
Structure-Guide Design and Optimization of Potential Druglikeness Inhibitors for TGFβRI with the Pyrrolopyrimidine Scaffold
 and
and 
Abstract
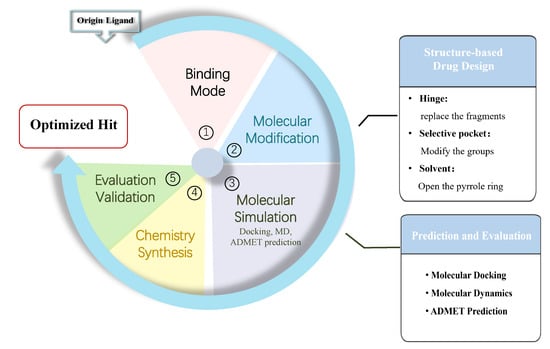
1. Introduction
2. Results and Discussion
2.1. The Binding Mode of the Co-Crystal of TGFβRI
2.2. Results of Molecular Modification
2.3. Molecular Docking Analysis
2.4. ADMET Prediction
2.5. MD Trajectories Analysis
2.6. Binding Energy Analysis and Energy Decomposition
2.7. Kinase Active Validation
3. Materials and Methods
3.1. Data Collection and Preparation
3.2. Molecular Docking
3.3. ADMET Prediction
3.4. Molecular Dynamics Simulation
3.5. Binding Free Energy Calculation and Decomposition
3.6. Experimental Sections
3.6.1. General Procedure of Synthetic Reaction
General Information
The Synthesis of 6-(6-(Trifluoromethyl)Pyridin-2-yl)-1,3,5-Triazine-2,4(1H,3H)-Dione (2 in Scheme 1)
The Synthesis of 2,4-Dichloro-6-(6-(Trifluoromethyl)Pyridin-2-yl)-1,3,5-Triazine (3 in Scheme 1)
The Synthesis of 1-((4-((3-Fluoropyridin-4-yl)Amino)-6-(6-(Trifluoromethyl)Pyridin-2-yl)-1,3,5-Triazin-2-yl)Amino)-2-Methylpropan-2-ol (W8 in Scheme 1)
3.6.2. Kinase Active Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Bonyadi, M.; Rusholme, S.A.; Cousins, F.M.; Su, H.C.; Biron, C.A.; Farrall, M.; Akhurst, R.J. Mapping of a major genetic modifier of embryonic lethality in TGF beta 1 knockout mice. Nat. Genet. 1997, 15, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Ten Dijke, P. Exploring anti-TGF-β therapies in cancer and fibrosis. Growth Factors 2011, 29, 140–152. [Google Scholar] [CrossRef]
- Heldin, C.H.; Landström, M.; Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Jachimczak, P.; Bogdahn, U.; Schneider, J.; Behl, C.; Meixensberger, J.; Apfel, R.; Dörries, R.; Schlingensiepen, K.H.; Brysch, W. The effect of transforming growth factor-beta 2-specific phosphorothioate-anti-sense oligodeoxynucleotides in reversing cellular immunosuppression in malignant glioma. J. Neurosurg. 1993, 78, 944–951. [Google Scholar] [CrossRef]
- Padua, D.; Massagué, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Pardali, E.; Ten Dijke, P. TGFβ signaling and cardiovascular diseases. Int. J. Biol. Sci. 2012, 8, 195–213. [Google Scholar] [CrossRef]
- de Caestecker, M.P.; Piek, E.; Roberts, A.B. Role of transforming growth factor-beta signaling in cancer. J. Natl. Cancer Inst. 2000, 92, 1388–1402. [Google Scholar] [CrossRef]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef]
- Papageorgis, P. TGFβ Signaling in Tumor Initiation, Epithelial-to-Mesenchymal Transition, and Metastasis. J. Oncol. 2015, 2015, 587193. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell. 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Mempel, T.R.; Pittet, M.J.; Khazaie, K.; Weninger, W.; Weissleder, R.; von Boehmer, H.; von Andrian, U.H. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity 2006, 25, 129–141. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Massagué, J.; Wotton, D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000, 19, 1745–1754. [Google Scholar] [CrossRef]
- Ooshima, A.; Park, J.; Kim, S.J. Phosphorylation status at Smad3 linker region modulates transforming growth factor-β-induced epithelial-mesenchymal transition and cancer progression. Cancer Sci. 2019, 110, 481–488. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Teicher, B.A. TGFβ-Directed Therapeutics: 2020. Pharmacol. Ther. 2021, 217, 107666. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Miao, M.; Schoeb, T.R.; Agarwal, A.; Murphy-Ullrich, J.E. Blockade of TSP1-dependent TGF-β activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am. J. Pathol. 2011, 178, 2573–2586. [Google Scholar] [CrossRef]
- Muraoka, R.S.; Dumont, N.; Ritter, C.A.; Dugger, T.C.; Brantley, D.M.; Chen, J.; Easterly, E.; Roebuck, L.R.; Ryan, S.; Gotwals, P.J.; et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J. Clin. Investig. 2002, 109, 1551–1559. [Google Scholar] [CrossRef]
- Akhurst, R.J. Large- and small-molecule inhibitors of transforming growth factor-beta signaling. Curr. Opin. Investig. Drugs 2006, 7, 513–521. [Google Scholar]
- Schlingensiepen, R.; Goldbrunner, M.; Szyrach, M.N.; Stauder, G.; Jachimczak, P.; Bogdahn, U.; Schulmeyer, F.; Hau, P.; Schlingensiepen, K.H. Intracerebral and intrathecal infusion of the TGF-beta 2-specific antisense phosphorothioate oligonucleotide AP 12009 in rabbits and primates: Toxicology and safety. Oligonucleotides 2005, 15, 94–104. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Fischer-Blass, B.; Schmaus, S.; Ludwig, S. Antisense therapeutics for tumor treatment: The TGF-beta2 inhibitor AP 12009 in clinical development against malignant tumors. Recent Results Cancer Res. 2008, 177, 137–150. [Google Scholar] [CrossRef]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro. Oncol. 2016, 18, 1146–1156. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, H.; Bu, H.; Zhou, J.; Zhang, H. Targeting the immunity protein kinases for immuno-oncology. Eur. J. Med. Chem. 2019, 163, 413–427. [Google Scholar] [CrossRef]
- Jung, S.Y.; Hwang, S.; Clarke, J.M.; Bauer, T.M.; Keedy, V.L.; Lee, H.; Park, N.; Kim, S.J.; Lee, J.I. Pharmacokinetic characteristics of vactosertib, a new activin receptor-like kinase 5 inhibitor, in patients with advanced solid tumors in a first-in-human phase 1 study. Investig New Drugs 2020, 38, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.S.; Anderson, B.D.; Beight, D.W.; Campbell, R.M.; Jones, M.L.; Herron, D.K.; Lampe, J.W.; McCowan, J.R.; McMillen, W.T.; Mort, N.; et al. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J. Med. Chem. 2003, 46, 3953–3956. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.S.; Beight, D.W.; Britt, K.S.; Anderson, B.D.; Campbell, R.M.; Goodson, T., Jr.; Herron, D.K.; Li, H.Y.; McMillen, W.T.; Mort, N.; et al. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. Bioorg. Med. Chem. Lett. 2004, 14, 3581–3584. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef]
- Li, H.Y.; McMillen, W.T.; Heap, C.R.; McCann, D.J.; Yan, L.; Campbell, R.M.; Mundla, S.R.; King, C.H.; Dierks, E.A.; Anderson, B.D.; et al. Optimization of a dihydropyrrolopyrazole series of transforming growth factor-beta type I receptor kinase domain inhibitors: Discovery of an orally bioavailable transforming growth factor-beta receptor type I inhibitor as antitumor agent. J. Med. Chem. 2008, 51, 2302–2306. [Google Scholar] [CrossRef]
- Koh, R.Y.; Lim, C.L.; Uhal, B.D.; Abdullah, M.; Vidyadaran, S.; Ho, C.C.; Seow, H.F. Inhibition of transforming growth factor-β via the activin receptor-like kinase-5 inhibitor attenuates pulmonary fibrosis. Mol. Med. Rep. 2015, 11, 3808–3813. [Google Scholar] [CrossRef]
- Callahan, J.F.; Burgess, J.L.; Fornwald, J.A.; Gaster, L.M.; Harling, J.D.; Harrington, F.P.; Heer, J.; Kwon, C.; Lehr, R.; Mathur, A.; et al. Identification of novel inhibitors of the transforming growth factor beta1 (TGF-beta1) type 1 receptor (ALK5). J. Med. Chem. 2002, 45, 999–1001. [Google Scholar] [CrossRef]
- Jiang, J.; Zhou, H.; Jiang, Q.; Sun, L.; Deng, P. Novel Transforming Growth Factor-Beta Receptor 1 Antagonists through a Pharmacophore-Based Virtual Screening Approach. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Jiang, J.H.; Deng, P. Discovery of New Inhibitors of Transforming Growth Factor-Beta Type 1 Receptor by Utilizing Docking and Structure-Activity Relationship Analysis. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, J.H.; Li, R.Y.; Deng, P. Docking-based virtual screening of TβR1 inhibitors: Evaluation of pose prediction and scoring functions. BMC Chem. 2020, 14, 52. [Google Scholar] [CrossRef]
- Harikrishnan, L.S.; Warrier, J.; Tebben, A.J.; Tonukunuru, G.; Madduri, S.R.; Baligar, V.; Mannoori, R.; Seshadri, B.; Rahaman, H.; Arunachalam, P.N.; et al. Heterobicyclic inhibitors of transforming growth factor beta receptor I (TGFβRI). Bioorg. Med. Chem. 2018, 26, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hou, T. Drug and drug candidate building block analysis. J. Chem. Inf. Model. 2010, 50, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Klug-Mcleod, J.; Rai, B.; Lunney, E.A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Pippin, D.A.; Salvino, J.M.; Mallamo, J.P. Knowledge based prediction of ligand binding modes and rational inhibitor design for kinase drug discovery. J. Med. Chem. 2008, 51, 5149–5171. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva-Jr, F.P. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–w14. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and homogeneous ADP monitoring assay for kinases. Assay Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef]
- Huse, M.; Chen, Y.G.; Massagué, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Zeng, G.; Shao, B.; Chen, M.; Li, Z.; Jiang, Y.; Liu, Y.; Zhang, Y.; Zhong, H. Application of molecular docking for the degradation of organic pollutants in the environmental remediation: A review. Chemosphere 2018, 203, 139–150. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Akobeng, A.K. Understanding diagnostic tests 3: Receiver operating characteristic curves. Acta Paediatr. 2007, 96, 644–647. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Menchon, G.; Maveyraud, L.; Czaplicki, G. Molecular Dynamics as a Tool for Virtual Ligand Screening. Methods Mol. Biol. 2018, 1762, 145–178. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inform. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A First-in-Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Medicinal Chemistry | Physicochemical Property | Absorption | Distribution | Metabolism | Toxicity | Synthetic Accessibility b | ||
|---|---|---|---|---|---|---|---|---|---|
| Lipinski Rules | LogP a | P-gp Substrate | GI Absorption | BBB Penetration | CYP2D6 Inhibitor | Carcinogenicity | hERG Blockers | ||
| H1 | Accepted | 4.434 | NO | LOW | YES | YES | NO | YES | 3.10 |
| H2 | Accepted | 4.473 | YES | LOW | NO | YES | NO | YES | 3.06 |
| H3 | Accepted | 4.003 | YES | HIGH | NO | YES | YES | YES | 2.82 |
| H4 | Accepted | 4.153 | YES | HIGH | NO | YES | NO | YES | 2.75 |
| H5 | Accepted | 0.834 | YES | HIGH | NO | YES | NO | YES | 2.98 |
| H6 | Accepted | 4.508 | YES | LOW | NO | YES | NO | YES | 3.11 |
| H7 | Accepted | 3.852 | YES | HIGH | NO | YES | NO | YES | 3.05 |
| H8 | Accepted | 0.493 | YES | HIGH | NO | YES | NO | YES | 2.91 |
| H9 | Accepted | 4.203 | YES | HIGH | NO | YES | NO | YES | 2.93 |
| H10 | Accepted | 2.344 | YES | LOW | NO | YES | NO | YES | 3.02 |
| S1 | Accepted | 0.944 | YES | LOW | NO | NO | NO | YES | 3.49 |
| S3 | Accepted | 1.720 | YES | LOW | NO | NO | YES | NO | 3.51 |
| S5 | Accepted | 1.080 | YES | LOW | NO | NO | NO | YES | 3.50 |
| S6 | Accepted | 1.630 | YES | LOW | NO | NO | NO | YES | 2.56 |
| W1 | Accepted | 1.358 | NO | LOW | NO | NO | NO | YES | 3.22 |
| W6 | Accepted | 3.430 | NO | LOW | NO | NO | NO | YES | 4.29 |
| W8 | Accepted | 2.386 | NO | HIGH | NO | NO | NO | NO | 3.10 |
| S1W8 | Accepted | 0.381 | YES | LOW | NO | YES | NO | NO | 3.87 |
| BMS22 | Accepted | 4.434 | YES | HIGH | NO | YES | NO | NO | 3.10 |
| Compound ID | ΔEvdw a | ΔEele b | ΔGMM c | ΔGPB d | ΔGSA e | ΔGbind f |
|---|---|---|---|---|---|---|
| W1 | −50.57 9 ± 3.021 | −43.260 ± 5.632 | −93.933 ± 5.471 | 52.089 ± 3.315 | −5.242 ± 0.203 | −46.992 ± 3.902 |
| H1 | −57.374 ± 3.601 | −60.696 ± 5.444 | −118.071 ± 8.208 | 78.628 ± 3.503 | −5.353 ± 0.201 | −44.797 ± 4.962 |
| W8 | −53.096 ± 2.804 | −28.215 ± 1.848 | −81.310 ± 3.683 | 44.303 ± 2.155 | −5.321 ± 0.195 | −42.330 ± 3.341 |
| S1W8 | −54.209 ± 3.598 | −32.781 ± 8.034 | −86.990 ± 6.865 | 56.018 ± 4.121 | −5.427 ± 0.204 | −36.399 ± 5.052 |
| BMS22 | −44.667 ± 3.130 | −47.651 ± 9.194 | −92.318 ± 8.797 | 66.294 ± 4.468 | −4.536 ± 0.185 | −30.560 ± 6.076 |
| S1 | −43.853 ± 2.726 | −29.833 ± 4.672 | −73.686 ± 4.405 | 49.486 ± 2.773 | −4.265 ±0.189 | −28.466 ± 4.026 |
| Pearson’s r g | 0.614 | 0.395 | 0.582 | −0.119 | 0.681 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, D.; Xie, J.; Li, Y.; Li, R.; Zhou, H.; Deng, P. Structure-Guide Design and Optimization of Potential Druglikeness Inhibitors for TGFβRI with the Pyrrolopyrimidine Scaffold. Pharmaceuticals 2022, 15, 1264. https://doi.org/10.3390/ph15101264
Meng D, Xie J, Li Y, Li R, Zhou H, Deng P. Structure-Guide Design and Optimization of Potential Druglikeness Inhibitors for TGFβRI with the Pyrrolopyrimidine Scaffold. Pharmaceuticals. 2022; 15(10):1264. https://doi.org/10.3390/ph15101264
Chicago/Turabian StyleMeng, Dan, Jiali Xie, Yihao Li, Ruoyu Li, Hui Zhou, and Ping Deng. 2022. "Structure-Guide Design and Optimization of Potential Druglikeness Inhibitors for TGFβRI with the Pyrrolopyrimidine Scaffold" Pharmaceuticals 15, no. 10: 1264. https://doi.org/10.3390/ph15101264
APA StyleMeng, D., Xie, J., Li, Y., Li, R., Zhou, H., & Deng, P. (2022). Structure-Guide Design and Optimization of Potential Druglikeness Inhibitors for TGFβRI with the Pyrrolopyrimidine Scaffold. Pharmaceuticals, 15(10), 1264. https://doi.org/10.3390/ph15101264