


Candesartan Attenuates Cisplatin-Induced Lung Injury by Modulating Oxidative Stress, Inflammation, and TLR-4/NF-κB, JAK1/STAT3, and Nrf2/HO-1 Signaling

 , , and
, , and
Abstract
1. Introduction
2. Results
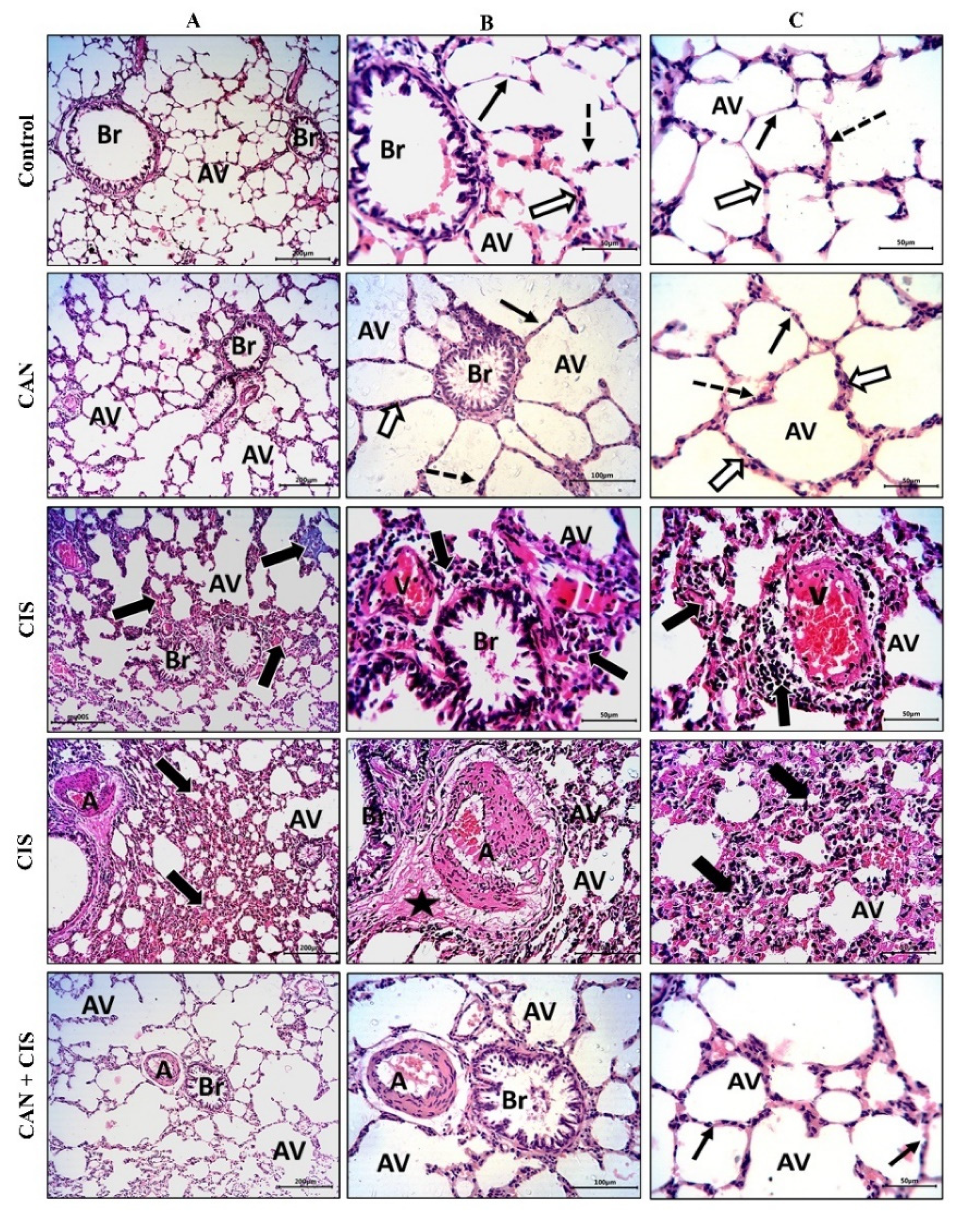
2.1. CAN Prevents CIS-Induced Lung Injury in Rats
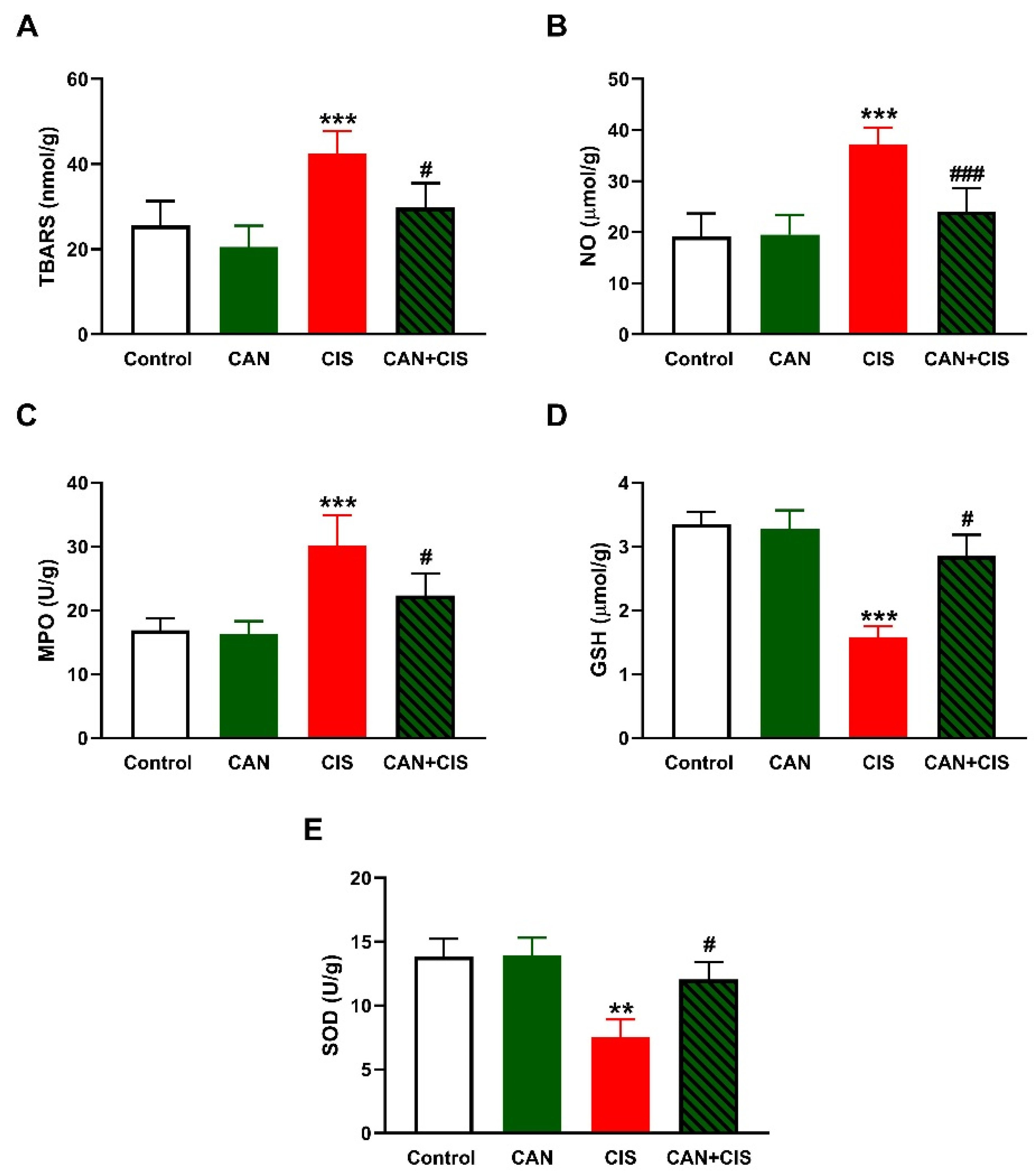
2.2. CAN Attenuates CIS-Induced Oxidative Stress in the Lungs of Rats
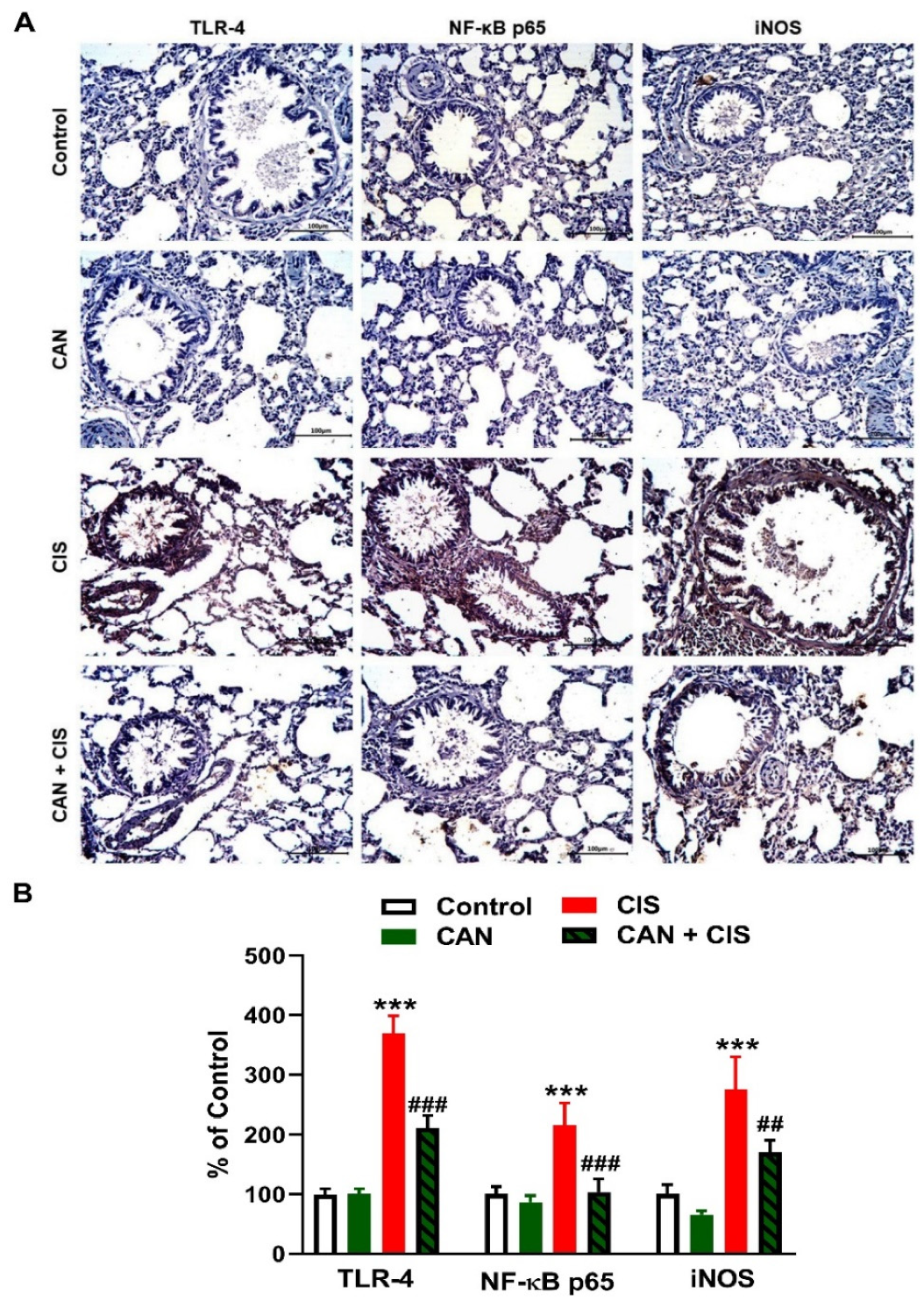
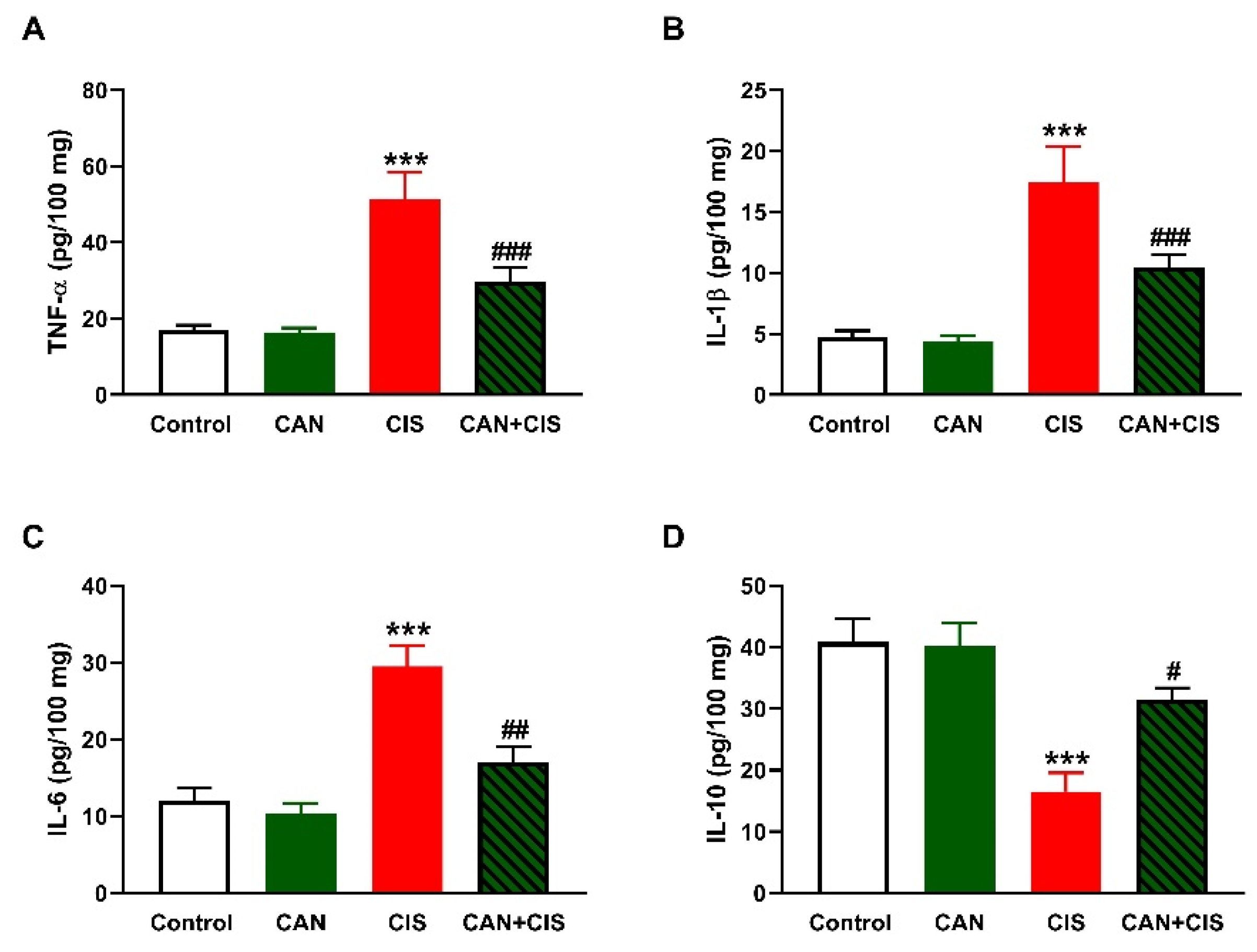
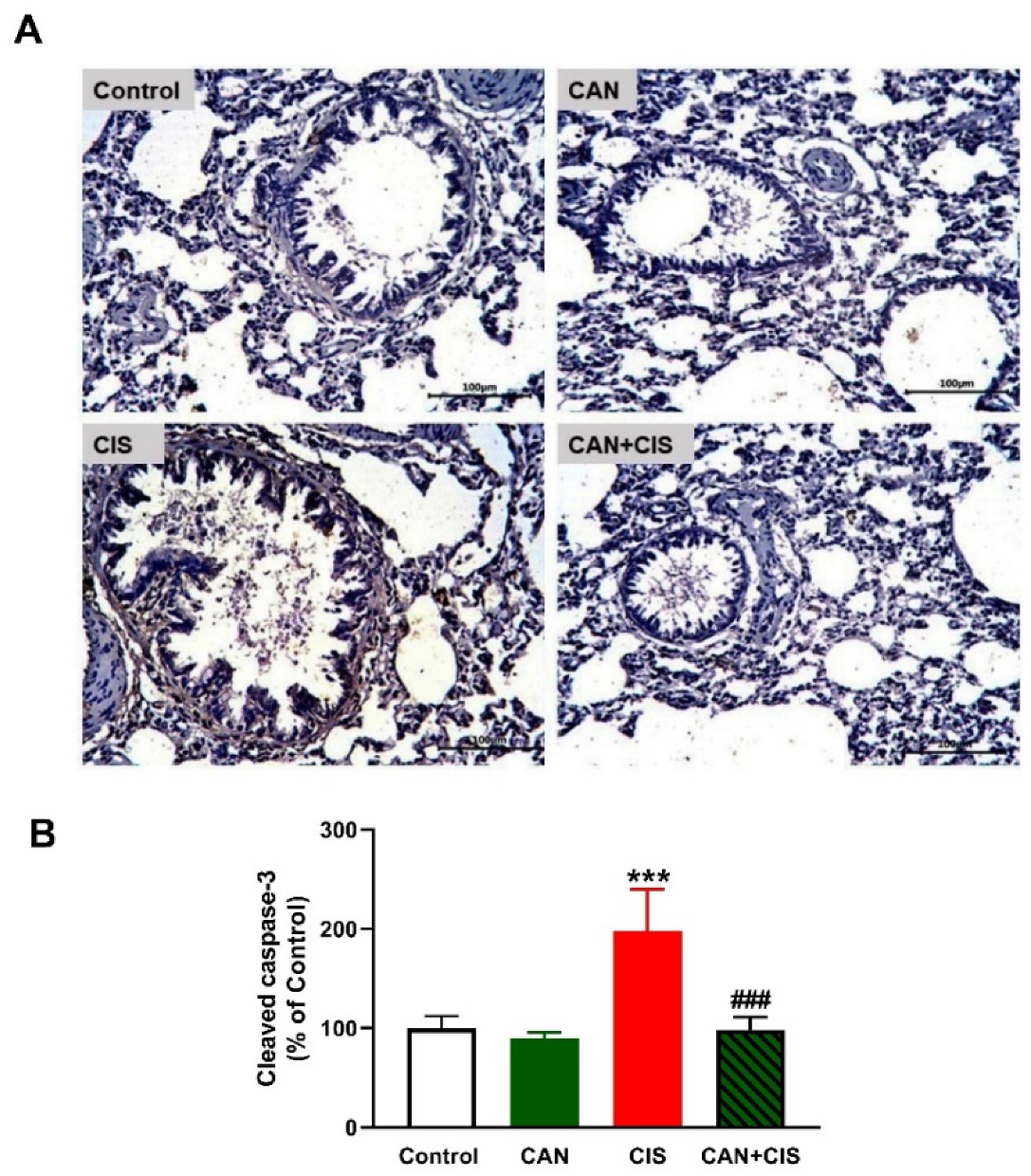
2.3. CAN Suppresses TLR-4/NF-κB Signaling and Prevents Inflammation and Apoptosis in CIS-Administered Rats
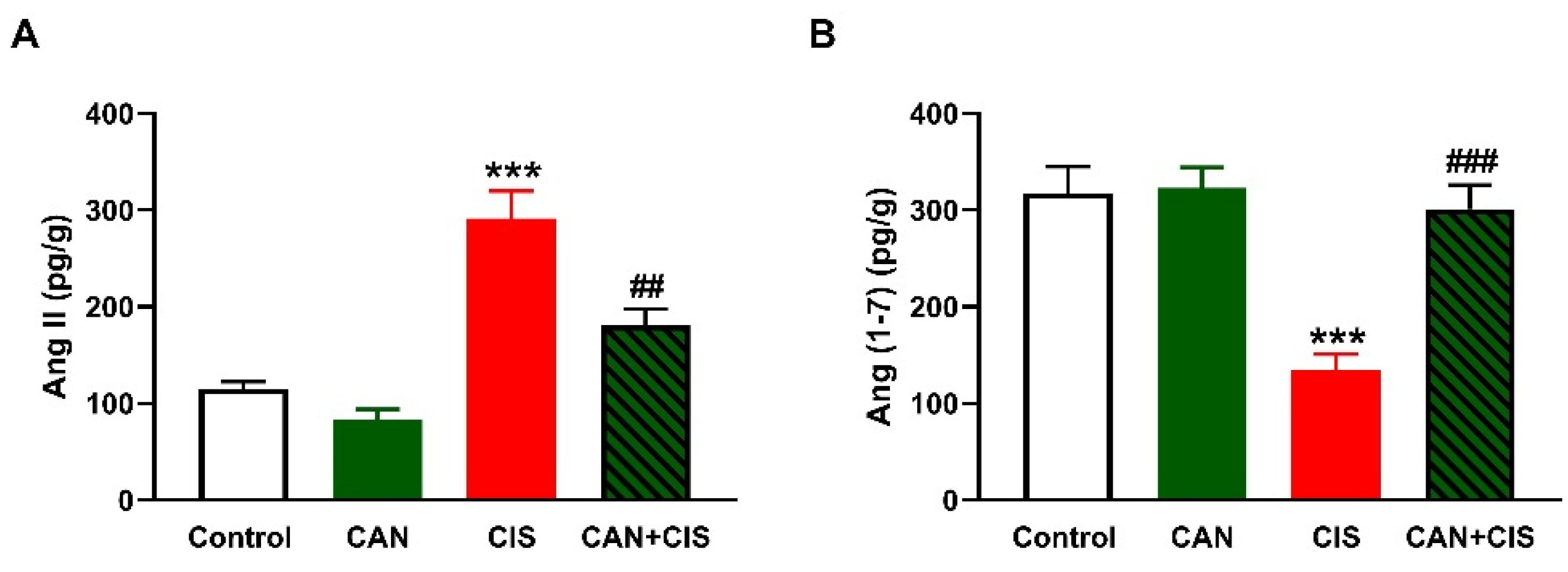
2.4. CAN Ameliorates Ang II and Ang (1–7) in CIS-Administered Rats
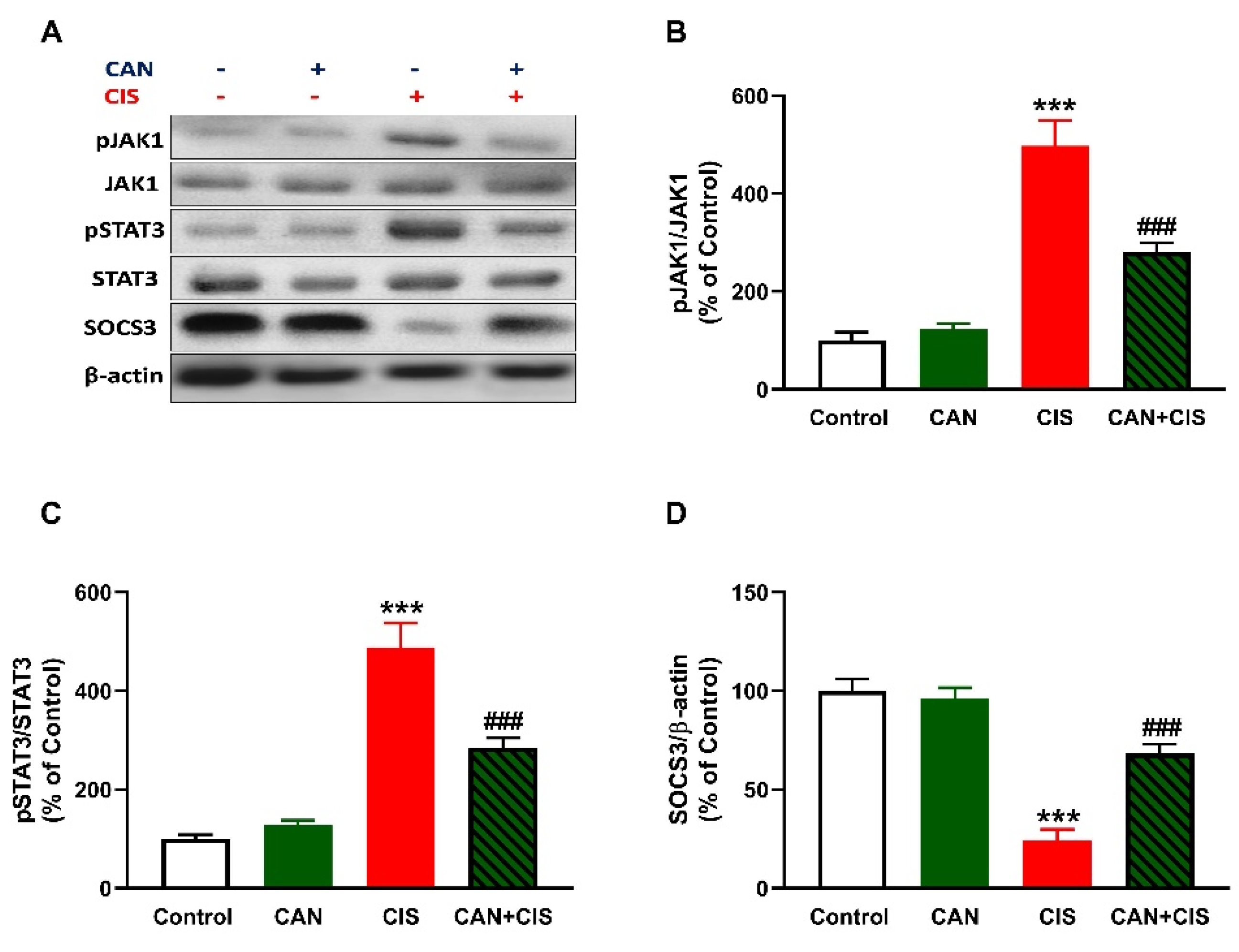
2.5. CAN Downregulates JAK1/STAT3 Signaling in CIS-Administered Rats
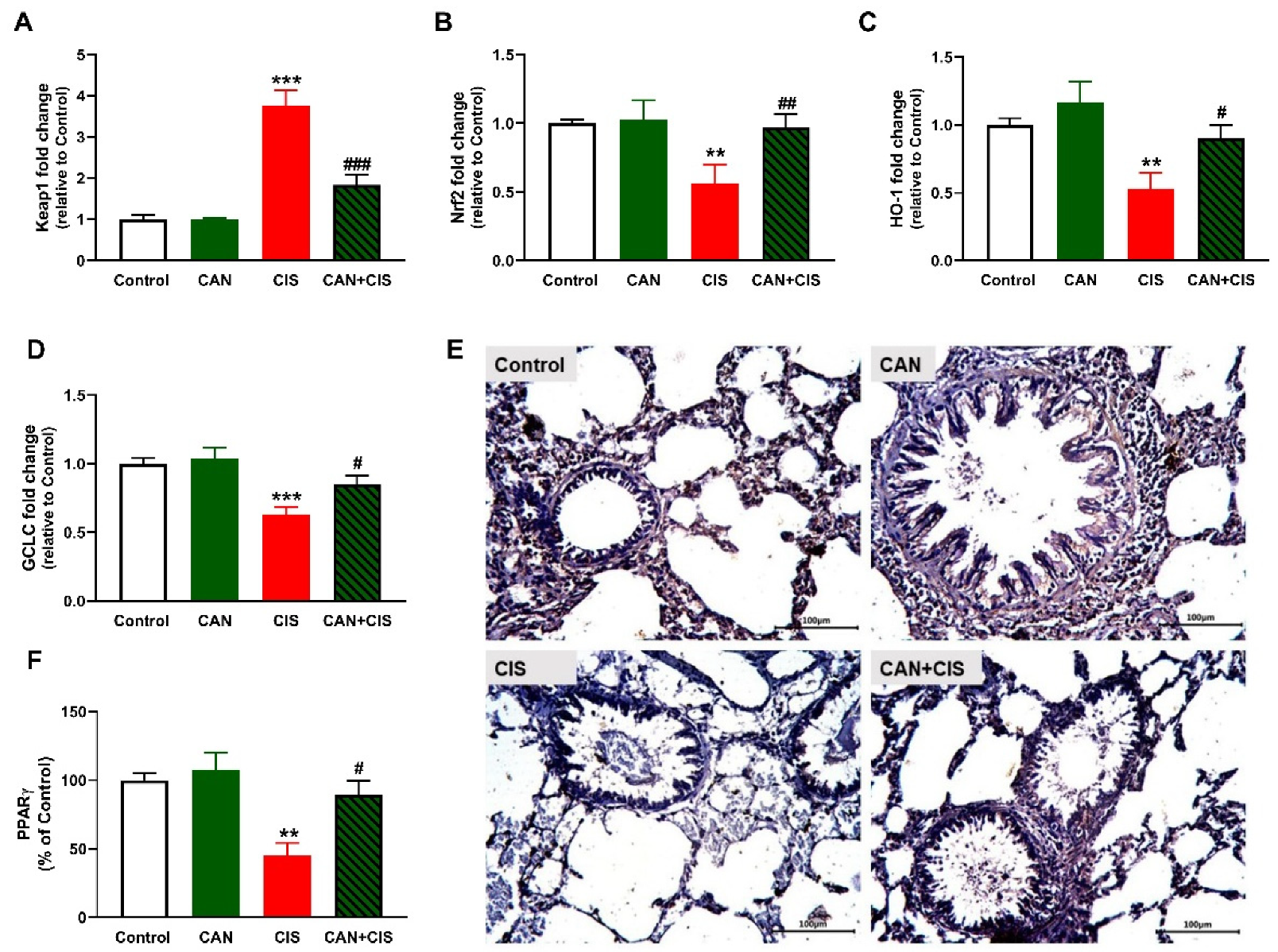
2.6. CAN Upregulates Nrf2/HO-1 Signaling and PPARγ in CIS-Administered Rats
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Experimental Animals and Treatments
4.3. Histological and Immunohistochemical (IHC) Assessment
4.4. Biochemical Assays
4.5. q-RT-PCR Analysis
4.6. Western Blotting
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorganic Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.J. Cisplatin overdose: Toxicities and management. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef]
- Li, L.; Mok, H.; Jhaveri, P.; Bonnen, M.D.; Sikora, A.G.; Eissa, N.T.; Komaki, R.U.; Ghebre, Y.T. Anticancer therapy and lung injury: Molecular mechanisms. Expert Rev. Anticancer. Ther. 2018, 18, 1041–1057. [Google Scholar] [CrossRef] [PubMed]
- Kovach, M.A.; Standiford, T.J. Toll like receptors in diseases of the lung. Int. Immunopharmacol. 2011, 11, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Aladaileh, S.H.; Al-Swailmi, F.K.; Abukhalil, M.H.; Ahmeda, A.F.; Mahmoud, A.M. Punicalagin prevents cisplatin-induced nephrotoxicity by attenuating oxidative stress, inflammatory response, and apoptosis in rats. Life Sci. 2021, 286, 120071. [Google Scholar] [CrossRef]
- Brozovic, A.; Ambriović-Ristov, A.; Osmak, M. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef]
- Unver, E.; Tosun, M.; Olmez, H.; Kuzucu, M.; Cimen, F.K.; Suleyman, Z. The Effect of Taxifolin on Cisplatin-Induced Pulmonary Damage in Rats: A Biochemical and Histopathological Evaluation. Mediat. Inflamm. 2019, 2019, 3740867. [Google Scholar] [CrossRef]
- Geyikoglu, F.; Isikgoz, H.; Onalan, H.; Colak, S.; Cerig, S.; Bakir, M.; Hosseinigouzdagani, M.; Koc, K.; Erol, H.S.; Saglam, Y.S.; et al. Impact of high-dose oleuropein on cisplatin-induced oxidative stress, genotoxicity and pathological changes in rat stomach and lung. J. Asian Nat. Prod. Res. 2017, 19, 1214–1231. [Google Scholar] [CrossRef]
- Ideguchi, H.; Kojima, K.; Hirosako, S.; Ichiyasu, H.; Fujii, K.; Kohrogi, H. Cisplatin-induced eosinophilic pneumonia. Case Rep. Pulmonol. 2014, 2014, 209732. [Google Scholar] [CrossRef]
- Han, Y.K.; Kim, J.S.; Jang, G.; Park, K.M. Cisplatin induces lung cell cilia disruption and lung damage via oxidative stress. Free. Radic. Biol. Med. 2021, 177, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baichwal, V.R. NF-kappa B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv. Immunol. 1997, 65, 111–137. [Google Scholar] [PubMed]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Asami, J.; Shimizu, T. Structural and functional understanding of the toll-like receptors. Protein Sci. A Publ. Protein Soc. 2021, 30, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Li, Y.; Noble, P.W. The role of Toll-like receptors in non-infectious lung injury. Cell Res. 2006, 16, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, S.T.; Zhang, X.; Aberg, E.; Bousette, N.; Giaid, A.; Shan, P.; Medzhitov, R.M.; Lee, P.J. Inducible activation of TLR4 confers resistance to hyperoxia-induced pulmonary apoptosis. J. Immunol. 2006, 176, 4950–4958. [Google Scholar] [CrossRef] [PubMed]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Planas, A.M.; Gorina, R.; Chamorro, Á. Signalling pathways mediating inflammatory responses in brain ischaemia. Biochem. Soc. Trans. 2006, 34, 1267–1270. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Abd El-Twab, S.M. Caffeic acid phenethyl ester protects the brain against hexavalent chromium toxicity by enhancing endogenous antioxidants and modulating the JAK/STAT signaling pathway. Biomed. Pharmacother. 2017, 91, 303–311. [Google Scholar] [CrossRef]
- Al-Samhari, M.M.; Al-Rasheed, N.M.; Al-Rejaie, S.; Al-Rasheed, N.M.; Hasan, I.H.; Mahmoud, A.M.; Dzimiri, N. Possible involvement of the JAK/STAT signaling pathway in N-acetylcysteine-mediated antidepressant-like effects. Exp. Biol. Med. 2016, 241, 509–518. [Google Scholar] [CrossRef]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Hasan, I.H.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mahmoud, A.M. Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats. Drug Des. Dev. Ther. 2016, 10, 2095–2107. [Google Scholar]
- Hassanein, E.H.M.; Sayed, A.M.; Hussein, O.E.; Mahmoud, A.M. Coumarins as Modulators of the Keap1/Nrf2/ARE Signaling Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 1675957. [Google Scholar] [CrossRef]
- Aladaileh, S.H.; Abukhalil, M.H.; Saghir, S.A.; Hanieh, H.; Alfwuaires, M.A.; Almaiman, A.A.; Bin-Jumah, M.; Mahmoud, A.M. Galangin activates Nrf2 signaling and attenuates oxidative damage, inflammation, and apoptosis in a rat model of cyclophosphamide-induced hepatotoxicity. Biomolecules 2019, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Germoush, M.O.; Alotaibi, M.F.; Hussein, O.E. Possible involvement of Nrf2 and PPARgamma up-regulation in the protective effect of umbelliferone against cyclophosphamide-induced hepatotoxicity. Biomed. Pharmacother. 2017, 86, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of Nrf2 in Cardiovascular Function and Disease. Oxidative Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef]
- El-Naa, M.M.; El-Refaei, M.F.; Nasif, W.A.; Abduljawad, S.H.; El-Brairy, A.I.; El-Readi, M.Z. In-vivo antioxidant and anti-inflammatory activity of rosiglitazone, a peroxisome proliferator-activated receptor-gamma (PPAR-γ) agonists in animal model of bronchial asthma. J. Pharm. Pharmacol. 2015, 67, 1421–1430. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Al Dera, H.S. 18β-Glycyrrhetinic acid exerts protective effects against cyclophosphamide-induced hepatotoxicity: Potential role of PPARγ and Nrf2 upregulation. Genes Nutr. 2015, 10, 1–13. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Abd El-Ghafar, O.A.M.; Alzoghaibi, M.A.; Hassanein, E.H.M. Agomelatine prevents gentamicin nephrotoxicity by attenuating oxidative stress and TLR-4 signaling, and upregulating PPARγ and SIRT1. Life Sci. 2021, 278, 119600. [Google Scholar] [CrossRef]
- Mateu, A.; Ramudo, L.; Manso, M.A.; De Dios, I. Cross-talk between TLR4 and PPARgamma pathways in the arachidonic acid-induced inflammatory response in pancreatic acini. Int. J. Biochem. Cell Biol. 2015, 69, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [PubMed]
- Lelis, D.F.; Freitas, D.F.; Machado, A.S.; Crespo, T.S.; Santos, S.H.S. Angiotensin-(1-7), Adipokines and Inflammation. Metab. Clin. Exp. 2019, 95, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, H.; Sahin, T.T.; Dikmen, K.; Dikmen, A.U.; Yuksel, O.; Gulbahar, O.; Poyraz, A.; Tekin, E. Candesartan mediates microcirculation in acute necrotizing pancreatitis. Bratisl. Lek. Listy 2015, 116, 270–275. [Google Scholar] [CrossRef]
- Lin, X.; Wu, M.; Liu, B.; Wang, J.; Guan, G.; Ma, A.; Zhang, Y. Candesartan ameliorates acute myocardial infarction in rats through inducible nitric oxide synthase, nuclear factor-κB, monocyte chemoattractant protein-1, activator protein-1 and restoration of heat shock protein 72. Mol. Med. Rep. 2015, 12, 8193–8200. [Google Scholar] [CrossRef][Green Version]
- Arumugam, S.; Thandavarayan, R.A.; Palaniyandi, S.S.; Giridharan, V.V.; Arozal, W.; Sari, F.R.; Soetikno, V.; Harima, M.; Suzuki, K.; Kodama, M.; et al. Candesartan cilexetil protects from cardiac myosin induced cardiotoxicity via reduction of endoplasmic reticulum stress and apoptosis in rats: Involvement of ACE2-Ang (1-7)-mas axis. Toxicology 2012, 291, 139–145. [Google Scholar] [CrossRef]
- Wang, X.; Ye, Y.; Gong, H.; Wu, J.; Yuan, J.; Wang, S.; Yin, P.; Ding, Z.; Kang, L.; Jiang, Q.; et al. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1-7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J. Mol. Cell. Cardiol. 2016, 97, 180–190. [Google Scholar] [CrossRef]
- Qie, S.; Ran, Y.; Lu, X.; Su, W.; Li, W.; Xi, J.; Gong, W.; Liu, Z. Candesartan modulates microglia activation and polarization via NF-κB signaling pathway. Int. J. Immunopathol. Pharmacol. 2020, 34, 2058738420974900. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X. Candesartan targeting of angiotensin II type 1 receptor demonstrates benefits for hypertension in pregnancy via the NF-κB signaling pathway. Mol. Med. Rep. 2018, 18, 705–714. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Zhang, R.; Brennan, M.L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem. 2002, 277, 46116–46122. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Keat, K. Myeloperoxidase and associated lung disease: Review of the latest developments. Int. J. Rheum. Dis. 2021, 24, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, J.; Weng, C.; Chen, R.; Zheng, Y.; Chen, Q.; Tang, H. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem. Biophys. Res. Commun. 2003, 305, 989–996. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Saitoh, Y.; Hongwei, W.; Ueno, H.; Mizuta, M.; Nakazato, M. Candesartan attenuates fatty acid-induced oxidative stress and NAD(P)H oxidase activity in pancreatic beta-cells. Diabetes Res. Clin. Pract. 2010, 90, 54–59. [Google Scholar] [CrossRef]
- Hofni, A.; El-Moselhy, M.A.; Taye, A.; Khalifa, M.M. Combination therapy with spironolactone and candesartan protects against streptozotocin-induced diabetic nephropathy in rats. Eur. J. Pharmacol. 2014, 744, 173–182. [Google Scholar] [CrossRef]
- Yang, H.; Song, X.; Wei, Z.; Xia, C.; Wang, J.; Shen, L.; Wang, J. TLR4/MyD88/NF-κB Signaling in the Rostral Ventrolateral Medulla Is Involved in the Depressor Effect of Candesartan in Stress-Induced Hypertensive Rats. ACS Chem. Neurosci. 2020, 11, 2978–2988. [Google Scholar] [CrossRef]
- Dasu, M.R.; Riosvelasco, A.C.; Jialal, I. Candesartan inhibits Toll-like receptor expression and activity both in vitro and in vivo. Atherosclerosis 2009, 202, 76–83. [Google Scholar] [CrossRef]
- Hitomi, H.; Kiyomoto, H.; Nishiyama, A. Angiotensin II and oxidative stress. Curr. Opin. Cardiol. 2007, 22, 311–315. [Google Scholar] [CrossRef]
- Mateo, T.; Abu Nabah, Y.N.; Abu Taha, M.; Mata, M.; Cerdá-Nicolás, M.; Proudfoot, A.E.; Stahl, R.A.; Issekutz, A.C.; Cortijo, J.; Morcillo, E.J.; et al. Angiotensin II-induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J. Immunol. 2006, 176, 5577–5586. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Ichiki, T.; Shimokawa, H.; Egashira, K.; Takeda, K.; Kaibuchi, K.; Takeya, M.; Yoshimura, T.; Takeshita, A. Rho-kinase mediates angiotensin II-induced monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Hypertension 2001, 38, 100–104. [Google Scholar] [CrossRef]
- Nair, A.R.; Ebenezer, P.J.; Saini, Y.; Francis, J. Angiotensin II-induced hypertensive renal inflammation is mediated through HMGB1-TLR4 signaling in rat tubulo-epithelial cells. Exp. Cell Res. 2015, 335, 238–247. [Google Scholar] [CrossRef]
- Ferreira, A.J.; Bader, M.; Santos, R.A. Therapeutic targeting of the angiotensin-converting enzyme 2/Angiotensin-(1-7)/Mas cascade in the renin-angiotensin system: A patent review. Expert Opin. Ther. Pat. 2012, 22, 567–574. [Google Scholar] [CrossRef]
- Moodley, Y.P.; Misso, N.L.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.P.; Scaffidi, A.K.; Misso, N.L.; Keerthisingam, C.; McAnulty, R.J.; Laurent, G.J.; Mutsaers, S.E.; Thompson, P.J.; Knight, D.A. Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin-6/gp130-mediated cell signaling and proliferation. Am. J. Pathol. 2003, 163, 345–354. [Google Scholar] [CrossRef]
- Wang, Y.J.; Li, Y.; Wang, X.L.; Li, X.Z.; Chen, Y.W.; Yang, L.L.; Ming, H.X. Effect of Total Flavonoids of Oxytropis falcata Bunge on the Expression of p-JAK1-and p-STAT1-Related Proteins in Idiopathic Pulmonary Fibrosis. Evid. Based Complement. Altern. Med. eCAM 2020, 2020, 2407239. [Google Scholar] [CrossRef] [PubMed]
- Digicaylioglu, M.; Lipton, S.A. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-κB signalling cascades. Nature 2001, 412, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, N.J.; Murphy, J.M.; Liau, N.P.; Varghese, L.N.; Laktyushin, A.; Whitlock, E.L.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 2013, 20, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, J.; Zhang, L.; Sun, Y.; Jiang, J.; Huang, Y.; Xu, H.; Jiang, H.; Hu, R. Nrf2 protects against acute lung injury and inflammation by modulating TLR4 and Akt signaling. Free. Radic. Biol. Med. 2018, 121, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Matsuda, M.; Miyata, Y.; Fukuhara, A.; Komuro, R.; Shimabukuro, M.; Shimomura, I. Human catalase gene is regulated by peroxisome proliferator activated receptor-gamma through a response element distinct from that of mouse. Endocr. J. 2010, 57, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Necela, B.M.; Su, W.; Thompson, E.A. Toll-like receptor 4 mediates cross-talk between peroxisome proliferator-activated receptor gamma and nuclear factor-kappaB in macrophages. Immunology 2008, 125, 344–358. [Google Scholar] [CrossRef]
- Yin, Y.; Hou, G.; Li, E.; Wang, Q.; Kang, J. PPAR Gamma agonists regulate tobacco smoke-induced toll like receptor 4 expression in alveolar macrophages. Respir. Res. 2014, 15, 28. [Google Scholar] [CrossRef]
- Park, E.J.; Park, S.Y.; Joe, E.-h.; Jou, I. 15d-PGJ2 and Rosiglitazone Suppress Janus Kinase-STAT Inflammatory Signaling through Induction of Suppressor of Cytokine Signaling 1 (SOCS1) and SOCS3 in Glia*. J. Biol. Chem. 2003, 278, 14747–14752. [Google Scholar] [CrossRef]
- Thakur, K.S.; Prakash, A.; Bisht, R.; Bansal, P.K. Beneficial effect of candesartan and lisinopril against haloperidol-induced tardive dyskinesia in rat. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2015, 16, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, J.D.; Gamble, M. Theory and Practice of Histological Techniques; Elsevier Health Sciences: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Mihara, M.; Uchiyama, M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal. Biochem. 1978, 86, 271–278. [Google Scholar]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Montgomery, H.; Dymock, J.F.J.A. Determination of nitrite in water. Analyst 1961, 86, 414–416. [Google Scholar]
- Marklund, S.L. Superoxide dismutase isoenzymes in tissues and plasma from New Zealand black mice, nude mice and normal BALB/c mice. Mutat. Res. 1985, 148, 129–134. [Google Scholar] [CrossRef]
- Krawisz, J.; Sharon, P.; Stenson, W.J.G. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity: Assessment of inflammation in rat and hamster models. Gastroenterology 1984, 87, 1344–1350. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D.J.m. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| Nrf2 | TTGTAGATGACCATGAGTCGC | TGTCCTGCTGTATGCTGCTT |
| GCLC | GTTGTTACTGAATGGCGGCG | CGGCGTTTCCTCATGTTGTC |
| HO-1 | GTAAATGCAGTGTTGGCCCC | ATGTGCCAGGCATCTCCTTC |
| Keap1 | TCAGCTAGAGGCGTACTGGA | TTCGGTTACCATCCTGCGAG |
| GAPDH | TGCTGGTGCTGAGTATGTCG | TTGAGAGCAATGCCAGCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atwa, A.M.; Abd El-Ghafar, O.A.M.; Hassanein, E.H.M.; Mahdi, S.E.; Sayed, G.A.; Alruhaimi, R.S.; Alqhtani, H.A.; Alotaibi, M.F.; Mahmoud, A.M. Candesartan Attenuates Cisplatin-Induced Lung Injury by Modulating Oxidative Stress, Inflammation, and TLR-4/NF-κB, JAK1/STAT3, and Nrf2/HO-1 Signaling. Pharmaceuticals 2022, 15, 1222. https://doi.org/10.3390/ph15101222
Atwa AM, Abd El-Ghafar OAM, Hassanein EHM, Mahdi SE, Sayed GA, Alruhaimi RS, Alqhtani HA, Alotaibi MF, Mahmoud AM. Candesartan Attenuates Cisplatin-Induced Lung Injury by Modulating Oxidative Stress, Inflammation, and TLR-4/NF-κB, JAK1/STAT3, and Nrf2/HO-1 Signaling. Pharmaceuticals. 2022; 15(10):1222. https://doi.org/10.3390/ph15101222
Chicago/Turabian StyleAtwa, Ahmed M., Omnia A. M. Abd El-Ghafar, Emad H. M. Hassanein, Somya E. Mahdi, Ghadir A. Sayed, Reem S. Alruhaimi, Haifa A. Alqhtani, Mohammed F. Alotaibi, and Ayman M. Mahmoud. 2022. "Candesartan Attenuates Cisplatin-Induced Lung Injury by Modulating Oxidative Stress, Inflammation, and TLR-4/NF-κB, JAK1/STAT3, and Nrf2/HO-1 Signaling" Pharmaceuticals 15, no. 10: 1222. https://doi.org/10.3390/ph15101222
APA StyleAtwa, A. M., Abd El-Ghafar, O. A. M., Hassanein, E. H. M., Mahdi, S. E., Sayed, G. A., Alruhaimi, R. S., Alqhtani, H. A., Alotaibi, M. F., & Mahmoud, A. M. (2022). Candesartan Attenuates Cisplatin-Induced Lung Injury by Modulating Oxidative Stress, Inflammation, and TLR-4/NF-κB, JAK1/STAT3, and Nrf2/HO-1 Signaling. Pharmaceuticals, 15(10), 1222. https://doi.org/10.3390/ph15101222