory/Analgesic Agents. Google Patent EP1104760A1, 12 March 2003. [Google Scholar]
- Bakr, R.B.; Azouz, A.A.; Abdellatif, K.R. Synthesis, cyclooxygenase inhibition, anti-inflammatory evaluation and ulcerogenic liability of new 1-phenylpyrazolo [3, 4-d] pyrimidine derivatives. J. Enzy. Inhib. Med. Chem. 2016, 31 (Suppl. S2), 6–12. [Google Scholar] [CrossRef] [Green Version]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Faouzi, M.E.A. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 2020, 97, 103470. [Google Scholar] [CrossRef] [PubMed]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Penning, T.D.; Seibert, K.; Isakson, P.C. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Sai Ram, K.; Rambabu, G.; Sarma, J.; Desiraju, G.R. Ligand coordinate analysis of SC-558 from the active site to the surface of COX-2: A molecular dynamics study. J. Chem. Inform. Model. 2006, 46, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inceler, N.; Ozkan, Y.; Turan, N.N.; Kahraman, D.C.; Cetin-Atalay, R.; Baytas, S.N. Design, synthesis and biological evaluation of novel 1, 3-diarylpyrazoles as cyclooxygenase inhibitors, antiplatelet and anticancer agents. Med. Chem. Comm. 2018, 9, 795–811. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Mukarram, S.; Bandgar, B.P.; Shaikh, R.U.; Ganapure, S.D.; Chavan, H.V. Synthesis of novel α, α-difluoro-β-hydroxycarbonyl pyrazole derivatives as antioxidant, anti-inflammatory and anticancer agents. Med. Chem. Res. 2017, 26, 262–273. [Google Scholar] [CrossRef]
- Ren, S.-Z.; Wang, Z.-C.; Zhu, D.; Zhu, X.-H.; Shen, F.-Q.; Wu, S.-Y.; Chen, J.-J.; Xu, C.; Zhu, H.-L. Design, synthesis and biological evaluation of novel ferrocene-pyrazole derivatives containing nitric oxide donors as COX-2 inhibitors for cancer therapy. Eur. J. Med. Chem. 2018, 157, 909–924. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Ece, A.; Bua, S.; Angeli, A.; Sakagami, H.; Sahin, E.; Supuran, C.T. Synthesis, biological evaluation and in silico modelling studies of 1, 3, 5-trisubstituted pyrazoles carrying benzenesulfonamide as potential anticancer agents and selective cancer-associated hCA IX isoenzyme inhibitors. Bioorg. Chem. 2019, 92, 103222. [Google Scholar] [CrossRef]
- Zhang, B.; Hu, X.-T.; Zhou, K.-M.; Yang, Y.-S.; Zhu, H.-L. Discovery of novel aminophosphonate derivatives containing pyrazole moiety as potential selective COX-2 inhibitors. Bioorg. Chem. 2020, 102, 104096. [Google Scholar] [CrossRef]
- Ansari, M.; Khan, S.A. Synthesis and antimicrobial activity of some novel quinoline-pyrazoline-based coumarinyl thiazole derivatives. Med. Chem. Res. 2017, 26, 1481–1496. [Google Scholar] [CrossRef]
- Haider, K.; Shafeeque, M.; Yahya, S.; Yar, M.S. A comprehensive review on pyrazoline based heterocyclic hybrids as potent anticancer agents. Eur. J. Med. Chem. 2022, 5, 100042. [Google Scholar] [CrossRef]
- Matiadis, D.; Sagnou, M. Pyrazoline hybrids as promising anticancer agents: An up-to-date overview. Int. J. Mol. Sci. 2020, 21, 5507. [Google Scholar] [CrossRef]
- Qiu, H.-Y.; Wang, P.-F.; Li, Z.; Ma, J.-T.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. Synthesis of dihydropyrazole sulphonamide derivatives that act as anti-cancer agents through COX-2 inhibition. Pharmacol. Res. 2016, 104, 86–96. [Google Scholar] [CrossRef]
- Yan, X.-Q.; Wang, Z.-C.; Zhang, B.; Qi, P.-F.; Li, G.-G.; Zhu, H.-L. Dihydropyrazole derivatives containing benzo oxygen heterocycle and sulfonamide moieties selectively and potently inhibit COX-2: Design, synthesis, and anti-colon cancer activity evaluation. Molecules 2019, 24, 1685. [Google Scholar] [CrossRef] [Green Version]
- Ivasiv, V.; Albertini, C.; Gonçalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular hybridization as a tool for designing multitarget drug candidates for complex diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef]
- Gontijo, V.S.; Viegas, F.P.D.; Ortiz, C.J.C.; de Freitas Silva, M.; Damasio, C.M.; Rosa, M.C.; Campos, T.G.; Couto, D.S.; Tranches Dias, K.S.; Viegas, C. Molecular hybridization as a tool in the design of multi-target directed drug candidates for neurodegenerative diseases. Curr. Neuropharmacol. 2020, 18, 348–407. [Google Scholar] [CrossRef]
- Abdellatif, K.R.; Fadaly, W.A.; Mostafa, Y.A.; Zaher, D.M.; Omar, H.A. Thiohydantoin derivatives incorporating a pyrazole core: Design, synthesis and biological evaluation as dual inhibitors of topoisomerase-I and cycloxygenase-2 with anti-cancer and anti-inflammatory activities. Bioorg. Chem. 2019, 91, 103132. [Google Scholar] [CrossRef]
- Akhtar, W.; Marella, A.; Alam, M.M.; Khan, M.F.; Akhtar, M.; Anwer, T.; Khan, F.; Naematullah, M.; Azam, F.; Rizvi, M.A. Design and synthesis of pyrazole-pyrazoline hybrids as cancer-associated selective COX-2 inhibitors. Arch. Pharm. 2021, 354, 2000116. [Google Scholar] [CrossRef]
- Belal, A.; Abdelgawad, M.A. New benzothiazole/benzoxazole-pyrazole hybrids with potential as COX inhibitors: Design, synthesis and anticancer activity evaluation. Res. Chem. Intermed. 2017, 43, 3859–3872. [Google Scholar] [CrossRef]
- Elzahhar, P.A.; Abd El Wahab, S.M.; Elagawany, M.; Daabees, H.; Belal, A.S.; El-Yazbi, A.F.; Eid, A.H.; Alaaeddine, R.; Hegazy, R.R.; Allam, R.M. Expanding the anticancer potential of 1, 2, 3-triazoles via simultaneously targeting Cyclooxygenase-2, 15-lipoxygenase and tumor-associated carbonic anhydrases. Eur. J. Med. Chem. 2020, 200, 112439. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Schweizer, J.; Wang, Y.; Cerna, C.; Wong, H.; Revilla, M. Effect of nitric oxide on cytotoxicity of Taxol: Enhanced Taxol transcellular permeability. Biochem. Pharmacol. 2003, 66, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Fadaly, W.A.; Elshaier, Y.A.; Hassanein, E.H.; Abdellatif, K.R. New 1, 2, 4-triazole/pyrazole hybrids linked to oxime moiety as nitric oxide donor celecoxib analogs: Synthesis, cyclooxygenase inhibition anti-inflammatory, ulcerogenicity, anti-proliferative activities, apoptosis, molecular modeling and nitric oxide release studies. Bioorg. Chem. 2020, 98, 103752. [Google Scholar] [PubMed]
- Kulkarni, R.C.; Madar, J.M.; Shastri, S.L.; Shaikh, F.; Naik, N.S.; Chougale, R.B.; Shastri, L.A.; Joshi, S.D.; Dixit, S.R.; Sunagar, V.A. Green synthesis of coumarin-pyrazolone hybrids: In vitro anticancer and anti-inflammatory activities and their computational study on COX-2 enzyme. Chem. Data Collect. 2018, 17, 497–506. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Z.-C.; Li, X.; Abbas, M.; Wu, S.-Y.; Ren, S.-Z.; Liu, Q.-X.; Liu, Y.; Chen, P.-W.; Duan, Y.-T. Design, synthesis and evaluation of novel diaryl-1, 5-diazoles derivatives bearing morpholine as potent dual COX-2/5-LOX inhibitors and antitumor agents. Eur. J. Med. Chem. 2019, 169, 168–184. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Musa, A.; Almalki, A.H.; Alzarea, S.I.; Mostafa, E.M.; Hegazy, M.M.; Mostafa-Hedeab, G.; Ghoneim, M.M.; Parambi, D.G.; Bakr, R.B. Novel Phenolic Compounds as Potential Dual EGFR and COX-2 Inhibitors: Design, Semisynthesis, in vitro Biological Evaluation and in silico Insights. Drug Des. Develop. Ther. 2021, 15, 2325. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Omar, H.A. Design, synthesis and biological evaluation of some novel benzothiazole/benzoxazole and/or benzimidazole derivatives incorporating a pyrazole scaffold as antiproliferative agents. Bioorg. Chem. 2017, 74, 82–90. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Ahmad, W.; Al-Sanea, M.M.; Elshemy, H.A. New pyrimidine-benzoxazole/benzimidazole hybrids: Synthesis, antioxidant, cytotoxic activity, in vitro cyclooxygenase and phospholipase A2-V inhibition. Bioorg. Chem. 2019, 92, 103218. [Google Scholar] [CrossRef]
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Indus. Microbiol. Biotech. 2016, 43, 155–176. [Google Scholar] [CrossRef]
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Calixto, J.B. The role of natural products in modern drug discovery. An. Acad. Bras. Cienc. 2019, 91, e20190105. [Google Scholar] [CrossRef]
- Ren, S.-Z.; Wang, Z.-C.; Zhu, X.-H.; Zhu, D.; Li, Z.; Shen, F.-Q.; Duan, Y.-T.; Cao, H.; Zhao, J.; Zhu, H.-L. Design and biological evaluation of novel hybrids of 1, 5-diarylpyrazole and Chrysin for selective COX-2 inhibition. Bioorg. Med. Chem. 2018, 26, 4264–4275. [Google Scholar] [CrossRef]
- Shen, F.-Q.; Wang, Z.-C.; Wu, S.-Y.; Ren, S.-Z.; Man, R.-J.; Wang, B.-Z.; Zhu, H.-L. Synthesis of novel hybrids of pyrazole and coumarin as dual inhibitors of COX-2 and 5-LOX. Bioorg. Med. Chem. Lett. 2017, 27, 3653–3660. [Google Scholar] [CrossRef]
- El-Miligy, M.M.; Al-Kubeisi, A.K.; El-Zemity, S.R.; Nassra, R.A.; Abu-Serie, M.M.; Hazzaa, A.A. Discovery of small molecule acting as multitarget inhibitor of colorectal cancer by simultaneous blocking of the key COX-2, 5-LOX and PIM-1 kinase enzymes. Bioorg. Chem. 2021, 115, 105171. [Google Scholar] [CrossRef]
- Banerjee, P.; Mandhare, A.; Bagalkote, V. Marine natural products as source of new drugs: An updated patent review (July 2018–July 2021). Expert Opin. Ther. Pat. 2022, 32, 317–363. [Google Scholar] [CrossRef]
- Papon, N.; Copp, B.R.; Courdavault, V. Marine drugs: Biology, pipelines, current and future prospects for production. Biotechnol. Adv. 2022, 54, 107871. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Huang, Y.; Hong, J.; Wei, X.; Zeng, F.; Li, J.; Ye, G.; Yuan, J.; Long, Y. Preparation, COX-2 inhibition and anticancer activity of sclerotiorin derivatives. Mar. Drugs 2020, 19, 12. [Google Scholar] [CrossRef]
- El-Naggar, M.H.; Abdel Bar, F.M.; Harsha, C.; Monisha, J.; Shimizu, K.; Kunnumakkara, A.B.; Badria, F.A. Synthesis of new selective cytotoxic ricinine analogues against oral squamous cell carcinoma. Nat. Prod. Res. 2021, 35, 2145–2156. [Google Scholar] [CrossRef]
- Musa, A.; Mostafa, E.M.; Bukhari, S.N.A.; Alotaibi, N.H.; El-Ghorab, A.H.; Farouk, A.; Nayl, A.A.; Ghoneim, M.M.; Abdelgawad, M.A. EGFR and COX-2 Dual Inhibitor: The Design, Synthesis, and Biological Evaluation of Novel Chalcones. Molecules 2022, 27, 1158. [Google Scholar] [CrossRef]
- Melge, A.R.; Manzoor, K.; Nair, S.V.; Mohan, C.G. Chapter 19—In Silico Modeling of FDA-Approved Drugs for Discovery of Anti-Cancer Agents: A Drug-Repurposing Approach. In In Silico Drug Design; Roy, K., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 577–608. [Google Scholar]
- Manohar, C.S.; Manikandan, A.; Sridhar, P.; Sivakumar, A.; Kumar, B.S.; Reddy, S.R. Drug repurposing of novel quinoline acetohydrazide derivatives as potent COX-2 inhibitors and anti-cancer agents. J. Mol. Struct. 2018, 1154, 437–444. [Google Scholar] [CrossRef]
- Pallavi, B.; Sharma, P.; Baig, N.; Kumar Madduluri, V.; Sah, A.K.; Saumya, U.; Dubey, U.S.; Shukla, P. Quinoline Glycoconjugates as Potentially Anticancer and Anti-Inflammatory Agents: An Investigation Involving Synthesis, Biological Screening, and Docking. Chem. Sel. 2020, 5, 9878–9882. [Google Scholar] [CrossRef]
- Sakr, A.; Rezq, S.; Ibrahim, S.M.; Soliman, E.; Baraka, M.M.; Romero, D.G.; Kothayer, H. Design and synthesis of novel quinazolinones conjugated ibuprofen, indole acetamide, or thioacetohydrazide as selective COX-2 inhibitors: Anti-inflammatory, analgesic and anticancer activities. J. Enzy. Inhib. Med. Chem. 2021, 36, 1810–1828. [Google Scholar] [CrossRef]
- El-Sayed, N.N.; Al-Otaibi, T.M.; Alonazi, M.; Masand, V.H.; Barakat, A.; Almarhoon, Z.M.; Ben Bacha, A. Synthesis and Characterization of Some New Quinoxalin-2 (1H) one and 2-Methyl-3H-quinazolin-4-one Derivatives Targeting the Onset and Progression of CRC with SRA, Molecular Docking, and ADMET Analyses. Molecules 2021, 26, 3121. [Google Scholar] [CrossRef]
- Sever, B.; Altıntop, M.D.; Kuş, G.; Özkurt, M.; Özdemir, A.; Kaplancıklı, Z.A. Indomethacin based new triazolothiadiazine derivatives: Synthesis, evaluation of their anticancer effects on T98 human glioma cell line related to COX-2 inhibition and docking studies. Eur. J. Med. Chem. 2016, 113, 179–186. [Google Scholar] [CrossRef]
- Kumari, P.; Mishra, V.S.; Narayana, C.; Khanna, A.; Chakrabarty, A.; Sagar, R. Design and efficient synthesis of pyrazoline and isoxazole bridged indole C-glycoside hybrids as potential anticancer agents. Sci. Rep. 2020, 10, 6660. [Google Scholar] [CrossRef] [Green Version]
- Sever, B.; Altıntop, M.D.; Çiftçi, G.A.; Özdemir, A. A new series of triazolothiadiazines as potential anticancer agents for targeted therapy of non-small cell lung and colorectal cancers: Design, synthesis, in silico and in vitro studies providing mechanistic insight into their anticancer potencies. Med. Chem. 2021, 17, 1104–1128. [Google Scholar] [CrossRef] [PubMed]
- Naaz, F.; Pallavi, M.P.; Shafi, S.; Mulakayala, N.; Yar, M.S.; Kumar, H.S. 1, 2, 3-triazole tethered Indole-3-glyoxamide derivatives as multiple inhibitors of 5-LOX, COX-2 & tubulin: Their anti-proliferative & anti-inflammatory activity. Bioorg. Chem. 2018, 81, 1–20. [Google Scholar]
- Abolhasani, H.; Zarghi, A.; Movahhed, T.K.; Abolhasani, A.; Daraei, B.; Dastmalchi, S. Design, synthesis and biological evaluation of novel indanone containing spiroisoxazoline derivatives with selective COX-2 inhibition as anticancer agents. Bioorg. Med. Chem. 2021, 32, 115960. [Google Scholar] [CrossRef] [PubMed]
- Tavolari, S.; Bonafe, M.; Marini, M.; Ferreri, C.; Bartolini, G.; Brighenti, E.; Manara, S.; Tomasi, V.; Laufer, S.; Guarnieri, T. Licofelone, a dual COX/5-LOX inhibitor, induces apoptosis in HCA-7 colon cancer cells through the mitochondrial pathway independently from its ability to affect the arachidonic acid cascade. Carcinogenesis 2008, 29, 371–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, N.K.; Nargi, D.; Attur, M.; Abramson, S.B.; Narayanan, B.A. Anticancer effects of licofelone (ML-3000) in prostate cancer cells. Anticancer Res. 2007, 27, 2393–2402. [Google Scholar]
- Gouda, A.M.; Abdelazeem, A.H.; Omar, H.A.; Abdalla, A.N.; Abourehab, M.A.; Ali, H.I. Pyrrolizines: Design, synthesis, anticancer evaluation and investigation of the potential mechanism of action. Bioorg. Med. Chem. 2017, 25, 5637–5651. [Google Scholar] [CrossRef]
- Attalah, K.M.; Abdalla, A.N.; Aslam, A.; Ahmed, M.; Abourehab, M.A.; ElSawy, N.A.; Gouda, A.M. Ethyl benzoate bearing pyrrolizine/indolizine moieties: Design, synthesis and biological evaluation of anti-inflammatory and cytotoxic activities. Bioorg. Chem. 2020, 94, 103371. [Google Scholar] [CrossRef]
- Protti, Í.F.; Rodrigues, D.R.; Fonseca, S.K.; Alves, R.J.; de Oliveira, R.B.; Maltarollo, V.G. Do Drug-likeness Rules Apply to Oral Prodrugs? Chem. Med. Chem. 2021, 16, 1446–1456. [Google Scholar] [CrossRef]
- Shawky, A.M.; Abourehab, M.A.; Abdalla, A.N.; Gouda, A.M. Optimization of pyrrolizine-based Schiff bases with 4-thiazolidinone motif: Design, synthesis and investigation of cytotoxicity and anti-inflammatory potency. Eur. J. Med. Chem. 2020, 185, 111780. [Google Scholar] [CrossRef]
- Miladiyah, I.; Jumina, J.; Haryana, S.M.; Mustofa, M. Biological activity, quantitative structure–activity relationship analysis, and molecular docking of xanthone derivatives as anticancer drugs. Drug Des. Devel. Ther. 2018, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Kavaliauskas, P.; Opazo, F.S.; Acevedo, W.; Petraitiene, R.; Grybaitė, B.; Anusevičius, K.; Mickevičius, V.; Belyakov, S.; Petraitis, V. Synthesis, biological activity, and molecular modelling studies of naphthoquinone derivatives as promising anticancer candidates targeting COX-2. Pharmaceuticals 2022, 15, 541. [Google Scholar] [CrossRef]
- Rai, A.; Kumar, U.; Raj, V.; Singh, A.K.; Kumar, P.; Keshari, A.K.; Kumar, D.; Maity, B.; De, A.; Samanta, A. Novel 1, 4-benzothazines obliterate COX-2 mediated JAK-2/STAT-3 signals with potential regulation of oxidative and metabolic stress during colorectal cancer. Pharmacol. Res. 2018, 132, 188–203. [Google Scholar] [CrossRef]
- Abd El Razik, H.A.; Mroueh, M.; Faour, W.H.; Shebaby, W.N.; Daher, C.F.; Ashour, H.M.; Ragab, H.M. Synthesis of new pyrazolo [3, 4-d] pyrimidine derivatives and evaluation of their anti-inflammatory and anticancer activities. Chem. Biol. Drug Des. 2017, 90, 83–96. [Google Scholar] [CrossRef]
- Hawash, M.; Jaradat, N.; Hameedi, S.; Mousa, A. Design, synthesis and biological evaluation of novel benzodioxole derivatives as COX inhibitors and cytotoxic agents. BMC Chem. 2020, 14, 54. [Google Scholar] [CrossRef]
- Azami Movahed, M.; Daraei, B.; Shahosseini, S.; Esfahanizadeh, M.; Zarghi, A. Design, synthesis, and biological evaluation of new pyrazino [1, 2-a] benzimidazole derivatives as selective cyclooxygenase (COX-2) inhibitors. Anticancer Agents Med. Chem. 2019, 352, 1800265. [Google Scholar] [CrossRef]
- Kirwen, E.M.; Batra, T.; Karthikeyan, C.; Deora, G.S.; Rathore, V.; Mulakayala, C.; Mulakayala, N.; Nusbaum, A.C.; Chen, J.; Amawi, H. 2, 3-Diaryl-3H-imidazo [4, 5-b] pyridine derivatives as potential anticancer and anti-inflammatory agents. Acta Pharm. Sin. B 2017, 7, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Jampilek, J. Heterocycles in medicinal chemistry. Molecules 2019, 24, 3839. [Google Scholar] [CrossRef] [Green Version]
- Lamie, P.F.; Philoppes, J.N.; Rárová, L. Design, synthesis, and biological evaluation of novel 1, 2-diaryl-4-substituted-benzylidene-5 (4H)-imidazolone derivatives as cytotoxic agents and COX-2/LOX inhibitors. Arch. Pharm. 2018, 351, 1700311. [Google Scholar] [CrossRef]
- Abdelazeem, A.H.; El-Saadi, M.T.; Said, E.G.; Youssif, B.G.; Omar, H.A.; El-Moghazy, S.M. Novel diphenylthiazole derivatives with multi-target mechanism: Synthesis, docking study, anticancer and anti-inflammatory activities. Bioorg. Chem. 2017, 75, 127–138. [Google Scholar] [CrossRef]
- El-Barghouthi, M.I.; Hasan, A.S.; Al-Awaida, W.; Al-Ameer, H.J.; Kaur, J.; Hayashibara, K.J.; Fleming, J.; Waknin, J.; Hayashibara, S.; Slewa, M. Novel therapeutic heterocycles as selective cyclooxygenase-2 inhibitors and anti-cancer agents: Synthesis, in vitro bioassay screenings, and molecular docking studies. J. Mol. Struct. 2022, 1263, 133141. [Google Scholar] [CrossRef]
- El-Sayed, N.A.; Nour, M.S.; Salem, M.A.; Arafa, R.K. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: Design, synthesis, cytotoxicity evaluation and in silico studies. Eur. J. Med. Chem. 2019, 183, 111693. [Google Scholar] [CrossRef]
- Akhtar, W.; Nainwal, L.M.; Khan, M.F.; Verma, G.; Chashoo, G.; Bakht, A.; Iqbal, M.; Akhtar, M.; Shaquiquzzaman, M.; Alam, M.M. Synthesis, COX-2 inhibition and metabolic stability studies of 6-(4-fluorophenyl)-pyrimidine-5-carbonitrile derivatives as anticancer and anti-inflammatory agents. J. Fluor. Chem. 2020, 236, 109579. [Google Scholar] [CrossRef]
- Akhtar, W.; Nainwal, L.M.; Kaushik, S.K.; Akhtar, M.; Shaquiquzzaman, M.; Almalki, F.; Saifullah, K.; Marella, A.; Alam, M. Methylene-bearing sulfur-containing cyanopyrimidine derivatives for treatment of cancer: Part-II. Arch. Pharm. 2020, 353, e1900333. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.M.; Abd El Razik, H.A.; Hazzaa, A.A.; El-Attar, M.A.; El Demellawy, M.A.; Abdel Wahab, A.E.; El Hawash, S.A. New pyrimidines and triazolopyrimidines as antiproliferative and antioxidants with cyclooxygenase-1/2 inhibitory potential. Future Med. Chem. 2019, 11, 1583–1603. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; El-Gaby, M.S.A.; Alsaid, M.S.; Elshaier, Y.A.M.M.; Soliman, A.M.; El-Senduny, F.F.; Badria, F.A.; Sherif, A.Y.A. Novel thiourea derivatives bearing sulfonamide moiety as anticancer agents through COX-2 inhibition. Anti-Cancer Agents Med. Chem. 2017, 17, 1411–1425. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.; Bhadauria, A.S.; Singh, A.K.; Kumar, U.; Rai, A.; Keshari, A.K.; Kumar, P.; Kumar, D.; Maity, B.; Nath, S. Novel 1, 3, 4-thiadiazoles inhibit colorectal cancer via blockade of IL-6/COX-2 mediated JAK2/STAT3 signals as evidenced through data-based mathematical modeling. Cytokine 2019, 118, 144–159. [Google Scholar] [CrossRef] [PubMed]
- El-Husseiny, W.M.; El-Sayed, M.A.-A.; El-Azab, A.S.; AlSaif, N.A.; Alanazi, M.M.; Abdel-Aziz, A.A.-M. Synthesis, antitumor activity, and molecular docking study of 2-cyclopentyloxyanisole derivatives: Mechanistic study of enzyme inhibition. J. Enzym. Inhib. Med. Chem. 2020, 35, 744–758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, F.; Shang, L. Advances in antitumor effects of NSAIDs. Cancer Manag. Res. 2018, 10, 4631. [Google Scholar] [CrossRef]
- Özdemir, Ö.; Marinelli, L.; Cacciatore, I.; Ciulla, M.; Emsen, B.; Di Stefano, A.; Mardinoglu, A.; Turkez, H. Anticancer effects of novel NSAIDs derivatives on cultured human glioblastoma cells. Z. Naturforsch. 2021, 76, 329–335. [Google Scholar] [CrossRef]
- Cairat, M.; Al Rahmoun, M.; Gunter, M.J.; Severi, G.; Dossus, L.; Fournier, A. Use of nonsteroidal anti-inflammatory drugs and breast cancer risk in a prospective cohort of postmenopausal women. Breast Cancer Res. 2020, 22, 118. [Google Scholar] [CrossRef]
- Veitonmäki, T.; Murtola, T.J.; Talala, K.; Taari, K.; Tammela, T.; Auvinen, A. Non-steroidal anti-inflammatory drugs and cancer death in the finnish prostate cancer screening trial. PLoS ONE 2016, 11, e0153413. [Google Scholar] [CrossRef] [Green Version]
- Koki, A.T.; Masferrer, J.L. Celecoxib: A specific COX-2 inhibitor with anticancer properties. Cancer Control 2002, 9 (Suppl. S2), 28–35. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Aziz, A.A.-M.; Abou-Zeid, L.A.; El-Husseiny, W.M.; El_Morsy, A.M.; El-Gendy, M.A.; El-Sayed, M.A.-A. Synthesis, antitumour activities and molecular docking of thiocarboxylic acid ester-based NSAID scaffolds: COX-2 inhibition and mechanistic studies. J. Enzym. Inhib. Med. Chem. 2018, 33, 989–998. [Google Scholar] [CrossRef]
- Coşkun, G.P.; Djikic, T.; Hayal, T.B.; Türkel, N.; Yelekçi, K.; Şahin, F.; Küçükgüzel, Ş.G. Synthesis, molecular docking and anticancer activity of diflunisal derivatives as cyclooxygenase enzyme inhibitors. Molecules 2018, 23, 1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Husseiny, W.M.; Magda, A.-A.; Abdel-Aziz, N.I.; El-Azab, A.S.; Asiri, Y.A.; Alaa, A.-M. Structural alterations based on naproxen scaffold: Synthesis, evaluation of antitumor activity and COX-2 inhibition, and molecular docking. Eur. J. Med. Chem. 2018, 158, 134–143. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, M.T.; El-Sharief, M.A.S.; Zarie, E.S.; Morsy, N.M.; Elsheakh, A.R.; Nayel, M.; Voronkov, A.; Berishvili, V.; Sabry, N.M.; Hassan, G.S. Design, synthesis, anti-inflammatory antitumor activities, molecular modeling and molecular dynamics simulations of potential naprosyn® analogs as COX-1 and/or COX-2 inhibitors. Bioorg. Chem. 2018, 76, 188–201. [Google Scholar] [CrossRef]
- Mareddy, J.; Suresh, N.; Kumar, C.G.; Kapavarapu, R.; Jayasree, A.; Pal, S. 1, 2, 3-Triazole-nimesulide hybrid: Their design, synthesis and evaluation as potential anticancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 518–523. [Google Scholar] [CrossRef]
- Celik, S.; Albayrak, A.T.; Akyuz, S.; Ozel, A.E. Synthesis, molecular docking and ADMET study of ionic liquid as anticancer inhibitors of DNA and COX-2, TOPII enzymes. J. Biomol. Struct. 2019, 38, 1354–1364. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Madala, H.R.; Mikelis, C.M.; Dixit, A.; Arutla, V.; Srivenugopal, K.S. Conception, synthesis, and characterization of a rofecoxib-combretastatin hybrid drug with potent cyclooxygenase-2 (COX-2) inhibiting and microtubule disrupting activities in colon cancer cell culture and xenograft models. Oncotarget 2018, 9, 26109. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Milacic, V.; Frezza, M.; Dou, Q.P. Metal complexes, their cellular targets and potential for cancer therapy. Curr. Pharm. Des. 2009, 15, 777–791. [Google Scholar] [CrossRef]
- Samra, M.M.; Sadia, A.; Azam, M.; Imran, M.; Ahmad, I.; Basra, M.A. Engineering, synthesis, spectroscopic and biological investigation of a new Ca (II) complex of meloxicam as potential COX-2 inhibitor. Arab J. Sci. Eng. 2022, 47, 7105–7122. [Google Scholar] [CrossRef]
- Obermoser, V.; Baecker, D.; Schuster, C.; Braun, V.; Kircher, B.; Gust, R. Chlorinated cobalt alkyne complexes derived from acetylsalicylic acid as new specific antitumor agents. Dalton Trans. 2018, 47, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Baecker, D.; Obermoser, V.; Kirchner, E.A.; Hupfauf, A.; Kircher, B.; Gust, R. Fluorination as tool to improve bioanalytical sensitivity and COX-2-selective antitumor activity of cobalt alkyne complexes. Dalton Trans. 2019, 48, 15856–15868. [Google Scholar] [CrossRef]
- Baecker, D.; Sagasser, J.; Karaman, S.; Hörmann, A.A.; Gust, R. Development of methylated cobalt–alkyne complexes with selective cytotoxicity against COX-positive cancer cell lines. Arch. Pharm. 2022, 355, 2100408. [Google Scholar] [CrossRef] [PubMed]
- Meieranz, S.; Stefanopoulou, M.; Rubner, G.; Bensdorf, K.; Kubutat, D.; Sheldrick, W.S.; Gust, R. The biological activity of Zeise’s salt and its derivatives. Angew. Chem. Int. Ed. 2015, 54, 2834–2837. [Google Scholar] [CrossRef] [PubMed]
- Weninger, A.; Baecker, D.; Obermoser, V.; Egger, D.; Wurst, K.; Gust, R. Synthesis and Biological Evaluation of Zeise’s Salt Derivatives with Acetylsalicylic Acid Substructure. Int. J. Mol. Sci. 2018, 19, 1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regulski, M.; Piotrowska-Kempisty, H.; Prukała, W.; Dutkiewicz, Z.; Regulska, K.; Stanisz, B.; Murias, M. Synthesis, in vitro and in silico evaluation of novel trans-stilbene analogues as potential COX-2 inhibitors. Bioorg. Med. Chem. 2018, 26, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Şenkardeş, S.; Han, M.İ.; Kulabaş, N.; Abbak, M.; Çevik, Ö.; Küçükgüzel, İ.; Küçükgüzel, Ş.G. Synthesis, molecular docking and evaluation of novel sulfonyl hydrazones as anticancer agents and COX-2 inhibitors. Mol. Div. 2020, 24, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Popiołek, Ł.; Patrejko, P.; Gawrońska-Grzywacz, M.; Biernasiuk, A.; Berecka-Rycerz, A.; Natorska-Chomicka, D.; Piątkowska-Chmiel, I.; Gumieniczek, A.; Dudka, J.; Wujec, M. Synthesis and in vitro bioactivity study of new hydrazide-hydrazones of 5-bromo-2-iodobenzoic acid. Biomed. Pharmacother. 2020, 130, 110526. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Ahmaditaba, M.A.; Shahosseini, S.; Daraei, B.; Zarghi, A.; Houshdar Tehrani, M.H. Design, synthesis, and biological evaluation of new peptide analogues as selective COX-2 inhibitors. Arch. Pharm. 2017, 350, 1700158. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohsin, N.u.A.; Aslam, S.; Ahmad, M.; Irfan, M.; Al-Hussain, S.A.; Zaki, M.E.A. Cyclooxygenase-2 (COX-2) as a Target of Anticancer Agents: A Review of Novel Synthesized Scaffolds Having Anticancer and COX-2 Inhibitory Potentialities. Pharmaceuticals 2022, 15, 1471. https://doi.org/10.3390/ph15121471
Mohsin NuA, Aslam S, Ahmad M, Irfan M, Al-Hussain SA, Zaki MEA. Cyclooxygenase-2 (COX-2) as a Target of Anticancer Agents: A Review of Novel Synthesized Scaffolds Having Anticancer and COX-2 Inhibitory Potentialities. Pharmaceuticals. 2022; 15(12):1471. https://doi.org/10.3390/ph15121471
Chicago/Turabian StyleMohsin, Noor ul Amin, Sana Aslam, Matloob Ahmad, Muhammad Irfan, Sami A. Al-Hussain, and Magdi E. A. Zaki. 2022. "Cyclooxygenase-2 (COX-2) as a Target of Anticancer Agents: A Review of Novel Synthesized Scaffolds Having Anticancer and COX-2 Inhibitory Potentialities" Pharmaceuticals 15, no. 12: 1471. https://doi.org/10.3390/ph15121471
APA StyleMohsin, N. u. A., Aslam, S., Ahmad, M., Irfan, M., Al-Hussain, S. A., & Zaki, M. E. A. (2022). Cyclooxygenase-2 (COX-2) as a Target of Anticancer Agents: A Review of Novel Synthesized Scaffolds Having Anticancer and COX-2 Inhibitory Potentialities. Pharmaceuticals, 15(12), 1471. https://doi.org/10.3390/ph15121471