1. Introduction
Owing to the tremendous blossoming of nanoscience in recent years, nanomaterials have been considered as potential candidates in a large array of applications such as optical devices [
1], catalysis [
2], and sensing materials [
3]. Among the growing applications of nanomaterials, nanocarriers are of keen interest given their pivotal importance in the drug delivery process [
4,
5]. In the extending field of nanocarriers, fullerene-like structures were proved as potent drug nanocarriers, inspired by high biocompatibility and low cytotoxicity nature [
6,
7,
8]. In the literature, group III nitride nanocarriers were found with exceptional properties, including elevated chemical stability, favorable band gap, suitable dielectric constant, considerable thermal conductivity, and upright oxidation resistance [
9,
10].
Furthermore, different geometries of boron nitrides (B
xN
x, where x refers to the number of boron and nitrogen atoms) were studied by Toftlund and Jensen, and the high stability of the B
12N
12 nanocarrier was addressed [
11]. Soltani et al. confirmed that B
12N
12 was more preferable than B
16N
16 in the drug delivery process of a 5-aminolevulinic acid (5-AVA) drug [
12]. By an arc-melting method, B
12N
12 was first synthesized with a band gap of 5.1 eV [
13,
14]. Afterward, B
12N
12 was extensively investigated to serve as a nanocarrier of diverse anti-cancer drugs such as Isoniazid [
6], Amphetamine [
15], Lomustine [
16], etc. Consequently, the optical, electronic, and chemical features of the boron nitride (B
12N
12) nanocarrier were thoroughly under study. It was reported that a significant alteration in boron nitride features occurred through doping the B
12N
12 surface [
17]. Concurrently, the aluminum (Al) atom is considered one of the most appropriate doping candidates due to its high porosity and ample surface active sites [
18]. In fact, Al-doped boron nitride (AlB
11N
12) was reported as an excellent nanocarrier for several drugs, owing to the sharp changes in the band gap and the reversibility of the adsorption process [
19]. In parallel, the AlB
11N
12 nanocarrier had a massive increment in its adsorption tendency, which was previously interpreted as an advantage of the Al doping processs [
20,
21]. Moreover, the adsorption processes of 5-aminosalicylic acid and the Cisplatin anti-cancer drug over the exterior surface of pristine and Al-doped B
12N
12 nanocarriers were investigated based on DFT calculations [
22,
23].
Chlormethine (CM), an anti-cancer drug with C
5H
11Cl
2N chemical formula, was synthesized in 1935 and applied for cancer treatment in 1946. Pharmacologists proved the effectiveness of CM to treat prostate cancer, polycythemia vera, chronic myelocytic leukemia, and lymphosarcoma after clinical trials. Moreover, the topical formulation of CM was documented to be operative for different conditions such as skin diseases and cell lymphoma [
24,
25,
26]. Nevertheless, CM had severe adverse effects that would eventually develop tentative blindness and skin cancer. With the aim of reducing undesirable side effects, versatile studies were conducted for using nanocarriers in the delivery process of CM. Subsequently, the adsorption process of CM over C
24, B
12C
6N
6, and B
12N
12 nanocarriers was investigated, revealing the crucial favorability for the B
12N
12 nanocarrier [
27]. Moreover, the effect of transition metal-doped boron nitride was theoretically studied over the surfaces of the B
12N
12, ZnB
12N
12, NiB
12N
12, CoB
12N
12, FeB
12N
12, and CuB
12N
12 nanocarriers [
28].
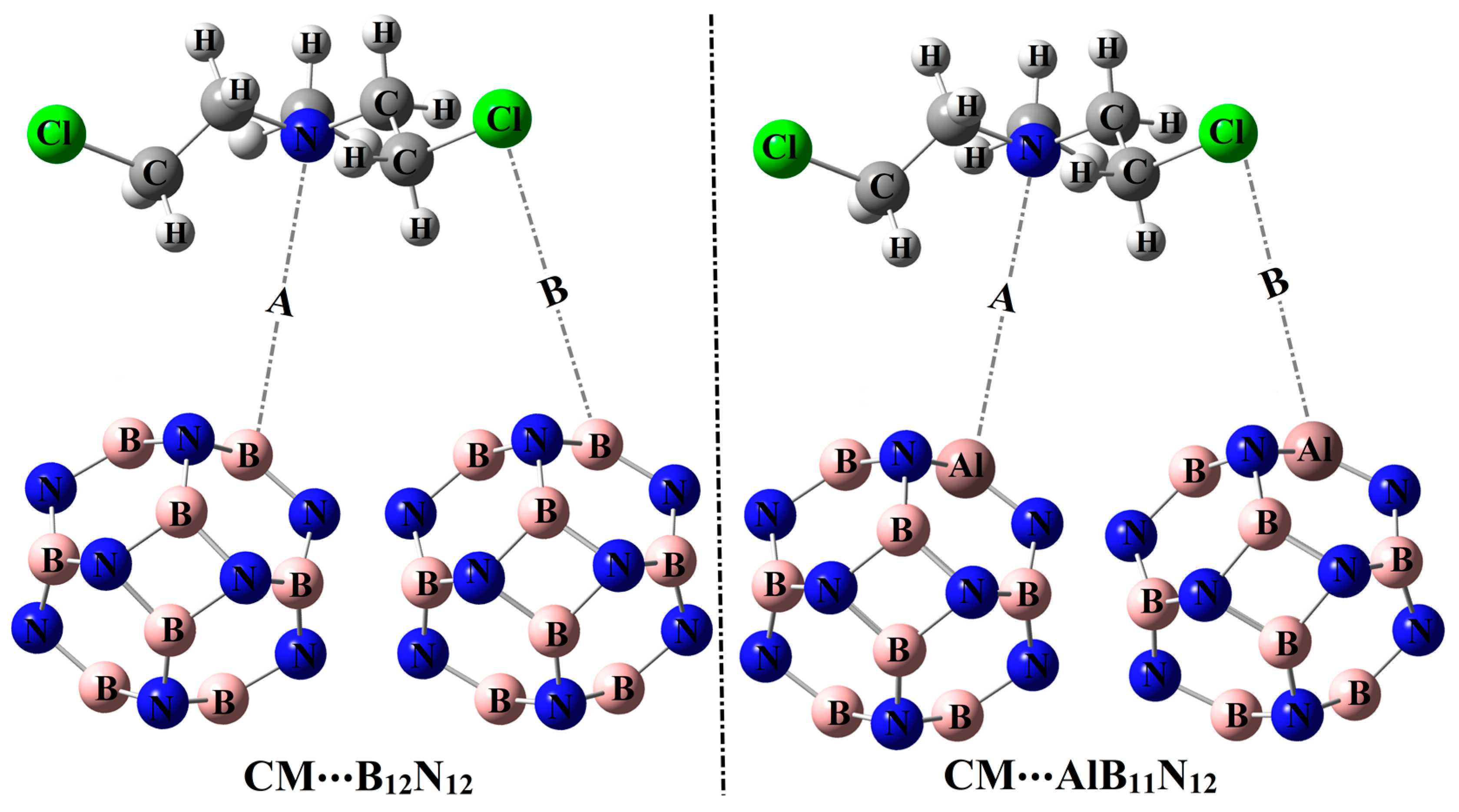
In the current investigation, the predilection of pure and Al-doped boron nitride nanocarriers (B
12N
12 and AlB
11N
12) to adsorb CM anti-cancer drug within configurations A and B was discussed (
Figure 1). By means of density functional theory (DFT) calculations, the occurrences of the adsorption process within the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes were detected in both gas and water phases. A series of thermodynamic parameters were also computed to elucidate the spontaneity and nature of the adsorption process. The obtained findings would be essential for the development of the applications of pure and doped boron nitride nanocarriers in the drug delivery process of anti-cancer drugs.
2. Results
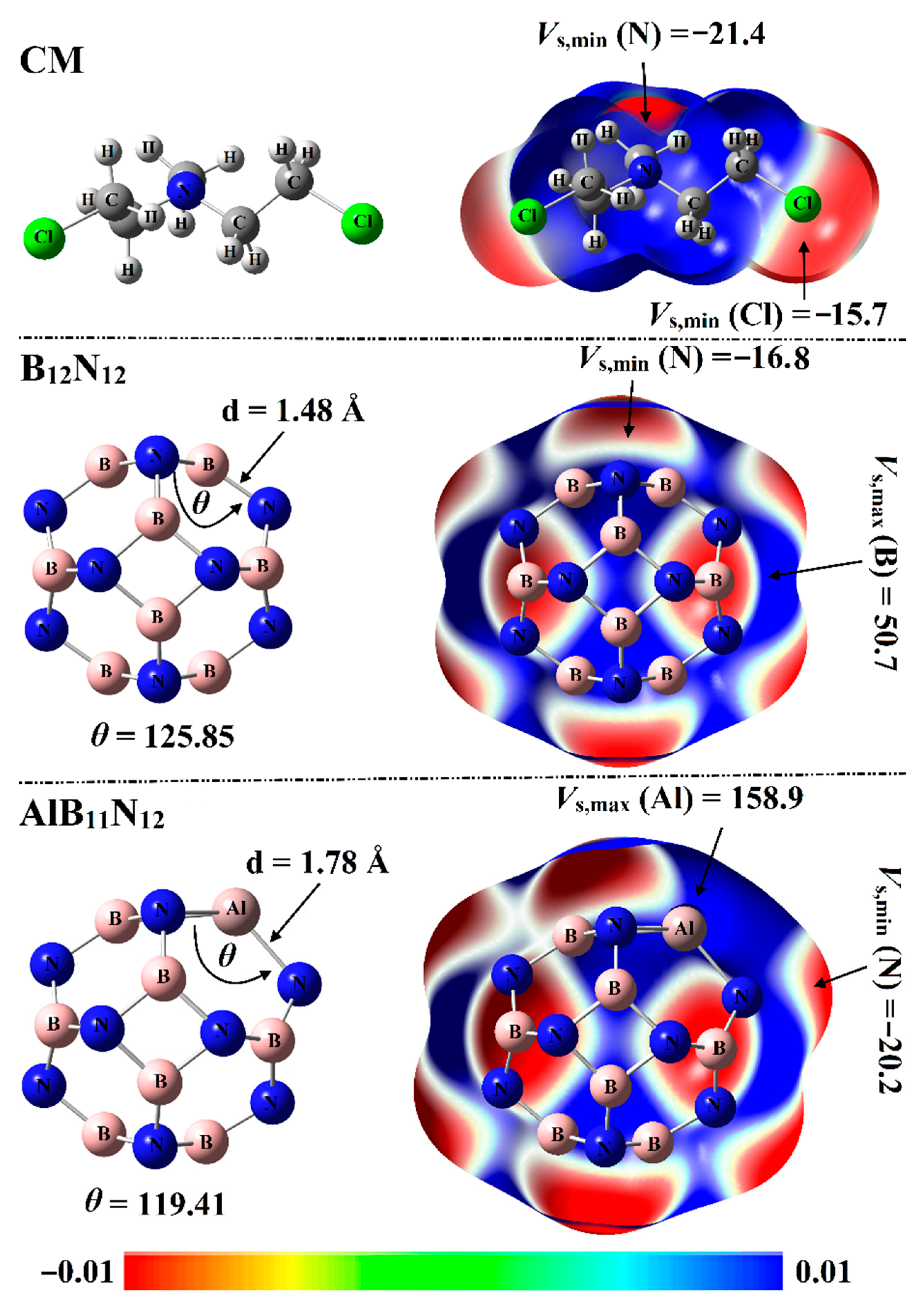
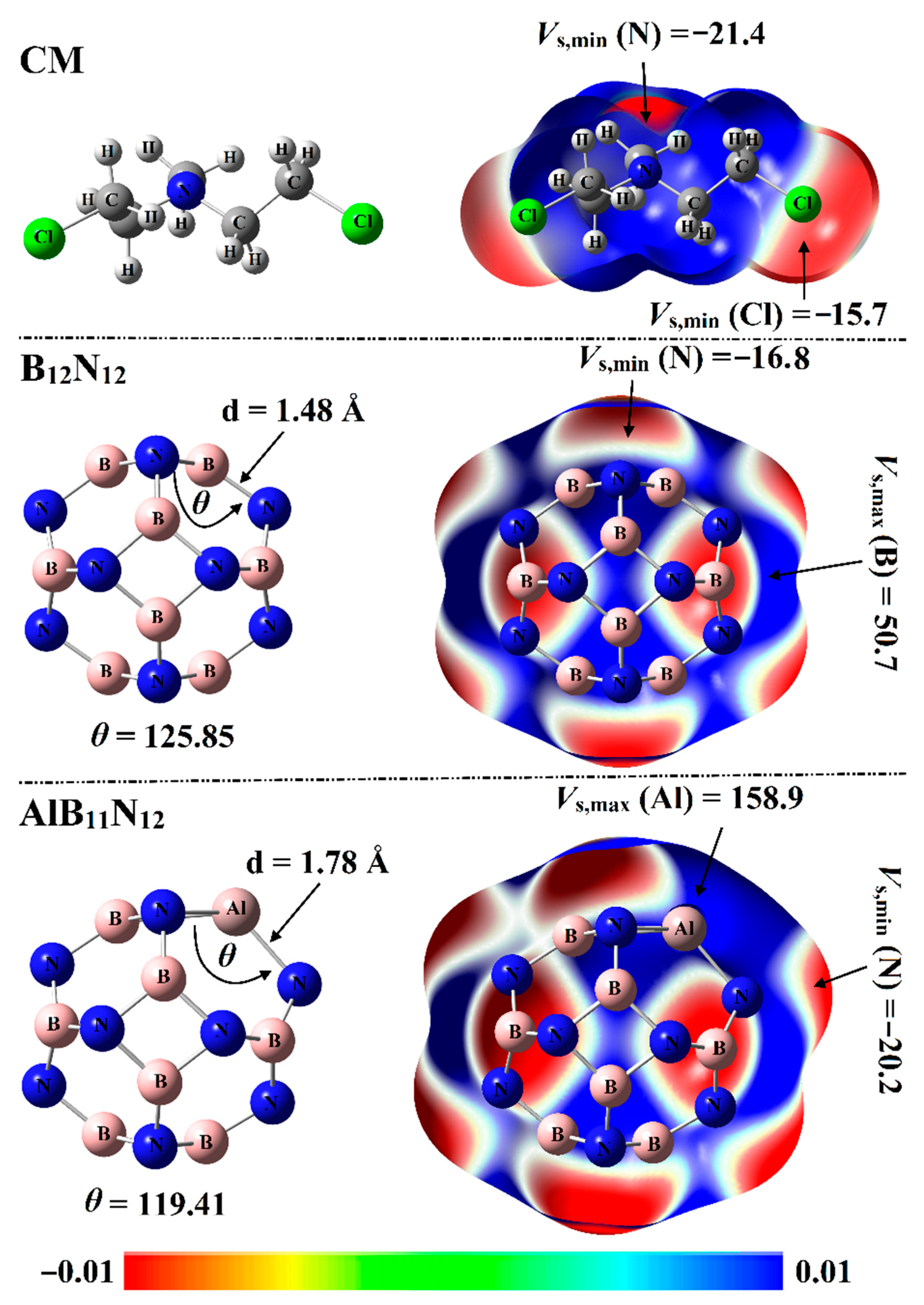
2.1. Electrostatic Potential (ESP) Analysis
Electrostatic potential (ESP) analysis has been introduced as a convenient and useful tool to qualitatively and quantitatively identify chemical systems [
29]. For the optimized systems, molecular electrostatic potential (MEP) maps were generated using 0.002 au electron density envelopes according to the previous recommendations [
30]. From a numerical point of view, surface electrostatic potential extrema were calculated in terms of maximum (
Vs,max) and minimum (
Vs,min) electrostatic potential values. The optimized structures with corresponding MEP maps of Chlormethine (CM) and pure and aluminum (Al)-doped boron nitride nanocarriers (B
12N
12 and AlB
11N
12) are graphed in
Figure 2.
As delineated in
Figure 2, the B
12N
12 nanocarrier with
Th symmetry was incorporated of identical eight hexagonal rings and six tetragonal rings. In terms of bond length, two nonequivalent B-N bond lengths were observed for the B
12N
12 nanocarrier: one between two hexagonal rings and the other one between a hexagonal and tetragonal ring. The B-N bond length was observed at 1.44 Å between two hexagonal rings and 1.48 Å between a hexagonal and tetragonal ring. Interestingly, the obtained bond length values were concomitant with previous studies [
31,
32].
For the AlB
11N
12 nanocarrier with the
Cs point group, the Al-N bond between two hexagonal rings was noticed with a length of 1.78 Å, while the Al-N bond between tetragonal and hexagonal was 1.82 Å. The bond length values illustrated in
Figure 2 were found to be compatible with previous studies pertinent to the influence of Al doping on the configuration of the B
12N
12 nanocarrier [
22].
From MEP maps visualized in
Figure 2, two nucleophilic sites labeled with red regions were noticed along the surface of the CM drug, numerically ensured by
Vs,min values of –15.7 and –21.4 kcal/mol along the surface of the Cl and N atoms, respectively. Turning to nanocarriers, positive ESP regions were observed over B and Al atoms, indicating the electrophilic nature of nanocarriers (
Figure 2). Illustratively,
Vs,max values over B and Al atoms of B
12N
12 and AlB
11N
12 nanocarriers were 50.7 and 158.9 kcal/mol, respectively. Evidently, the negative ESP on the N atoms in the AlB
11N
12 system was found with a higher
Vs,min value compared with that in the B
12N
12 system, ensuring the prominent electron donation ability of the Al atom more than the B atom. Quantitatively,
Vs,min values of N atoms were –16.8 and –20.2 kcal/mol for the B
12N
12 and AlB
11N
12 nanocarriers, respectively.
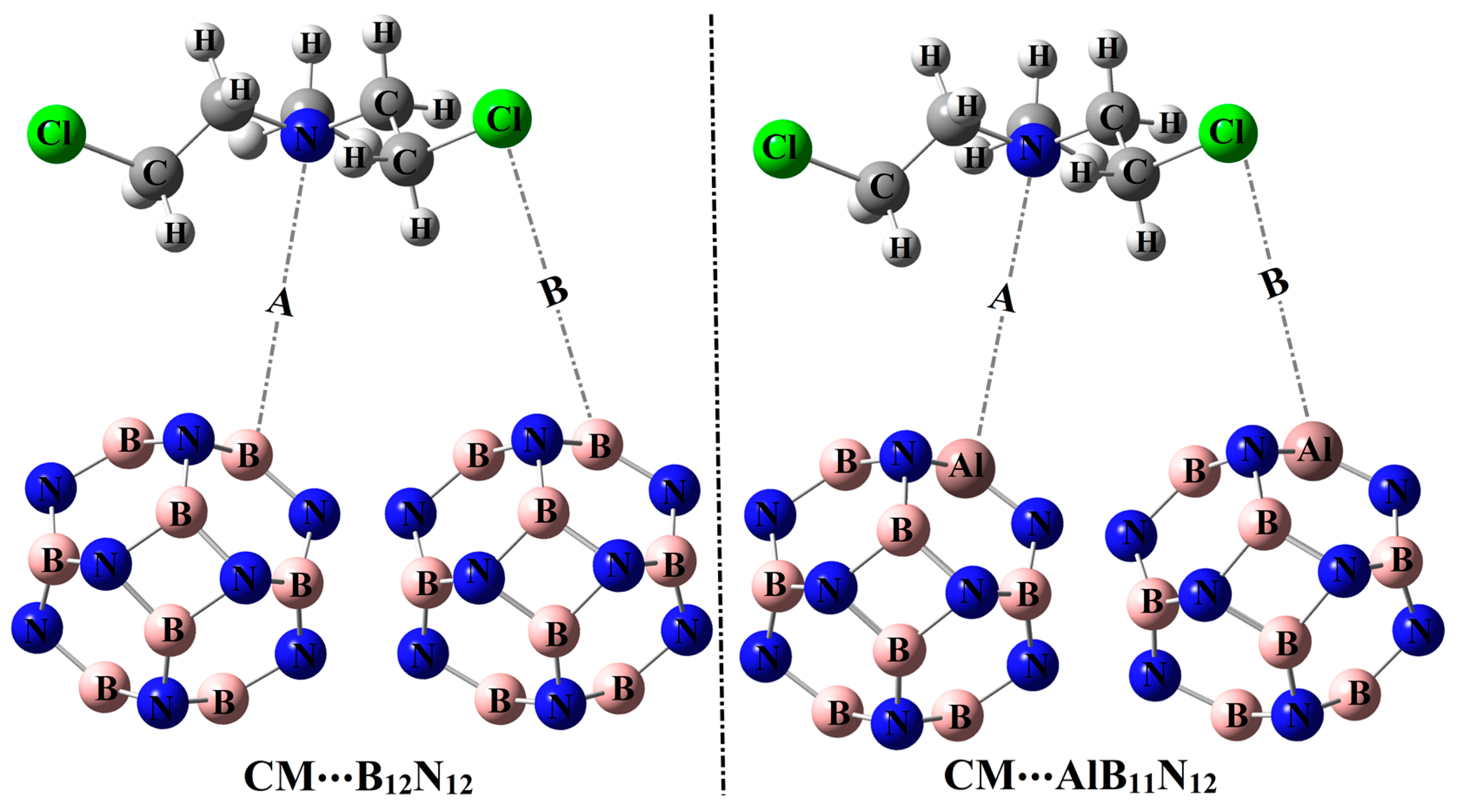
2.2. Adsorption Process
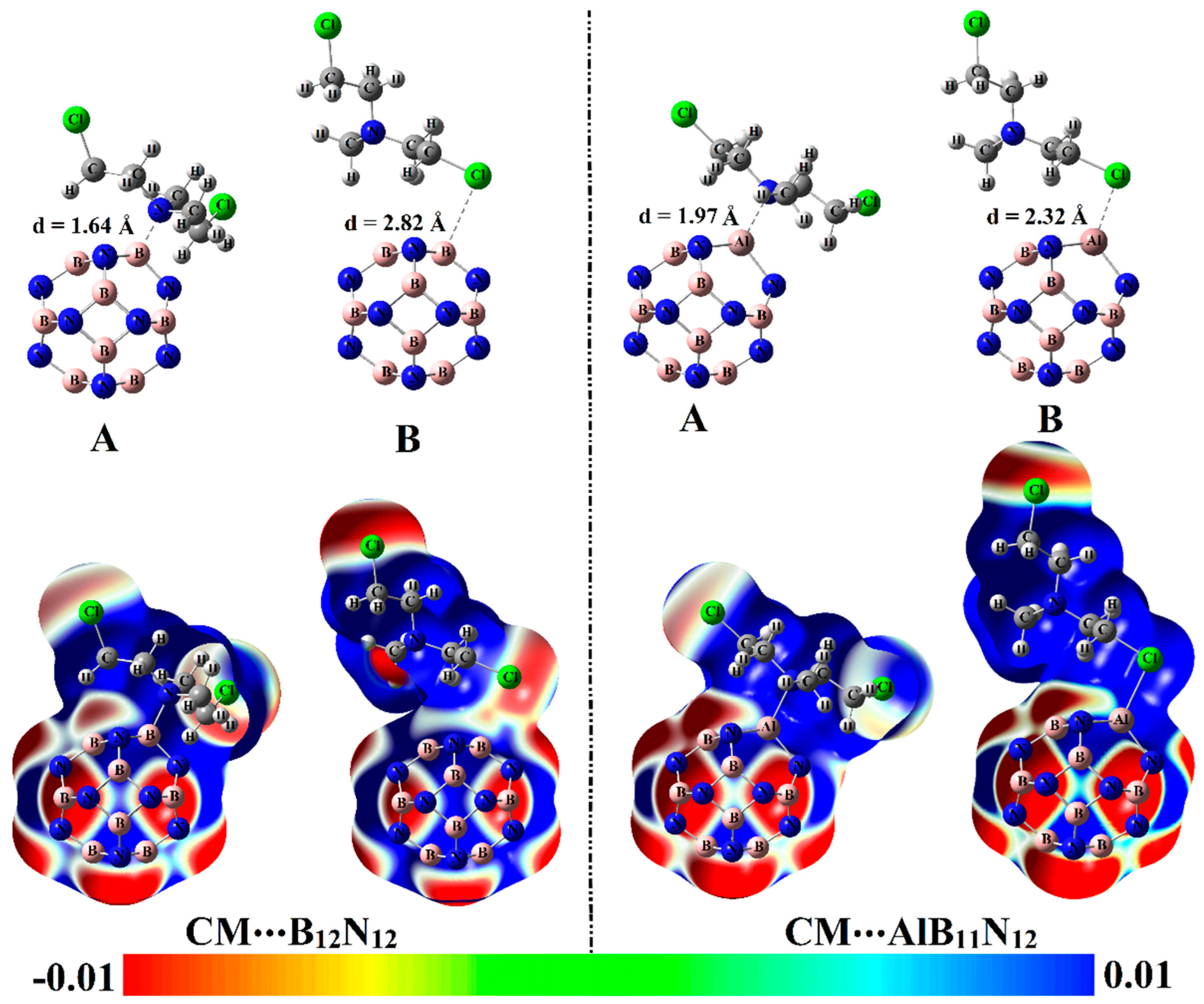
The potentiality of pure and Al-doped boron nitride (B
12N
12 and AlB
11N
12) nanocarriers to adsorb the Chlormethine (CM) anti-cancer drug was thoroughly studied within configurations A and B for CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes (
Figure 1). Geometrical optimization and MEP maps calculations were executed for the studied CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes, and the obtained structures are depicted in
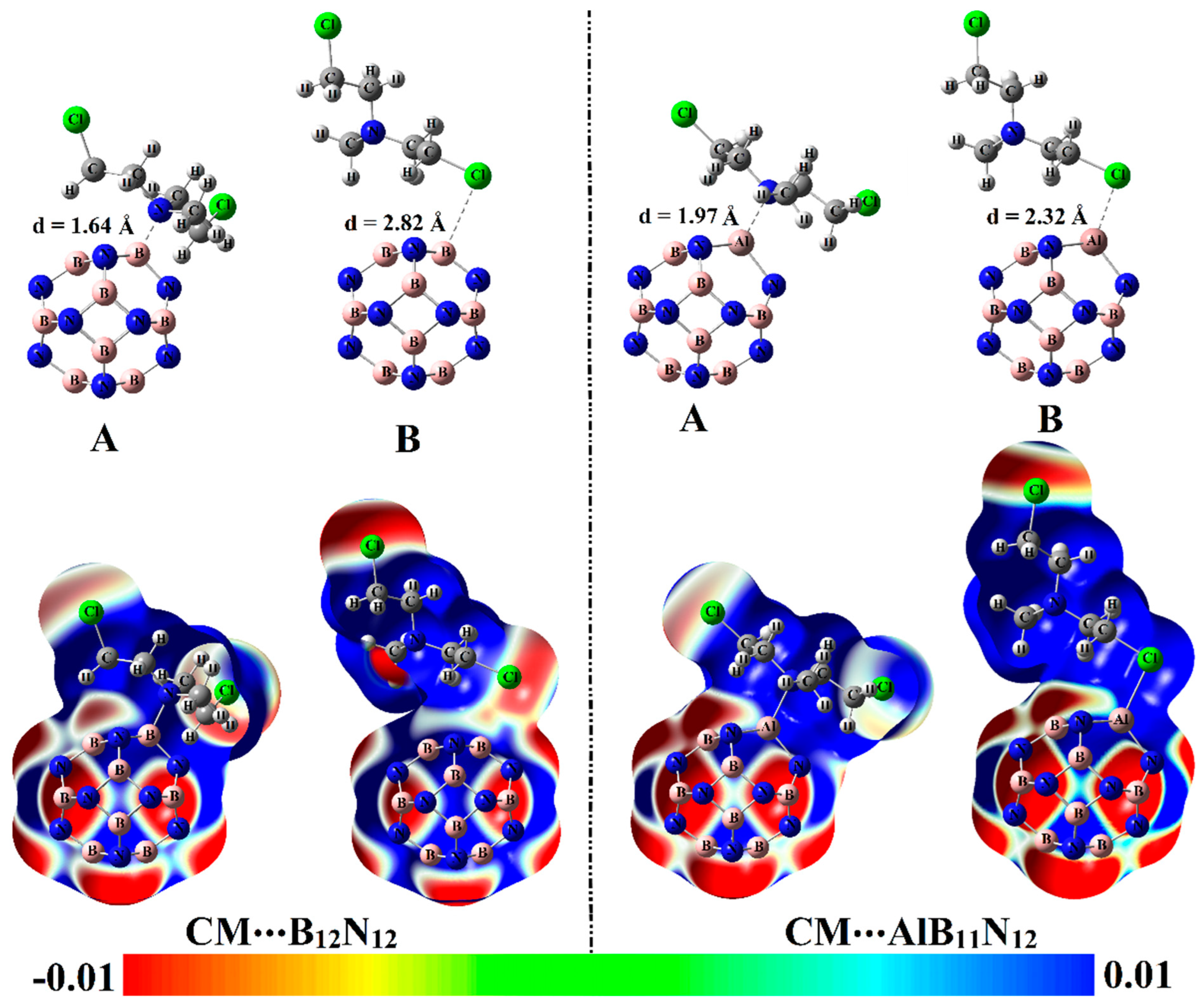
Figure 3.
Table 1 compiles the interaction and adsorption energies of the optimized complexes along with intermolecular distances between the interacted species.
From the data listed in
Table 1, substantial negative adsorption (
Eads) and interaction (
Eint) energies of the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes were observed, ensuring the occurrence of the adsorption process within modeled configurations. Notably, short CM∙∙∙B/∙∙∙Al intermolecular distances within the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes were detected, with values ranging from 1.64 Å to 2.82 Å (
Figure 3 and
Table 1).
Among the studied complexes, the CM∙∙∙AlB
11N
12 complex had higher negative interaction and adsorption energies compared with CM∙∙∙B
12N
12 complex and showed values of –59.68 and –52.40 kcal/mol, respectively, within configuration A. The favorability of CM∙∙∙AlB
11N
12 complex could be ascribed to the further prominent electrophilic character of the AlB
11N
12 nanocarrier compared with the B
12N
12 analog in line with the
Vs,max affirmations, which showed values of 158.9 and 50.7 kcal/mol, respectively. The interpretation of the superior adsorption behavior of the AlB
11N
12 nanocarrier toward the CM drug via site A more than B could be considered as a consequence of the higher negativity of ESP over the N atom compared with the Cl one that showed
Vs,min values of –21.4 and –15.7 kcal/mol, respectively. The obtained results were quite similar in their foundations to the literature, which documented the highest adsorption favorability through the N site of the CM drug over X
3O (X = Li, Na, and K)-doped B
12N
12 nanocarriers [
33]. It is worth mentioning that the presented result of the CM∙∙∙B
12N
12 complex within configuration A is compatible with a previous study [
27]. It was reported that the B
12N
12 nanocarrier showed preferable potency toward adsorbing the CM drug with
Eads up to −24.33 kcal/mol. The energy difference between the obtained and previously reported
Eads values could be mainly relevant to the variant in the utilized levels of computations.
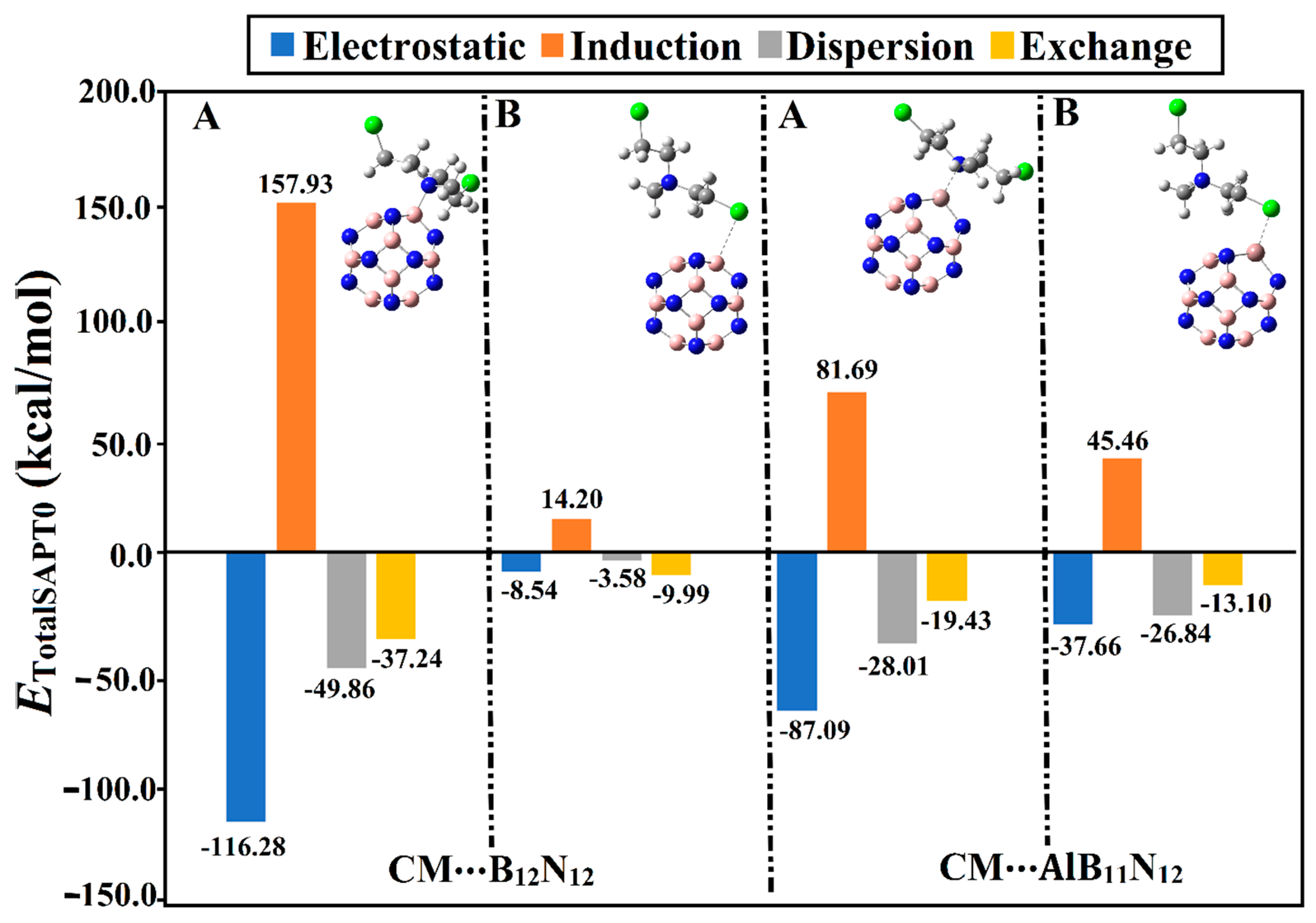
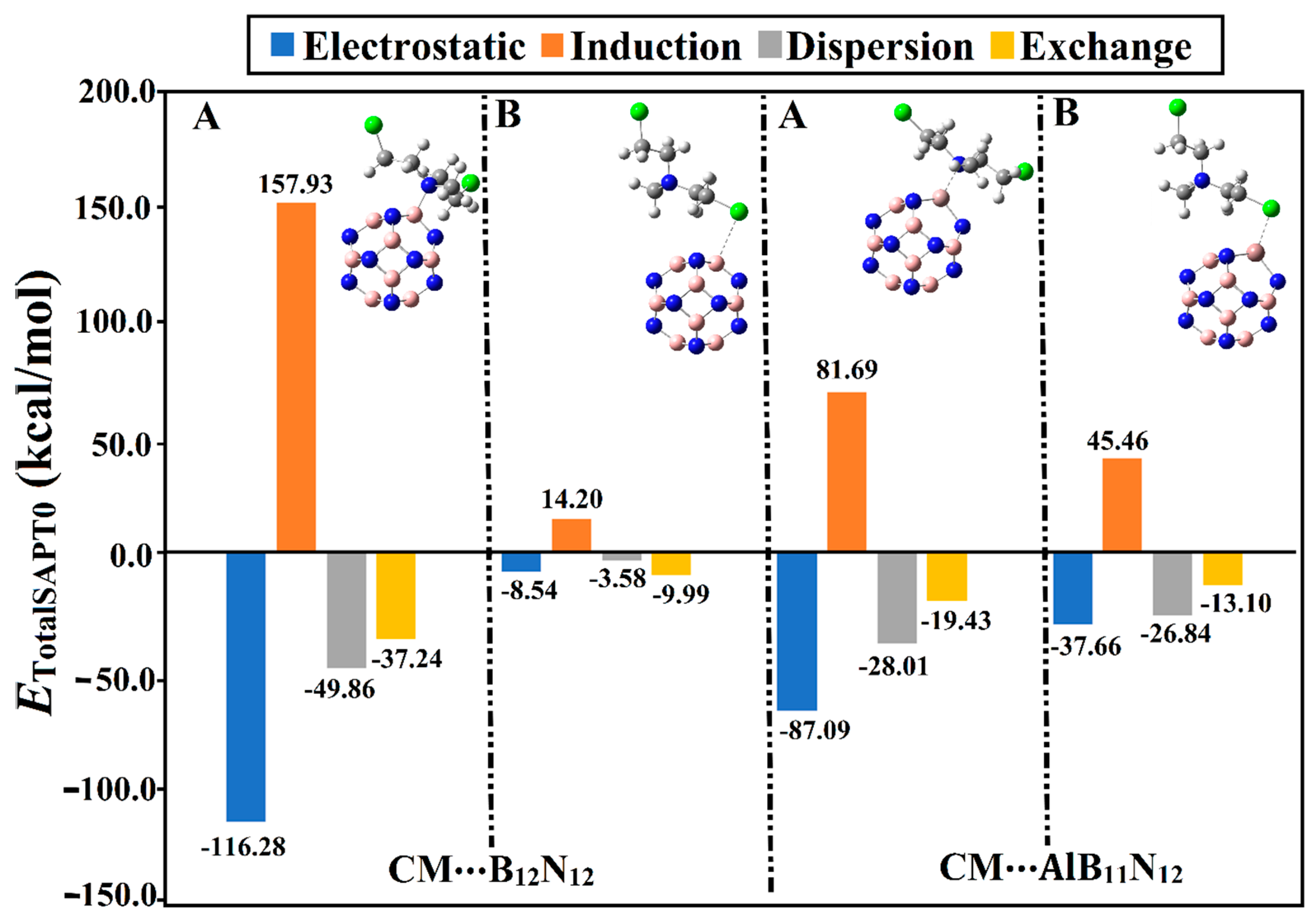
2.3. SAPT Calculations
Symmetry-adapted perturbation theory (SAPT) analysis was conducted to decompose the interaction energy into its four main physical forces, namely, electrostatic (
Eelst), exchange (
Eexch), dispersion (
Edisp), and induction (
Eind) forces. For the optimized complexes, SAPT analysis was performed at the SAPT0 level of theory applying PSI4 code [
34]. The four physical components of the interaction energy of Chlormethine with B
12N
12 and AlB
11N
12 nanocarriers are graphically correlated in
Figure 4.
It can be observed from
Figure 4 that the electrostatic forces (
Eelst) governed the adsorption process within the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes. Notably, the dispersion (
Edis) and induction (
Eind) forces exhibited preferable contributions, reinforcing the occurrence of the adsorption process. On the other hand, exchange forces (
Eexch) were found with positive values, demonstrating their unfavorable contribution to the forces beyond the adsorption process. Illustratively, the energetic values of
Eelst,
Eind,
Edisp, and
Eexch were found to be –87.09, –28.01, –19.43, and 81.69 kcal/mol, respectively, for the CM∙∙∙AlB
11N
12 complex within configuration A. Apparently, the obtained total SAPT0 energies were observed to be in line with the adsorption and interaction energy values (
Table 1). Numerically, the total SAPT0 energies of the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes within configurations A/B were –45.46/–7.91 and –59.74/–32.74 kcal/mol, respectively.
Although the Eelst and Eexch components of the CM∙∙∙B12N12 complex within configuration A showed higher values compared with that of the CM∙∙∙AlB11N12 complex, the total SAPT0 energy of the latter was higher than the former. Although the total SAPT0 energy of the CM∙∙∙AlB11N12 complex within configuration A was higher than that of the CM∙∙∙B12N12 analog, the Eelst and Eexch components of the latter complex showed higher values compared with those of the former.
Although the Eelst components of the CM∙∙∙B12N12 complex within configuration A showed a higher negative value compared with that of the CM∙∙∙AlB11N12 complex, the total SAPT0 energy of the latter complex was higher than the former. This observation could be interpreted by considering the Eexch component, which exhibited a higher positive value for the latter complex than the former. Illustratively, Eelst values were –116.28 and –87.09 kcal/mol for the CM∙∙∙B12N12 and ∙∙∙AlB11N12 complexes, respectively, within configuration A. In contrast, Eexch values for CM∙∙∙B12N12 and ∙∙∙AlB11N12 complexes within configuration A were found to be 157.93 and 81.69 kcal/mol, respectively.
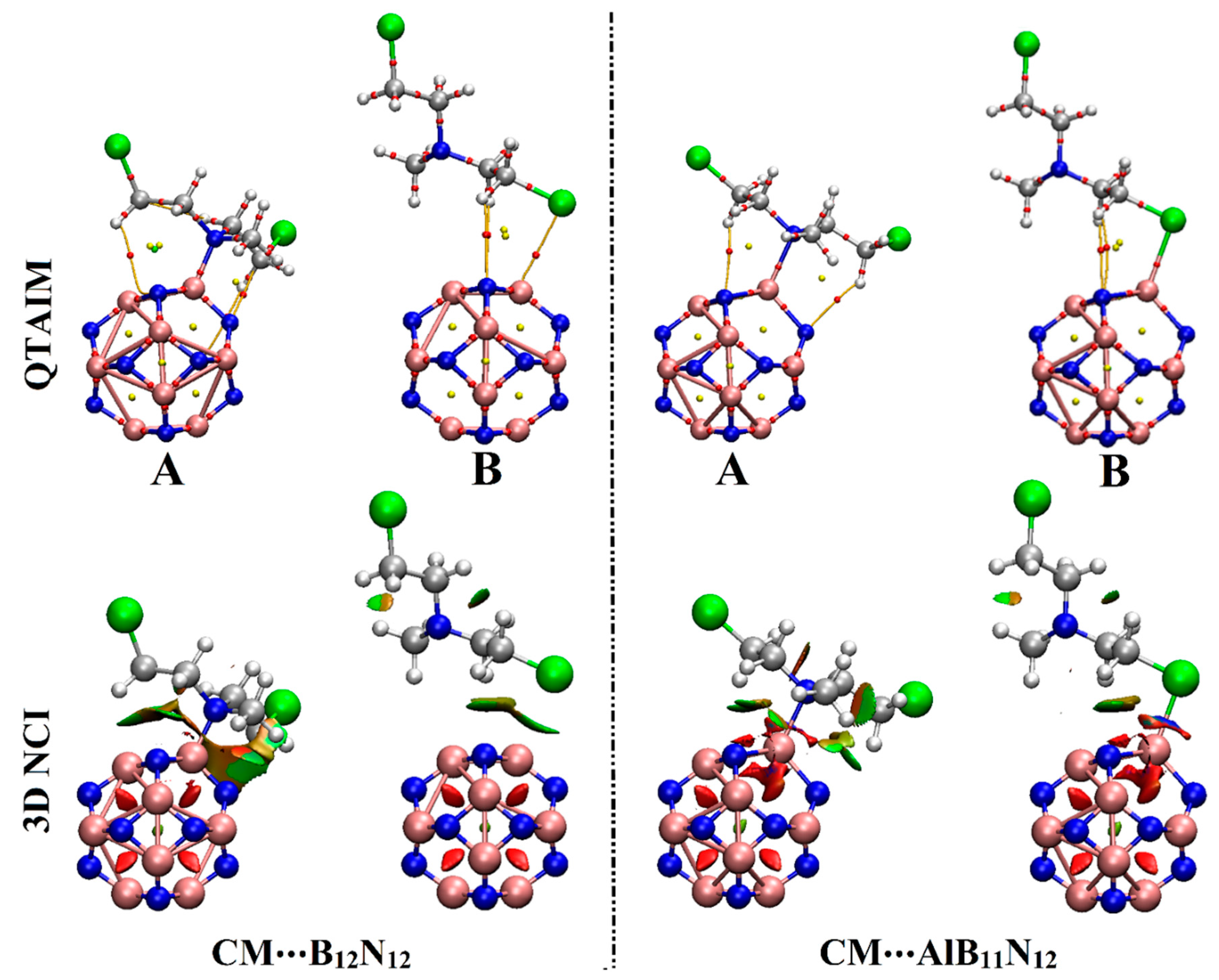
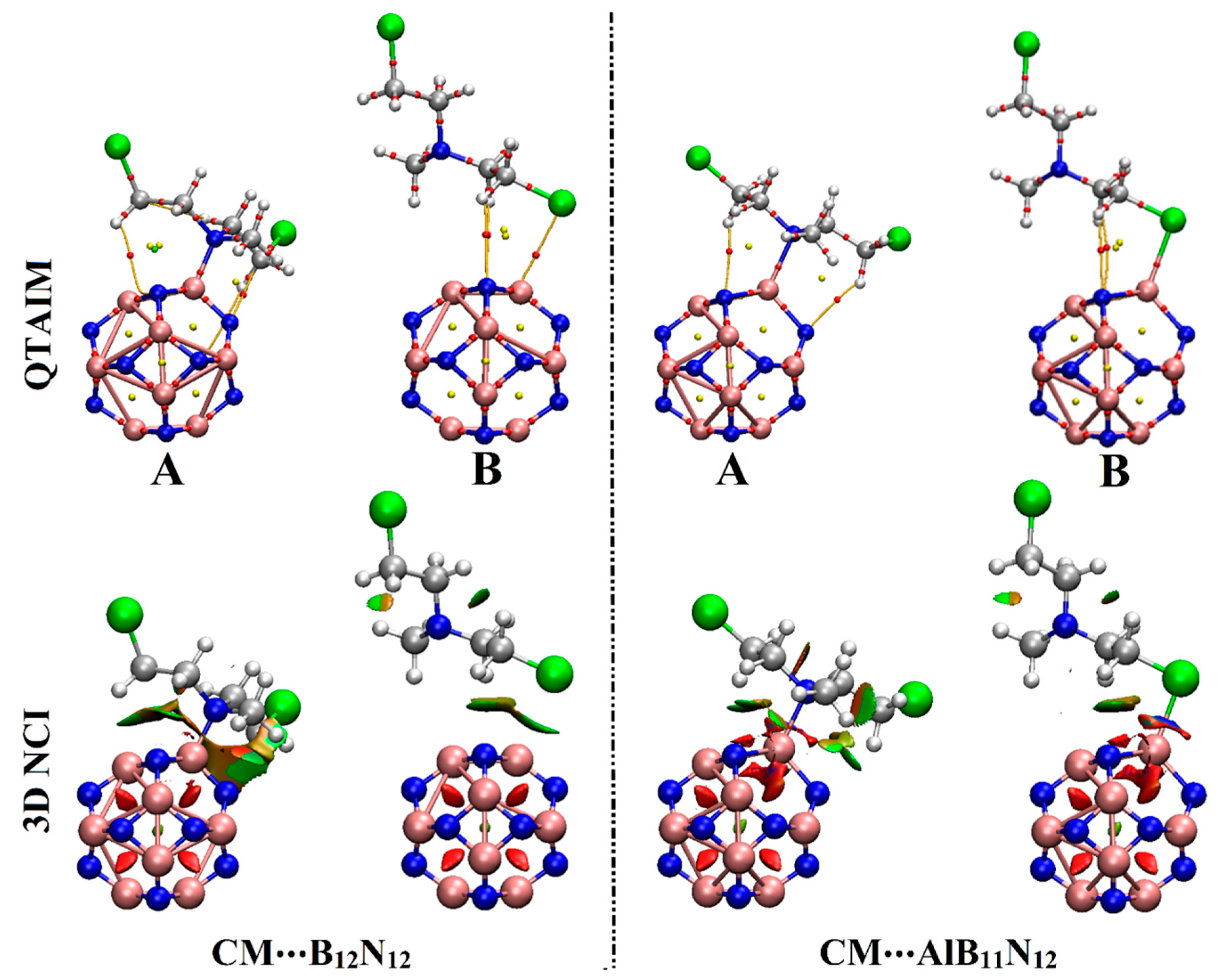
2.4. QTAIM and NCI Calculations
Quantum theory of atoms in molecules (QTAIM) and noncovalent interaction (NCI) index analyses were considered as informative investigations for visualizing inter- and intra-molecular interactions [
35]. Upon QTAIM and NCI, the adsorption of a drug over nanocarriers was endorsed by electron density and its derivatives. By applying QTAIM analysis, bond paths (BPs) and bond critical points (BCPs) were generated. The topological derivatives, inclusive total energy density (H
b), Laplacian (∇
2ρb), electron density (
ρb), kinetic electron density (G
b), local potential electron energy density (V
b), and negative ratio of kinetic and potential electron energy density (−G
b/V
b) were calculated and are gathered in
Table 2. Three-dimensional isosurface graphs were generated using sign (λ
2)
ρ with colors ranging from blue (–0.035 au) to red (0.020 au). QTAIM and 3D NCI diagrams of the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 optimized complexes are depicted in
Figure 5.
As explicitly shown in
Figure 5, the tendency of B
12N
12 and AlB
11N
12 nanocarriers to adsorb the CM drug was clearly confirmed by the visualized bond critical points (BCPs) and bond paths (BPs) between the interacted species (i.e., N/Cl of CM drug and B/Al of B
12N
12/AlB
11N
12 nanocarriers). Evidently, the contributions of secondary interactions that supported the adsorption process within the studied complexes were noticed via the existence of variant BCPs and BPs between the hydrogen atom of the CM drug and the N atom of B
12N
12/AlB
11N
12 nanocarriers (
Figure 5).
According to the data in
Table 2, the partial covalent and electrostatic nature of the adsorption process within the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes were generally unveiled. The obtained negative H
b values, positive ∇
2ρb values, and values less than the unity of –G
b/V
b outlined the partial covalent and electrostatic nature of the interactions within the CM∙∙∙B
12N
12 (configuration A) and ∙∙∙AlB
11N
12 (configurations A and B) complexes [
36]. For instance, H
b, ∇
2ρb, and –G
b/V
b of the CM∙∙∙AlB
11N
12 complex within configuration A were –0.0046, 0.3581, and 0.9530 au, respectively. The CM∙∙∙B
12N
12 within configuration B was noticed with a weak electrostatic nature that was detected by the positive H
b values, positive ∇
2ρb values, and values exceeding the unity for –G
b/V
b. Numerically, H
b, ∇
2ρb, and –G
b/V
b of the CM∙∙∙B
12N
12 complex within configuration B were 0.0007, 0.0387, and 1.0842 au, respectively.
On 3D NCI isosurfaces, the tendency of the B12N12 and AlB11N12 nanocarriers to adsorb the CM dug was elucidated by the occurrence of colored isosurfaces between the interacted species within the studied complexes. Moreover, the change in the color scale of the 3D NCI isosurfaces was found to be concurrent with the adsorption energy pattern; the strongest and weakest interactions were observed in blue and green colors, respectively.
2.5. Electronic Parameters
The impact of the adsorption process of Chlormethine (CM) over pure and Al-doped boron nitride (B
12N
12 and AlB
11N
12) nanocarriers was investigated by elucidating their densities and electronic energy levels. To this end, the highest occupied molecular orbital (
EHOMO), Fermi level (
EFL), and the lowest unoccupied molecular orbital (
ELUMO), were calculated before and after the adsorption process. Subsequently, the energy gap (
Egap) was computed as the difference between the
ELUMO and E
HOMO.
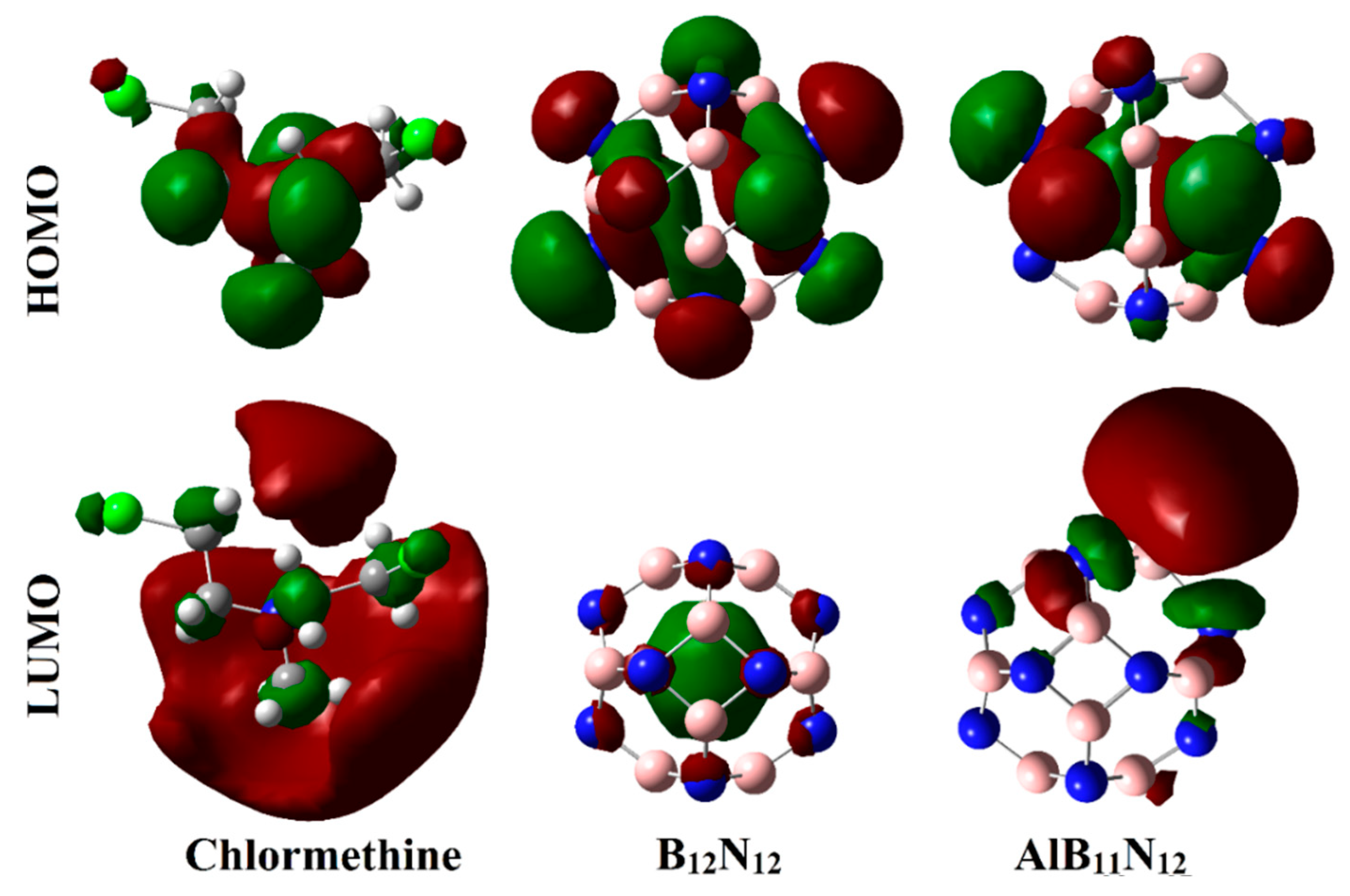
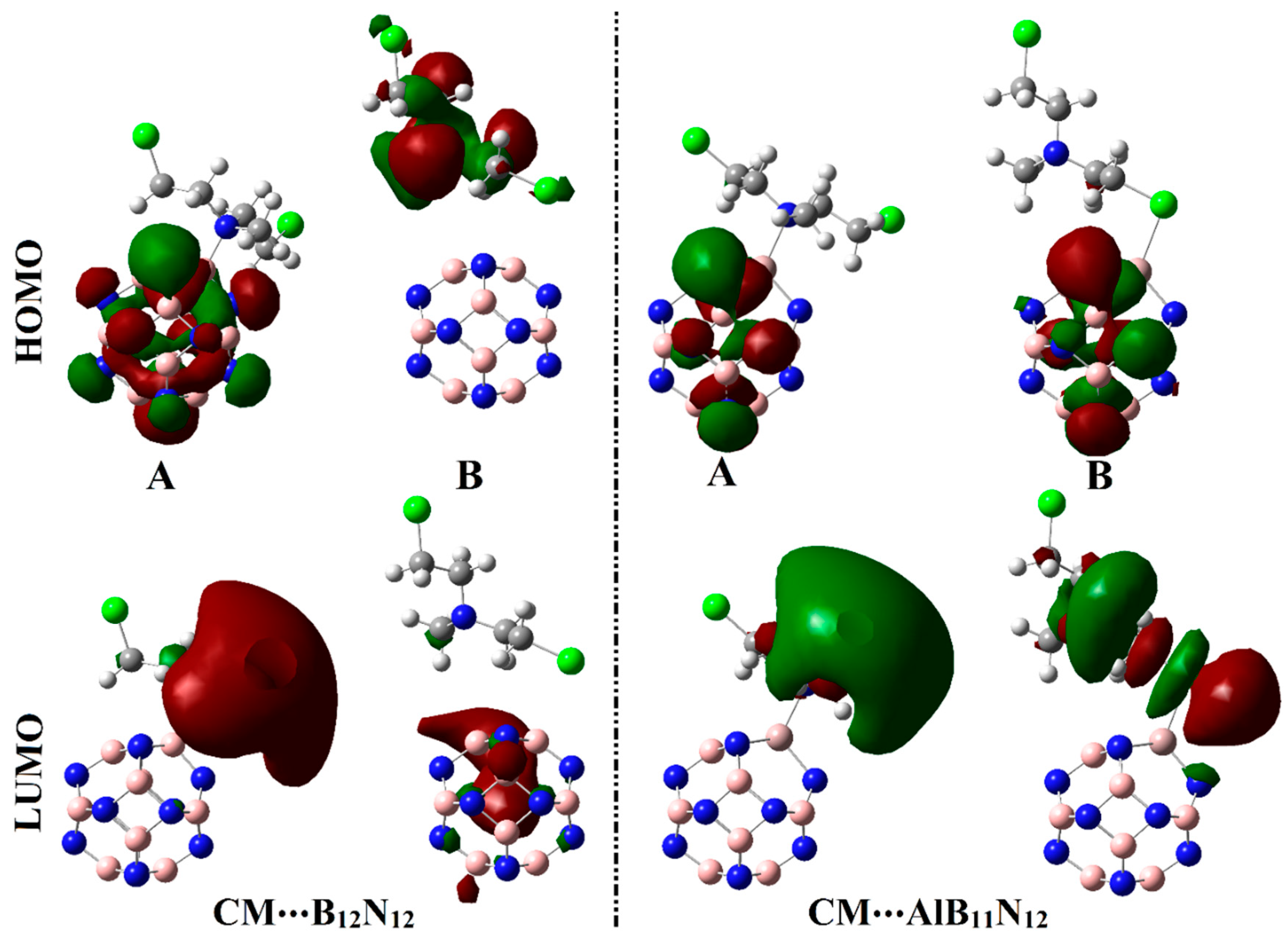
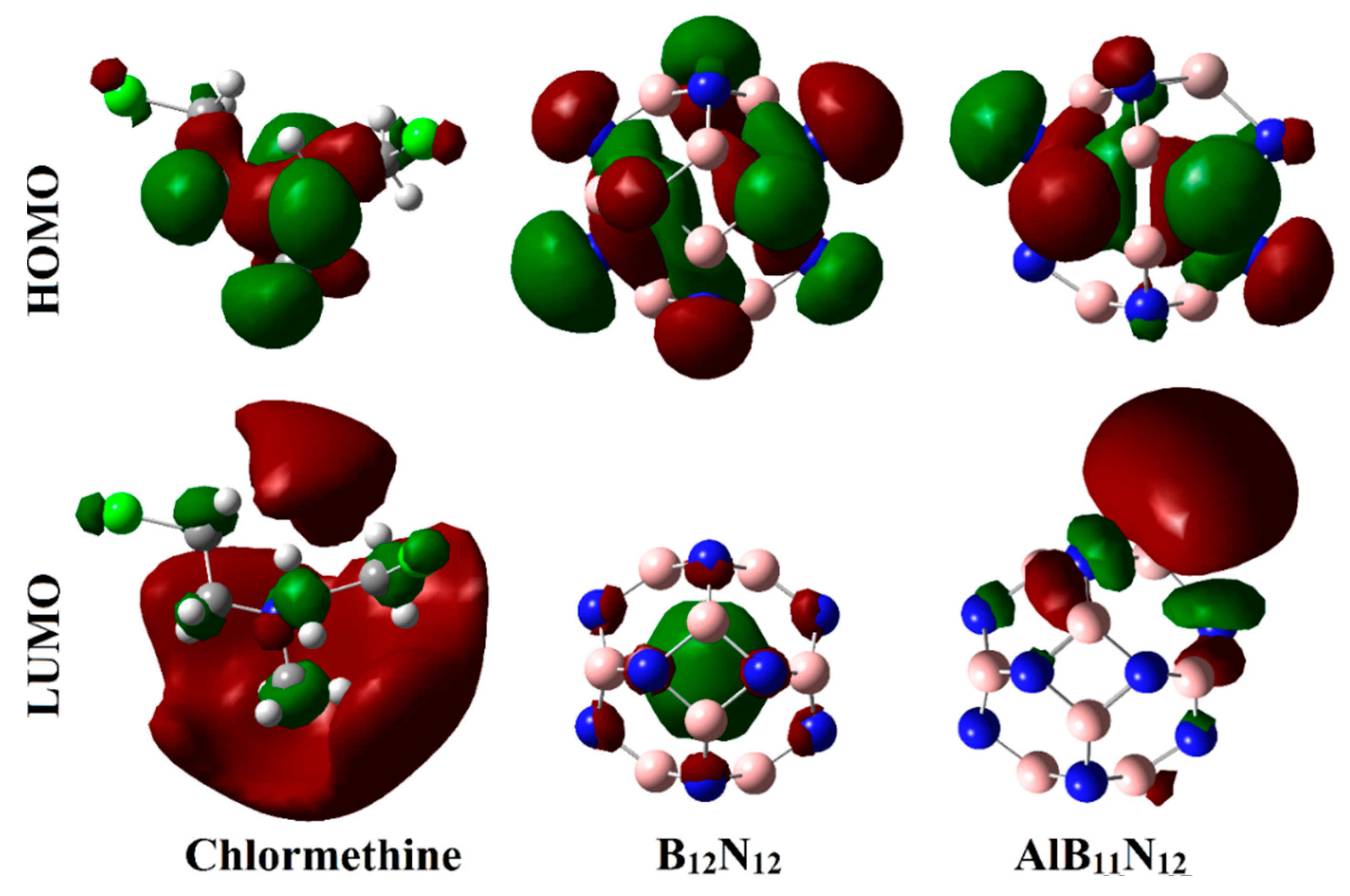
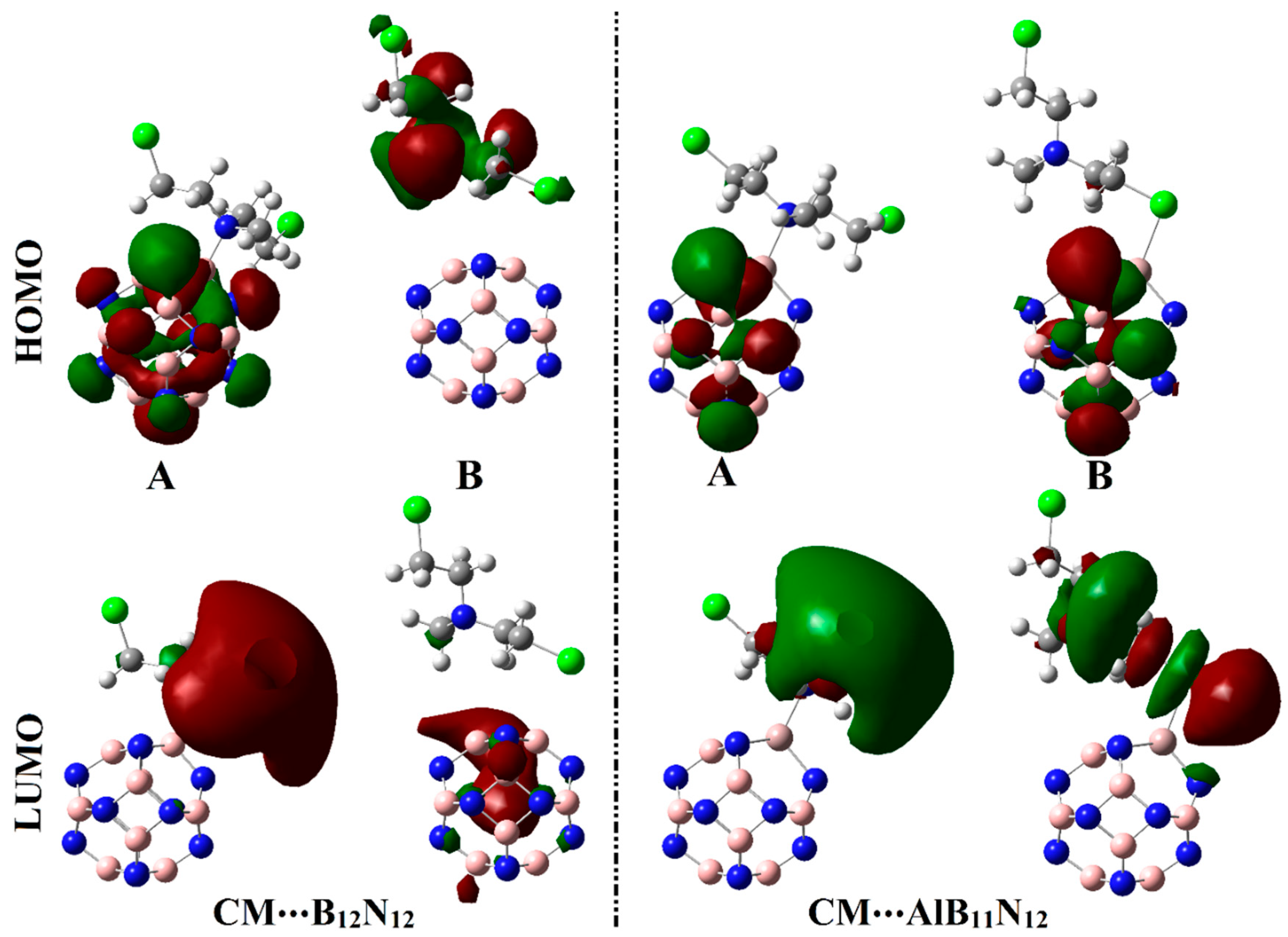
Figure 6 and
Figure 7 show the electron densities of HOMO/LUMO for the isolated systems and complexes, respectively. The obtained
EHOMO, E
FL,
ELUMO, and
Egap values are given in
Table 3.
As exemplified in
Figure 6, the distributions of HOMO and LUMO represented the densities purely around the nucleophilic and electrophilic sites, respectively. Apparently, HOMO distributions were concentrated around the N and Cl atoms of the CM, while the LUMO ones were denoted over B and Al atoms of B
12N
12 and AlB
11N
12, respectively. Upon contact with CM, obvious changes in HOMO and LUMO distributions were identified, as is apparent in
Figure 7. HOMO and LUMO levels were found over nanocarrier and drug, respectively, within the studied complexes, with an exception for the CM∙∙∙B
12N
12 complex within configuration B. This observation outlined that the occurrence of the adsorption process is attributed to the charge transfer between drug and nanocarriers in all the studied complexes. The HOMO and LUMO affirmations were consistent with the energetic ones summarized in
Table 1, which highlighted the lesser favorability of the CM∙∙∙B
12N
12 complex within configuration B compared with other studied complexes.
According to the data in
Table 3, the
EHOMO,
EFL,
ELUMO, and
Egap values of the pure B
12N
12 nanocarrier were evidently changed after doping by an Al atom in the AlB
11N
12 nanocarrier. From
Table 3, the
EHOMO,
EFL,
ELUMO, and
Egap values of B
12N
12/AlB
11N
12 nanocarriers were –9.54/–9.10, –5.12/–7.05, –0.35/–2.50, and 9.19/6.60 eV, respectively. These numerical values particularly addressed the contributions of Al doping in destabilizing and stabilizing
EHOMO and
ELUMO of the pure B
12N
12 nanocarrier.
It is also worth mentioning that an obvious variation in EHOMO and ELUMO values was unveiled after the adsorption process of the CM drug over the B12N12 and AlB11N12 nanocarriers. Notably, a slight decline in EHOMO and EFL values was found after the adsorption process. For instance, EHOMO of B12N12 nanocarrier was –9.54 eV, and it diminished to –8.92 and –8.30 eV for CM∙∙∙B12N12 complexes within configurations A and B, respectively. Generally, obvious downshifts and upshifts in the ELUMO/Egap values were observed after the adsorption process within CM∙∙∙B12N12 and ∙∙∙AlB11N12 complexes, respectively.
2.6. Global Indices of Reactivity
Global indices of reactivity were computed before and after the adsorption process to comprehensively clarify the impact of adsorption of the CM drug over B
12N
12 and AlB
11N
12 nanocarriers. Various parameters, including ionization potential (
IP), electron affinity (
EA), chemical potential (
μ), global hardness (
η), global softness (
S), electrophilicity index (
ω), and work function (
Φ), were calculated and are compiled in
Table 4.
With regard to global indices of reactivity, a significant difference was denoted in the values of
IP,
EA,
μ,
η,
S,
ω, and
Φ between the pure and Al-doped boron nitride (B
12N
12 and AlB
11N
12) nanocarriers. From summarized data in
Table 4, the
IP,
EA,
μ, η,
S,
ω, and
Φ values of B
12N
12/AlB
11N
12 were 9.54/9.10 eV, 0.35/2.50 eV, –4.94/–5.80 eV, 4.60/3.30 eV, 0.22/0.30 eV
–1, 2.66/5.10 eV, and 5.12/7.05 eV, respectively. Consequently, the Al doping enhanced the properties of the pure boron nitride nanocarrier to be a suitable surface for adsorbing the CM drug by declining and increasing the values of
IP/
η and
EA/
μ/
S/
ω/
Φ, respectively.
Moreover,
Table 4 offers insight into the substantial effect of the adsorption process of the CM drug over pure and Al-doped boron nitride (B
12N
12 and AlB
11N
12) nanocarriers via sizeable changes in the calculated reactivity parameters. As detailed in
Table 4,
IP values of the B
12N
12 and AlB
11N
12 nanocarriers decreased upon contact with the CM drug from 9.54 and 9.10 eV to 8.92 and 8.55 eV, respectively, within configuration A. The same pattern was noticed in the case of
μ, ω, and
Φ, and an irregular pattern was noticed for
EA,
η, and
S values, which could be ascribed to the main dependence of these values on
ELUMO. Overall, the noticeable difference in the global indices of reactivity parameters extensively ensured the adequacy of the B
12N
12 and AlB
11N
12 nanocarriers for adsorbing the CM drug, along with highlighting the preferential role of the Al doping process.
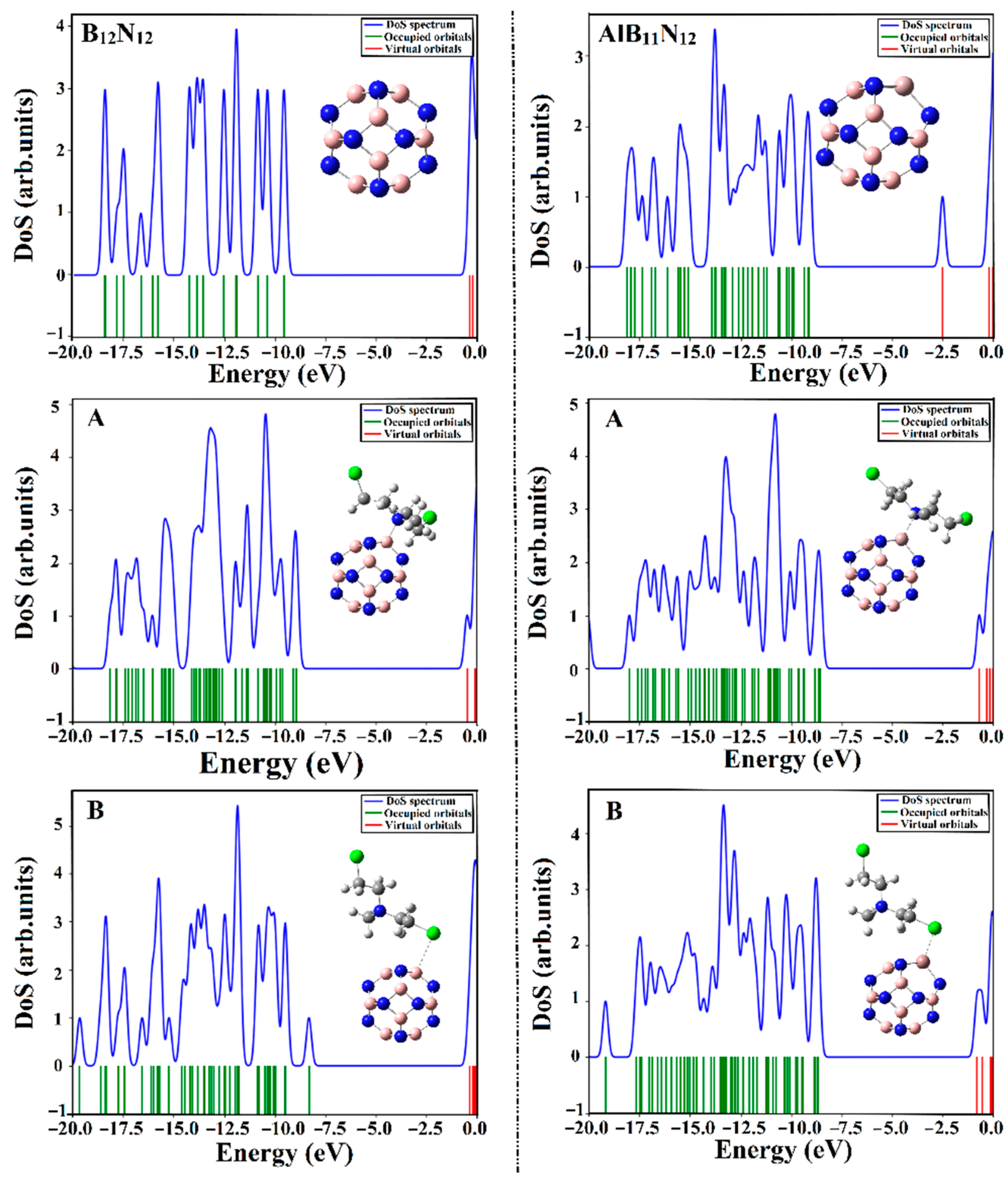
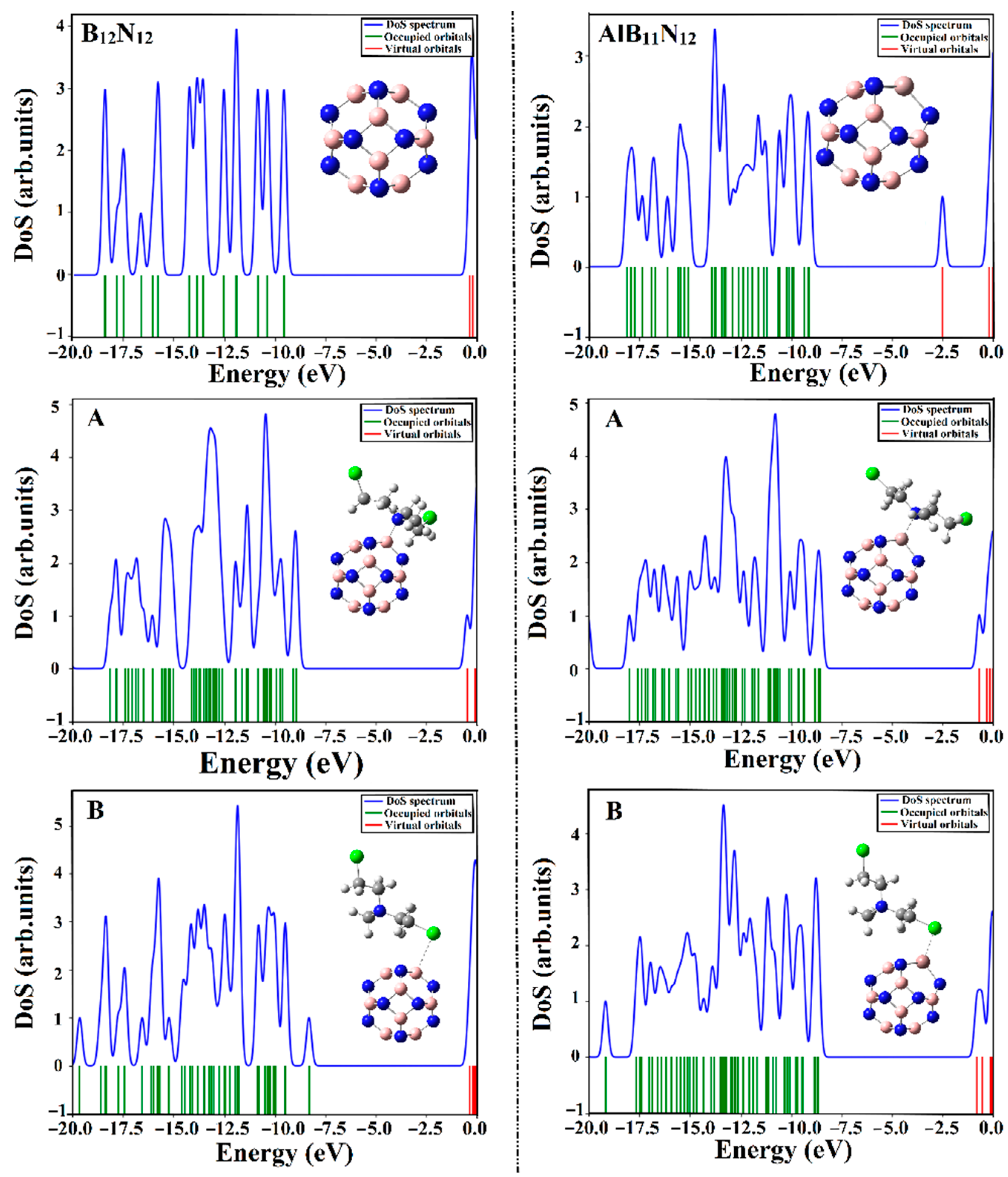
2.7. DOS Analysis
The density of states (DOS) analysis was apprehended for B
12N
12 and AlB
11N
12 nanocarriers before and after the adsorption process of the CM drug over B
12N
12 and AlB
11N
12 nanocarriers. DOS diagrams were generated and are expressed in
Figure 8.
As is evident in
Figure 8, the energy states within the DOS diagram of the AlB
11N
12 nanocarrier were found to be more concentrated than the B
12N
12 analog, revealing the favorable impact of the Al doping process on the electronic properties of the pure boron nitride (B
12N
12) nanocarrier.
It is apparent that the occurrence of the adsorption process within the CM∙∙∙B12N12 and ∙∙∙AlB11N12 complexes was thoroughly confirmed by the appearance of new energy states around the Fermi level in the extracted DOS diagrams. The resulted states were noticed more obviously in the case of CM∙∙∙AlB11N12 complexes compared with CM∙∙∙B12N12, in line with energetic patterns. Indeed, an obvious decrement in Egap was noticed following the adsorption of CM that in turn enhanced the conductivity (Equation (17)) of the investigated nanocarriers. Overall, the obtained results affirmed that the B12N12 and AlB11N12 nanocarriers are promising electrochemical biosensors for the Chlormethine (CM) anti-cancer drug.
2.8. Solvent Effect
Toward tackling the influence of the solvent on the adsorption process of the CM drug over B
12N
12 and AlB
11N
12 nanocarriers, the polarizable continuum model was utilized for water as a solvent. For the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes, the calculations of geometrical optimization were executed in the presence of water. Then, adsorption (
) and solvation (Δ
Esolv) energies were computed for the obtained optimized complexes and are given in
Table 5.
According to summarized data in
Table 5, negative values of
were noticed for all the considered complexes, clarifying the potential of the B
12N
12 and AlB
11N
12 nanocarriers to adsorb CM in the water phase. In line with the data in
Table 1, the energetics of the studied complexes in the water phase within configuration A were more preferable compared to B. For example,
values of the CM∙∙∙B
12N
12 complex within configurations A and B were –30.86 and –4.06 kcal/mol, respectively.
Furthermore, the CM∙∙∙AlB
11N
12 complexes were noticed with higher negative values of
compared with CM∙∙∙B
12N
12 analogs. This observation consistently ensured the favorable Al doping contributions to the adsorption process in the water phase, as
Table 5 shows. For instance, the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes within configuration A exhibited
with values of –30.86 and –51.30 kcal/mol, respectively. Further favorability of the occurrence of the adsorption process was affirmed in the water phase more than in the gas phase and confirmed by the negative values of the Δ
Esolv.
2.9. Thermodynamic Parameters
To deeply understand the nature of the adsorption process of the CM drug on the B
12N
12 and AlB
11N
12 nanocarriers, the thermodynamic parameters were computed. The thermodynamic parameters for CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 optimized complexes, at 1 atm and 298 K, Gibbs free energy (Δ
G), enthalpy change (Δ
H), and entropy change (Δ
S) were calculated and are given in
Table 6.
As shown in
Table 6, the spontaneity of the adsorption process within the CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes was confirmed via the negative values of Δ
G. Furthermore, negative Δ
H values were an indication of the exothermic nature of the adsorption process within all considered complexes. Small and neglectable values of Δ
S were noticed for all the studied complexes. Similar to the adsorption energy pattern, higher negative values of thermodynamic energetic quantities were mainly pertinent to the CM∙∙∙AlB
11N
12 complexes, compared with CM∙∙∙B
12N
12 complexes. From enrolled data in
Table 6, the Δ
G, Δ
H, and Δ
S of the CM∙∙∙AlB
11N
12 complexes within configuration A were –37.15, –50.14, and –0.04 kcal/mol, respectively.
2.10. Recovery Time
Recovery time (τ) is informative in the desorption process and is mainly correlated with the adsorption energy. For the CM∙∙∙B12N12 complexes within configurations A and B, τ values were 0.02 nS and 0.01 pS, respectively. In comparison, τ values of the CM∙∙∙AlB11N12 complexes within configurations A and B were 1.4 × 108 S and 0.11 μS, respectively. Apparently, the CM∙∙∙B12N12 complex within configuration B, the least favorable energetic complex, had the shortest recovery time value, indicating its preferential feasibility in the desorption process. Contrarily, the most favorable CM∙∙∙AlB11N12 complex within configuration A had the longest recovery time, which is consistent with its substantial negative adsorption energy. Based on these results, the B12N12 and AlB11N12 nanocarriers would be preferential for adsorbing the CM drug within the modeled configurations given their short recovery time.
3. Computational Methods
The adsorption process of the Chlormethine (CM) anti-cancer drug over the exterior surface of pure and aluminum (Al)-doped boron nitride nanocarriers (B
12N
12 and AlB
11N
12) was comparatively scrutinized. The CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 complexes were modeled within configurations A and B, in which the adsorption process occurred via N∙∙∙ and Cl∙∙∙B/Al interactions (see
Figure 1). To follow the drug loading process, CM, B
12N
12, and AlB
11N
12 structures were fully optimized at Minnesota 2006 with the double Hartree–Fock exchange function (M06-2X) [
37] along with the 6-311+G** basis set. Based on the optimized structures, electrostatic potential analysis (ESP) was conducted to clarify the electron-rich and -deficient sites over the molecular surface of the investigated systems. To acquire pictorial descriptions, molecular electrostatic potential (MEP) maps were created using electron density envelopes of 0.002 au [
30]. Furthermore, using ESP, the numerical description was obtained by executing and extracting surface electrostatic potential extrema (
Vs,min/
Vs,max) with the help of Multiwfn 3.7 software [
38].
Upon CM∙∙∙B
12N
12 and ∙∙∙AlB
11N
12 optimized complexes, adsorption (
Eads) and interaction (
Eint) energies were calculated, and the counterpoise correction method was considered to abolish the basis set superposition error (BSSE) as follows:
where
,
, and
represent energies of the complex, the isolated CM drug, and the isolated B
12N
12/AlB
11N
12 nanocarrier, respectively.
and
are the energies of the CM drug and the B
12N
12/AlB
11N
12 nanocarrier based on their respective coordinates in the optimized CM∙∙∙B
12N
12/∙∙∙AlB
11N
12 complexes.
To unveil the physical forces that controlled the adsorption process, symmetry-adapted perturbation theory (SAPT) analysis was executed with the help of the PSI4 package [
34]. Using SAPT analysis, total adsorption energies could be portioned into four physical components, namely, electrostatic (
Eelst), exchange (
Eexch), induction (
Eind), and dispersion (
Edisp) forces. Within SAPT analysis, the SAPT0 level of truncation was applied, and Total SAPT0 energies were calculated as described in the following equations [
39]:
where:
The calculations of the quantum theory of atoms in molecules (QTAIM) in addition to the noncovalent interaction (NCI) index were also performed. Using QTAIM analysis, bond critical points (BCPs) and bond paths (BPs) were induced. Topological features, including total energy density (H
b), Laplacian (∇
2ρb), electron density (
ρb), kinetic electron density (G
b), local potential electron energy density (V
b), and negative ratio of kinetic and potential electron energy density (−G
b/V
b), were also calculated. Three-dimensional NCI diagrams were generated and visualized using (λ
2)
ρ with values from –0.035 to 0.020 au and represented by colors ranging from blue to red, respectively. QTAIM and NCI analyses were achieved with the help of Multiwfn 3.7 [
38] and pictorially represented using the Visual Molecular Dynamics (VMD) program [
40].
To elucidate the electronic parameters, the frontier molecular orbitals (FMOs) theory was invoked. Within the context of FMOs, the electron density distribution of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were plotted before and after adsorption. In addition, energies of HOMO (
EHOMO) and LUMO (
ELUMO) were computed. Based on
EHOMO and
ELUMO values, the Fermi level (
EFL) and LUMO–HOMO energy gap (
Egap) were calculated by utilizing Equations (8) and (9), respectively.
Various electronic properties, including ionization potential (
IP), electron affinity (
EA), chemical potential (
μ), global hardness (
η), global softness (
S), electrophilicity index (
ω), and work function (
Φ), were computed for CM, B
12N
12, and AlB
11N
12 molecules before and after adsorption, as follows:
where V
eL(+∞) is the vacuum-level electrostatic potential that is assumed to be ≈ 0. In parallel, to provide further evidence of the attractive nature of pure and Al-doped (B
12N
12 and AlB
11N
12) nanocarriers toward CM adsorption, electrical conductivity was elucidated utilizing the following equation:
where
refers to electrical conductivity,
Egap is the energy gap, k is Boltzmann’s constant, and T characterizes temperature. The density of states (DOS) plots were generated using GaussSum software [
41]. To explicitly treat the effect of water as a solvent, the polarizable continuum model (PCM) method was applied [
42,
43]. For the optimized complexes, the solvation (
) energy was computed as the difference between the total energies in the water and gas phases according to Equation (18). The adsorption energies (
in the water phase were also assessed.
For a more quantitative view of favorability, the thermodynamic parameters including changes in Gibbs free energy (Δ
G), enthalpy (Δ
H), and entropy (Δ
S) were calculated for the studied complexes by applying frequency calculations [
44]. The change in thermodynamic parameters was presented as the following equation:
where Δ
M represents the quantity of Δ
G, Δ
H, and Δ
S. Furthermore,
, M
CM, and
introduce the
G/
H/
S parameters of the optimized complexes, the CM drug, and B
12N
12/AlB
11N
12 nanocarriers, respectively. In addition, an insightful perspective on the desorption process was gained through calculating recovery time for all considered complexes, as illustrated in the following equation:
where
addresses the attempt frequency of 10
–18 s
−1. K defines Boltzmann’s constant with a value of 0.00199 kcal/mol.K. T refers to temperature with a value of 310.15 K. All the employed DFT calculations were executed with the Gaussian 09 package [
45] using the M06-2X method with the 6-311+G** basis set.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}