
Molecular Mechanisms of Inflammasome in Ischemic Stroke Pathogenesis

 , ,
, , 
Abstract
1. Introduction
2. An Overview of the Pathogenesis of Cerebral Ischemia
3. NLRP3 Inflammasome
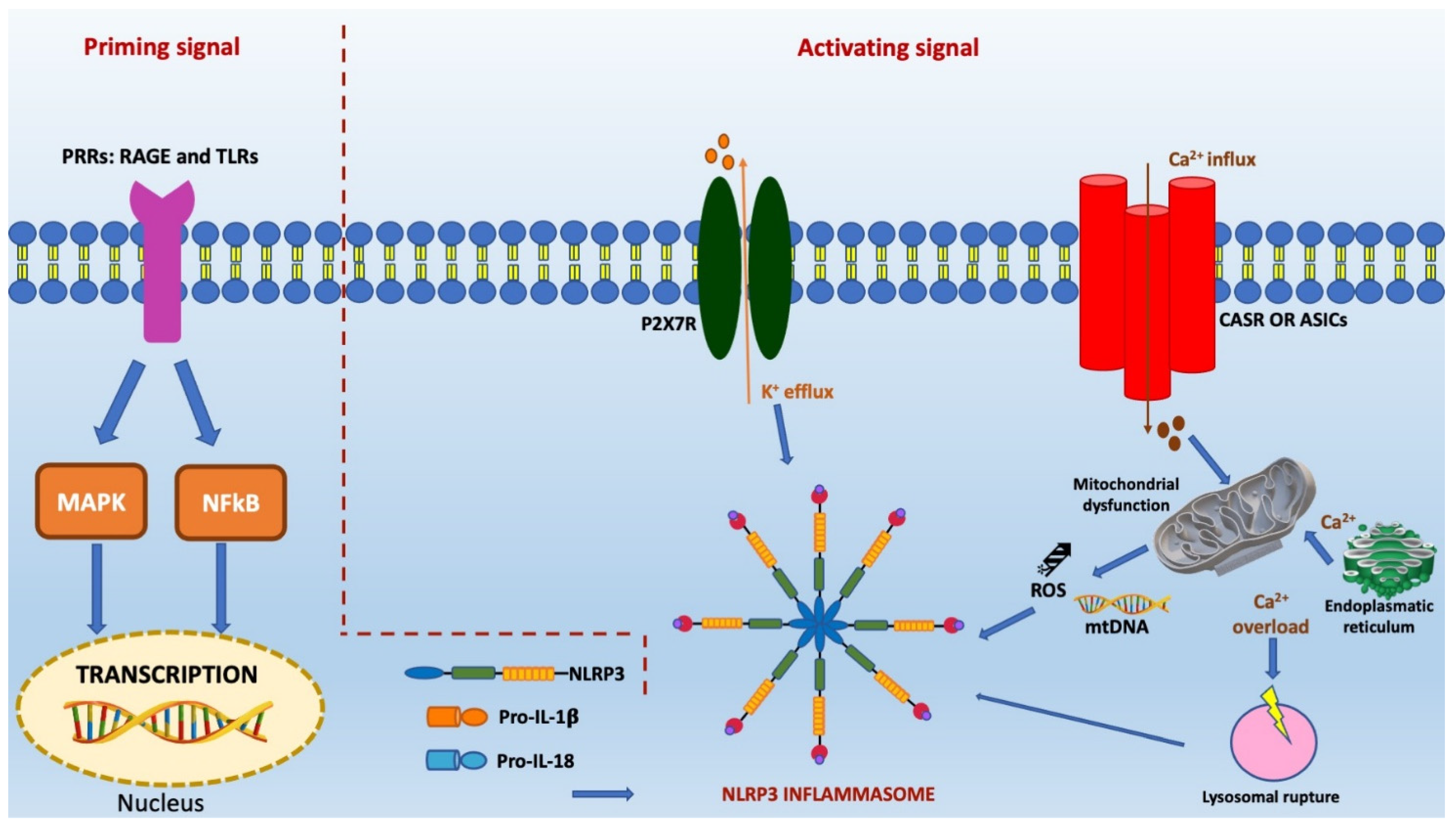
3.1. NLRP3 Inflammasome: Molecular Characteristics
3.2. Role of NLRP3 in Cerebral Ischemia
3.3. Expression of NLRP3 Inflammasome in Ischemic Stroke
3.4. Activation of NLRP3 Inflammasome Pathway in Ischemic Stroke
3.4.1. K+ Efflux and Ischemic Neuronal Damaged Mediated by Inflammation
3.4.2. Role of Mitochondrial Dysfunction and ROS in Inflammasome-Related Neuronal Damage
3.4.3. Ca2+ Mobilization in Inflammasome-Related Neuronal Damage
3.4.4. Lysosomal Detriment in Inflammasome-Related Neuronal Damage
3.4.5. Non-Canonical Inflammasome Pathway and Alternative Inflammasome Pathway
3.5. Direct and Indirect Inhibitors Targeting the NLRP3 Inflammasome Pathway for Ischemic Stroke Treatment
3.5.1. Small Molecules, Inflammasome Targeting, and Ischemic Neuroprotection
3.5.2. NRF2 as Possible Therapeutic Target in Ischemic Neuroprotection
3.5.3. Nitric Oxide (NO) as Possible Therapeutic Target in Ischemic Neuroprotection
3.5.4. IFN as Possible Therapeutic Target in Ischemic Neuroprotection
3.5.5. Micro-NAs Modulators of Inflammasome
3.5.6. Colchicine and Its Role against NLRP3 Inflammasome
3.5.7. Other Anti-Inflammasome Candidate Drugs
3.5.8. Future Perspectives
4. NLRP1 as a Target of a Possible Anti-Inflammasome Therapeutic Strategies
5. NLRP2 as a Target of a Possible Anti-Inflammasome Therapeutic Strategies
6. NLRC4 and Therapeutic Strategies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2APB | 2-aminoethoxy diphenylborinate |
| ADP | Adenosine diphosphate |
| ASC | Apoptosis-associated speck-like protein containing a caspase-recruitment domain |
| ASICs | Acid-sensing ion channels |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| ATP | Adenosine triphosphate |
| BBB | Blood brain barrier |
| BHB | Beta-hydroxybutyrate |
| BTK | Bruton’s tyrosine kinase |
| CARD | Caspase activation and recruitment domain |
| CASr | Calcium-sensing receptor |
| CIAS-1 | Cold-induced autoinflammatory syndrome 1 |
| CNS | Central nervous system |
| CPT1A | Carnitine palmitoyltransferase 1A |
| DAMPs | Damage-associated molecular patterns |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinases |
| FADD | Fas-associated protein with death domain |
| Fendrr | LncRNA FOXF1 adjacent non-coding developmental Regulatory RNA |
| GPRC6A | G protein-coupled receptor family C group 6 member A |
| GSDMD | Gasdermin D |
| HERC2 | HECT And RLD Domain Containing E3 Ubiquitin Protein Ligase 2 |
| IFN | Interferon |
| IFNAR | Interferon-α/β receptor |
| IL-18 | Interleukin-18 |
| IL-1β | Interleukin-1 β |
| IP3 | Inositol 1,4,5-triphosphate |
| IP3R | IP3 receptor |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinases |
| LPC | Lysophosphatidylcholine |
| LPS | Lipopolysaccharide |
| LRR | Leucine-rich repeat |
| MAM | Mitochondria-associated membrane |
| MAPK | Mitogen-activated protein kinase |
| MAVS | Mitochondrial antiviral-signaling protein |
| MCC950 | 1-(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)-3-[4-(2-hydroxypropan-2-yl)furan-2-yl]sulfonylurea |
| MCP-1/CCL2 | Monocyte Chemoattractant Protein-1 |
| MMPs | Matrix metalloproteinases |
| MSU | Monosodium urate |
| mtROS | Mitochondrial ROS |
| Myd88 | Myeloid differentiation primary response 88 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLR | NOD-like receptor |
| NLRC4 | NLR Family CARD-Domain-Containing 4 |
| NLRP1 | NLR family pyrin-domain-containing 1 |
| NLRP2 | NLR family pyrin-domain containing 2 |
| NLRP3 | NLR family pyrin-domain-containing 3 |
| NMDA | N methyl D aspartate |
| NO | Nitric oxide |
| NOD | Nucleotide-binding oligomerization domain |
| NOX2 | NADPH oxidase 2 |
| NOX4 | NADPH oxidase 4 |
| NRF2 | Nuclear factor erythroid-2 related factor 2 |
| NVU | Neurovascular unit |
| OGD | Oxygen-glucose deprivation |
| P2X7R | P2X purinoceptor 7 |
| PAMPs | Pathogen Associated Molecular Patterns |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PLA2 | Phospholipase A2 |
| PRRs | Pattern recognition receptors |
| PYD | Pyrin domain |
| PYHIN | Pyrin and HIN domain |
| RAGE | Receptor for advanced glycation end-products |
| ROS | Reactive oxygen species |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAK1 | Transforming growth factor beta-activated kinase 1 |
| tMCAO | Transient middle cerebral artery occlusion |
| TLRs | Toll-like receptors |
| TRAF3 | TNF-receptor-associated factor 3 |
| TRAF6 | TNF-receptor-associated factor 6 |
| TRPM | Transient receptor potential ion melastatin |
| TRPV | Transient receptor potential ion vanilloid |
| TXNIP | Thioredoxin-interacting protein |
| VDAC | Voltage-dependent anion channel |
References
- Jacob, M.A.; Ekker, M.S.; Allach, Y.; Cai, M.; Aarnio, K.; Arauz, A.; Arnold, M.; Bae, H.-J.; Bandeo, L.; Barboza, M.A.; et al. Global Differences in Risk Factors, Etiology, and Outcome of Ischemic Stroke in Young Adults—A Worldwide Meta-Analysis: The GOAL Initiative. Neurology 2022, 98, e573–e588. [Google Scholar] [CrossRef] [PubMed]
- Rabinstein, A.A. Update on Treatment of Acute Ischemic Stroke. Continuum 2020, 26, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral Vascular Disease and Neurovascular Injury in Ischemic Stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef] [PubMed]
- PubMed. Pericytes in Brain Injury and Repair after Ischemic Stroke. Available online: https://pubmed.ncbi.nlm.nih.gov/27837475/ (accessed on 15 March 2022).
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 Inflammasome: Its Regulation and Involvement in Atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The Role of Mitochondria in NLRP3 Inflammasome Activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Puleo, M.G.; Velardo, M.C.; Corpora, F.; Daidone, M.; Pinto, A. Molecular Biology of Atherosclerotic Ischemic Strokes. Int. J. Mol. Sci. 2020, 21, 9372. [Google Scholar] [CrossRef]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef]
- Won, W.; Choi, H.-J.; Yoo, J.-Y.; Kim, D.; Kim, T.Y.; Ju, Y.; Park, K.D.; Lee, H.; Jung, S.Y.; Lee, C.J. Inhibiting Peripheral and Central MAO-B Ameliorates Joint Inflammation and Cognitive Impairment in Rheumatoid Arthritis. Exp. Mol. Med. 2022, 54, 1188–1200. [Google Scholar] [CrossRef]
- Hu, H.-J.; Song, M. Disrupted Ionic Homeostasis in Ischemic Stroke and New Therapeutic Targets. J. Stroke Cerebrovasc. Dis. 2017, 26, 2706–2719. [Google Scholar] [CrossRef]
- Xu, L.; Emery, J.F.; Ouyang, Y.-B.; Voloboueva, L.A.; Giffard, R.G. Astrocyte Targeted Overexpression of Hsp72 or SOD2 Reduces Neuronal Vulnerability to Forebrain Ischemia. Glia 2010, 58, 1042–1049. [Google Scholar] [CrossRef]
- Bai, R.; Lang, Y.; Shao, J.; Deng, Y.; Refuhati, R.; Cui, L. The Role of NLRP3 Inflammasome in Cerebrovascular Diseases Pathology and Possible Therapeutic Targets. ASN Neuro 2021, 13, 17590914211018100. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-Y.; Gao, Z.-K.; Han, Y.; Yuan, M.; Guo, Y.-S.; Bi, X. Activation and Role of Astrocytes in Ischemic Stroke. Front. Cell. Neurosci. 2021, 15, 755955. [Google Scholar] [CrossRef] [PubMed]
- Bakaeva, Z.; Lizunova, N.; Tarzhanov, I.; Boyarkin, D.; Petrichuk, S.; Pinelis, V.; Fisenko, A.; Tuzikov, A.; Sharipov, R.; Surin, A. Lipopolysaccharide from E. coli Increases Glutamate-Induced Disturbances of Calcium Homeostasis, the Functional State of Mitochondria, and the Death of Cultured Cortical Neurons. Front. Mol. Neurosci. 2021, 14, 811171. [Google Scholar] [CrossRef] [PubMed]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Shen, S.; Zhang, S.; Huang, M.; Zhang, L.; Chen, X. Autophagy in Bone Remodeling: A Regulator of Oxidative Stress. Front. Endocrinol. 2022, 13, 898634. [Google Scholar] [CrossRef] [PubMed]
- Rehni, A.K.; Cho, S.; Dave, K.R. Ischemic Brain Injury in Diabetes and Endoplasmic Reticulum Stress. Neurochem. Int. 2022, 152, 105219. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Anderson, C.S. Precision Management of Brain Oedema after Acute Ischaemic Stroke. Precis. Clin. Med. 2022, 5, pbac019. [Google Scholar] [CrossRef]
- Kriska, J.; Hermanova, Z.; Knotek, T.; Tureckova, J.; Anderova, M. On the Common Journey of Neural Cells through Ischemic Brain Injury and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9689. [Google Scholar] [CrossRef]
- Yoshimura, S.; Sakai, N.; Uchida, K.; Yamagami, H.; Ezura, M.; Okada, Y.; Kitagawa, K.; Kimura, K.; Sasaki, M.; Tanahashi, N.; et al. Endovascular Therapy in Ischemic Stroke with Acute Large-Vessel Occlusion: Recovery by Endovascular Salvage for Cerebral Ultra-Acute Embolism Japan Registry 2. J. Am. Heart Assoc. 2018, 7, e008796. [Google Scholar] [CrossRef]
- Sušjan-Leite, P.; Ramuta, T.Ž.; Boršić, E.; Orehek, S.; Hafner-Bratkovič, I. Supramolecular Organizing Centers at the Interface of Inflammation and Neurodegeneration. Front. Immunol. 2022, 13, 940969. [Google Scholar] [CrossRef]
- Ji, X.; Tian, L.; Yao, S.; Han, F.; Niu, S.; Qu, C. A Systematic Review of Body Fluids Biomarkers Associated with Early Neurological Deterioration following Acute Ischemic Stroke. Front. Aging Neurosci. 2022, 14, 918473. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M. The Interplay of MicroRNAs in the Inflammatory Mechanisms Following Ischemic Stroke. J. Neuropathol. Exp. Neurol. 2017, 76, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P. Microglia as the Critical Regulators of Neuroprotection and Functional Recovery in Cerebral Ischemia. Cell. Mol. Neurobiol. 2021. [CrossRef] [PubMed]
- Alishahi, M.; Farzaneh, M.; Ghaedrahmati, F.; Nejabatdoust, A.; Sarkaki, A.; Khoshnam, S.E. NLRP3 Inflammasome in Ischemic Stroke: As Possible Therapeutic Target. Int. J. Stroke 2019, 14, 574–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Wu, W.; Liu, P.; Sun, S.; Hong, M.; Yuan, Y.; Xia, Q.; Chen, Z. Elevation of Neutrophil Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Associated with Multiple Inflammatory Mediators Was Related to Different Clinical Stages in Ischemic Stroke Patients. J. Clin. Lab. Anal. 2022, 36, e24526. [Google Scholar] [CrossRef]
- Cui, J.; Li, H.; Chen, Z.; Dong, T.; He, X.; Wei, Y.; Li, Z.; Duan, J.; Cao, T.; Chen, Q.; et al. Thrombo-Inflammation and Immunological Response in Ischemic Stroke: Focusing on Platelet-Tregs Interaction. Front. Cell. Neurosci. 2022, 16, 955385. [Google Scholar] [CrossRef]
- Geng, H.; Chen, L.; Tang, J.; Chen, Y.; Wang, L. The Role of CCL2/CCR2 Axis in Cerebral Ischemia-Reperfusion Injury and Treatment: From Animal Experiments to Clinical Trials. Int. J. Mol. Sci. 2022, 23, 3485. [Google Scholar] [CrossRef]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory Mechanisms of Blood-Brain Barrier Damage in Ischemic Stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- He, T.; Yang, G.-Y.; Zhang, Z. Crosstalk of Astrocytes and Other Cells during Ischemic Stroke. Life 2022, 12, 910. [Google Scholar] [CrossRef]
- Tian, X.; Liu, C.; Shu, Z.; Chen, G. Review: Therapeutic Targeting of HMGB1 in Stroke. Curr. Drug Deliv. 2017, 14, 785–790. [Google Scholar] [CrossRef]
- Richard, S.A.; Sackey, M.; Su, Z.; Xu, H. Pivotal Neuroinflammatory and Therapeutic Role of High Mobility Group Box 1 in Ischemic Stroke. Biosci. Rep. 2017, 37, BSR20171104. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qian, C.; Cao, X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.-W.; Lim, Y.-A.; Cheng, Y.-L.; Lok, K.-Z.; Chunduri, P.; Baik, S.-H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.-G.; et al. Evidence That NF-ΚB and MAPK Signaling Promotes NLRP Inflammasome Activation in Neurons following Ischemic Stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Liu, S.; Zhang, S.; Xu, T.; Yu, X.; Gao, Y.; Zhai, C.; Li, C.; Lei, C.; Fan, S.; et al. Evidence and Perspective for the Role of the NLRP3 Inflammasome Signaling Pathway in Ischemic Stroke and Its Therapeutic Potential (Review). Int. J. Mol. Med. 2018, 42, 2979–2990. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; de Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 Closes the Active Conformation of NLRP3 to an Inactive State. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef]
- Hafner-Bratkovič, I.; Sušjan, P.; Lainšček, D.; Tapia-Abellán, A.; Cerović, K.; Kadunc, L.; Angosto-Bazarra, D.; Pelegrin, P.; Jerala, R. NLRP3 Lacking the Leucine-Rich Repeat Domain Can Be Fully Activated via the Canonical Inflammasome Pathway. Nat. Commun. 2018, 9, 5182. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Kanneganti, T.-D. Inflammatory Bowel Disease and the NLRP3 Inflammasome. N. Engl. J. Med. 2017, 377, 694–696. [Google Scholar] [CrossRef]
- Toldo, S.; Abbate, A. The NLRP3 Inflammasome in Acute Myocardial Infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.-W.; Lee, S.-Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of Acute Stroke and the Role of Inflammasomes. Ageing Res. Rev. 2013, 12, 941–966. [Google Scholar] [CrossRef]
- Li, X.; Zhang, P.; Yin, Z.; Xu, F.; Yang, Z.-H.; Jin, J.; Qu, J.; Liu, Z.; Qi, H.; Yao, C.; et al. Caspase-1 and Gasdermin D Afford the Optimal Targets with Distinct Switching Strategies in NLRP1b Inflammasome-Induced Cell Death. Research 2022, 2022, 9838341. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Antonioli, L.; Lopez-Castejon, G.; Blandizzi, C.; Fornai, M. Canonical and Non-Canonical Activation of NLRP3 Inflammasome at the Crossroad between Immune Tolerance and Intestinal Inflammation. Front. Immunol. 2017, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef]
- Koizumi, J.; Yoshida, Y.; Nakazawa, T.; Ooneda, G. Experimental Studies of Ischemic Brain Edema. Nosotchu 1986, 8, 1–8. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, J.; Bi, F.; Li, H.; Chang, C.; Ji, C.; Liu, W. EK7 Regulates NLRP3 Inflammasome Activation and Neuroinflammation Post-Traumatic Brain Injury. Front. Mol. Neurosci. 2019, 12, 202. [Google Scholar] [CrossRef]
- Jagadapillai, R.; Qiu, X.; Ojha, K.; Li, Z.; El-Baz, A.; Zou, S.; Gozal, E.; Barnes, G.N. Potential Cross Talk between Autism Risk Genes and Neurovascular Molecules: A Pilot Study on Impact of Blood Brain Barrier Integrity. Cells 2022, 11, 2211. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Jalouli, M.; Rahman, M.A.; Jeandet, P.; Behl, T.; Alexiou, A.; Albadrani, G.M.; Abdel-Daim, M.M.; Perveen, A.; et al. Neuroinflammatory Signaling in the Pathogenesis of Alzheimer’s Disease. Curr. Neuropharmacol. 2022, 20, 126–146. [Google Scholar] [CrossRef]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925. [Google Scholar] [CrossRef]
- Gao, L.; Dong, Q.; Song, Z.; Shen, F.; Shi, J.; Li, Y. NLRP3 Inflammasome: A Promising Target in Ischemic Stroke. Inflamm. Res. 2017, 66, 17–24. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chi, L.; He, Z.; Gao, Y.; Gao, Y.; Huang, Y.; Nan, G. NLRP3 Inflammasome Contributes to Neurovascular Unit Damage in Stroke. J. Drug Target. 2019, 27, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 Deficiency Ameliorates Neurovascular Damage in Experimental Ischemic Stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.-W.; Lee, S.Y.; Manzanero, S.; Tang, S.C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.L.; Thundyil, J.; et al. Intravenous Immunoglobulin Suppresses NLRP1 and NLRP3 Inflammasome-Mediated Neuronal Death in Ischemic Stroke. Cell Death Dis. 2013, 4, e790. [Google Scholar] [CrossRef]
- Nagyőszi, P.; Nyúl-Tóth, Á.; Fazakas, C.; Wilhelm, I.; Kozma, M.; Molnár, J.; Haskó, J.; Krizbai, I.A. Regulation of NOD-like Receptors and Inflammasome Activation in Cerebral Endothelial Cells. J. Neurochem. 2015, 135, 551–564. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like Receptors in the Pathogenesis of Neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Chen, L.-L.; Morcelle, C.; Cheng, Z.-L.; Chen, X.; Xu, Y.; Gao, Y.; Song, J.; Li, Z.; Smith, M.D.; Shi, M.; et al. Itaconate Inhibits TET DNA Dioxygenases to Dampen Inflammatory Responses. Nat. Cell Biol. 2022, 24, 353–363. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Murthy, P.; Durco, F.; Miller-Ocuin, J.L.; Takedai, T.; Shankar, S.; Liang, X.; Liu, X.; Cui, X.; Sachdev, U.; Rath, D.; et al. The NLRP3 Inflammasome and Bruton’s Tyrosine Kinase in Platelets Co-Regulate Platelet Activation, Aggregation, and In Vitro Thrombus Formation. Biochem. Biophys. Res. Commun. 2017, 483, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, P. Activation and Pharmacological Regulation of Inflammasomes. Biomolecules 2022, 12, 1005. [Google Scholar] [CrossRef] [PubMed]
- Buendia, I.; Tenti, G.; Michalska, P.; Méndez-López, I.; Luengo, E.; Satriani, M.; Padín-Nogueira, F.; López, M.G.; Ramos, M.T.; García, A.G.; et al. ITH14001, a CGP37157-Nimodipine Hybrid Designed to Regulate Calcium Homeostasis and Oxidative Stress, Exerts Neuroprotection in Cerebral Ischemia. ACS Chem. Neurosci. 2017, 8, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, J.; Xu, Y.; Cao, W.; Wang, J.; Wang, B.; Zhao, L.; Zhang, X.; Liao, W. Enriched Environment Attenuates Pyroptosis to Improve Functional Recovery after Cerebral Ischemia/Reperfusion Injury. Front. Aging Neurosci. 2021, 13, 717644. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 Receptor: A Main Player in Inflammation. Biochem. Pharmacol. 2018, 151, 234–244. [Google Scholar] [CrossRef]
- Zhao, N.; Li, C.; Di, B.; Xu, L. Recent Advances in the NEK7-Licensed NLRP3 Inflammasome Activation: Mechanisms, Role in Diseases and Related Inhibitors. J. Autoimmun. 2020, 113, 102515. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Kozlov, A.V.; Lancaster, J.R.; Meszaros, A.T.; Weidinger, A. Mitochondria-Meditated Pathways of Organ Failure upon Inflammation. Redox Biol. 2017, 13, 170–181. [Google Scholar] [CrossRef]
- Kulakauskienė, D.; Narauskaitė, D.; Gečys, D.; Juknaitė, O.; Jankauskaitė, L.; Masaitytė, A.; Šventoraitienė, J.; Inokaitis, H.; Miknienė, Z.; Sadauskienė, I.; et al. Virus Mimetic Poly (I:C)-Primed Airway Exosome-like Particles Enter Brain and Induce Inflammatory Cytokines and Mitochondrial Reactive Oxygen Species in Microglia. Biology 2021, 10, 1359. [Google Scholar] [CrossRef]
- Han, Y.; He, X.; Wei, C.; Song, T.; Zou, L.; Li, Z.; Ye, J.; Qi, L.; Li, L.; Zhong, H.; et al. Negative Regulation of MAVS-Mediated Innate Immune Response by ASC. Mol. Cell. Biochem. 2018, 445, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhou, Z.; Liu, W.; Chang, Q.; Sun, G.; Dai, Y. Vascular Endothelial Cells Senescence Is Associated with NOD-like Receptor Family Pyrin Domain-Containing 3 (NLRP3) Inflammasome Activation via Reactive Oxygen Species (ROS)/Thioredoxin-Interacting Protein (TXNIP) Pathway. Int. J. Biochem. Cell Biol. 2017, 84, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Shao, G.; Chen, J.; Gong, J. The NLRP3 Inflammasome: An Important Driver of Neuroinflammation in Hemorrhagic Stroke. Cell. Mol. Neurobiol. 2017, 38, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, S.; Yang, G.; Wang, N. Spotlight on NLRP3 Inflammasome: Role in Pathogenesis and Therapies of Atherosclerosis. J. Inflamm. Res. 2021, 14, 7143–7172. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Dhandapani, K.M.; Brann, D.W. NADPH Oxidase 2 Regulates NLRP3 Inflammasome Activation in the Brain after Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2017, 2017, 6057609. [Google Scholar] [CrossRef]
- Moon, J.-S.; Nakahira, K.; Chung, K.-P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.-H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-Dependent Fatty Acid Oxidation Promotes NLRP3 Inflammasome Activation in Macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef]
- Yu, S.; Fu, J.; Wang, J.; Zhao, Y.; Liu, B.; Wei, J.; Yan, X.; Su, J. The Influence of Mitochondrial-DNA-Driven Inflammation Pathways on Macrophage Polarization: A New Perspective for Targeted Immunometabolic Therapy in Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2021, 23, 135. [Google Scholar] [CrossRef]
- Molagoda, I.M.N.; Lee, K.T.; Choi, Y.H.; Jayasingha, J.A.C.C.; Kim, G.-Y. Anthocyanins from Hibiscus syriacus L. Inhibit NLRP3 Inflammasome in BV2 Microglia Cells by Alleviating NF-ΚB- and ER Stress-Induced Ca2+ Accumulation and Mitochondrial ROS Production. Oxid. Med. Cell. Longev. 2021, 2021, 1246491. [Google Scholar] [CrossRef]
- Humeau, J.; Bravo-San Pedro, J.M.; Vitale, I.; Nuñez, L.; Villalobos, C.; Kroemer, G.; Senovilla, L. Calcium Signaling and Cell Cycle: Progression or Death. Cell Calcium 2018, 70, 3–15. [Google Scholar] [CrossRef]
- D’Espessailles, A.; Mora, Y.A.; Fuentes, C.; Cifuentes, M. Calcium-Sensing Receptor Activates the NLRP3 Inflammasome in LS14 Preadipocytes Mediated by ERK1/2 Signaling. J. Cell. Physiol. 2018, 233, 6232–6240. [Google Scholar] [CrossRef]
- Rodríguez, L.R.; Lapeña-Luzón, T.; Benetó, N.; Beltran-Beltran, V.; Pallardó, F.V.; Gonzalez-Cabo, P.; Navarro, J.A. Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases? Antioxidants 2022, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tong, Z.; Jiang, S.; Zheng, W.; Zhao, J.; Zhou, X. The Roles of Endoplasmic Reticulum in NLRP3 Inflammasome Activation. Cells 2020, 9, 1219. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Yang, S.; Hu, X. Activation of Canonical Inflammasome Complex by Acute Silica Exposure in Experimental Rat Model. Toxicol. Res. 2022, 11, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 Inflammasome Activation and Cell Death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Karki, R.; Berwin, B.; Yamamoto, M.; Kanneganti, T.-D. Guanylate Binding Proteins Facilitate Caspase-11-Dependent Pyroptosis in Response to Type 3 Secretion System-Negative Pseudomonas Aeruginosa. Cell Death Discov. 2018, 4, 3. [Google Scholar] [CrossRef]
- Zamyatina, A.; Heine, H. Lipopolysaccharide Recognition in the Crossroads of TLR4 and Caspase-4/11 Mediated Inflammatory Pathways. Front. Immunol. 2020, 11, 585146. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglié, D.; Fossati, G.; Rubartelli, A. ATP Is Released by Monocytes Stimulated with Pathogen-Sensing Receptor Ligands and Induces IL-1beta and IL-18 Secretion in an Autocrine Way. Proc. Natl. Acad. Sci. USA 2008, 105, 8067–8072. [Google Scholar] [CrossRef]
- Mauro, A.G.; Bonaventura, A.; Abbate, A. Drugs to Inhibit the NLRP3 Inflammasome: Not Always on Target. J. Cardiovasc. Pharmacol. 2019, 74, 225–227. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, B.; Ye, Y.; Li, Y.; Zhang, Y.; Xiong, X.; Gu, L. Relevant Mediators Involved in and Therapies Targeting the Inflammatory Response Induced by Activation of the NLRP3 Inflammasome in Ischemic Stroke. J. Neuroinflamm. 2021, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Deng, X.; Huang, W.; Yu, J.-H.; Wang, J.-X.; Wang, J.-P.; Yang, S.-B.; Liu, X.; Wang, L.; Zhang, Y.; et al. Irisin Protects against Neuronal Injury Induced by Oxygen-Glucose Deprivation in Part Depends on the Inhibition of ROS-NLRP3 Inflammatory Signaling Pathway. Mol. Immunol. 2017, 91, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-Inflammasome as a Potential Approach for Neuroprotection after Stroke. Sci. Rep. 2018, 8, 5971. [Google Scholar] [CrossRef]
- Ye, X.; Shen, T.; Hu, J.; Zhang, L.; Zhang, Y.; Bao, L.; Cui, C.; Jin, G.; Zan, K.; Zhang, Z.; et al. Purinergic 2X7 Receptor/NLRP3 Pathway Triggers Neuronal Apoptosis after Ischemic Stroke in the Mouse. Exp. Neurol. 2017, 292, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone Bodies Mimic the Life Span Extending Properties of Caloric Restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a Selective and Direct NLRP3 Inhibitor to Treat Inflammatory Disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced MtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 Inhibits NLRP3 Inflammasome Activation through Regulating Trx1/TXNIP Complex in Cerebral Ischemia Reperfusion Injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef]
- Poh, X.Y.; Loh, F.K.; Friedland, J.S.; Ong, C.W.M. Neutrophil-Mediated Immunopathology and Matrix Metalloproteinases in Central Nervous System—Tuberculosis. Front. Immunol. 2021, 12, 788976. [Google Scholar] [CrossRef]
- Chen, Z.-Q.; Mou, R.-T.; Feng, D.-X.; Wang, Z.; Chen, G. The Role of Nitric Oxide in Stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P. Multiple Sclerosis: Focus on Extracellular and Artificial Vesicles, Nanoparticles as Potential Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 8866. [Google Scholar] [CrossRef] [PubMed]
- Moasses Ghafary, S.; Soriano-Teruel, P.M.; Lotfollahzadeh, S.; Sancho, M.; Serrano-Candelas, E.; Karami, F.; Barigye, S.J.; Fernández-Pérez, I.; Gozalbes, R.; Nikkhah, M.; et al. Identification of NLRP3PYD Homo-Oligomerization Inhibitors with Anti-Inflammatory Activity. Int. J. Mol. Sci. 2022, 23, 1651. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wang, Y.; Ding, X.; He, Y.; Lu, Z.; Wu, P.; Tian, L.; Yuan, H.; Liu, D.; Shi, G.; et al. Long Non-Coding RNA XLOC_000647 Suppresses Progression of Pancreatic Cancer and Decreases Epithelial-Mesenchymal Transition-Induced Cell Invasion by Down-Regulating NLRP3. Mol. Cancer 2018, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Byeon, H.-E.; Jeon, J.Y.; Kim, H.J.; Kim, D.J.; Lee, K.-W.; Kang, Y.; Han, S.J. MicroRNA-132 Negatively Regulates Palmitate-Induced NLRP3 Inflammasome Activation through FOXO3 Down-Regulation in THP-1 Cells. Nutrients 2017, 9, 1370. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Q.; Wang, H.; Zhang, H.; Chen, F. MicroRNA-22 Suppresses Cell Proliferation, Migration and Invasion in Oral Squamous Cell Carcinoma by Targeting NLRP3. J. Cell. Physiol. 2018, 233, 6705–6713. [Google Scholar] [CrossRef]
- Zhou, C.; Zheng, J.; Fan, Y.; Wu, J. TI: NLRP3 Inflammasome-Dependent Pyroptosis in CNS Trauma: A Potential Therapeutic Target. Front. Cell Dev. Biol. 2022, 10, 821225. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Z.; Fan, Y.; Zhang, J.; Chen, K.; Gao, L.; Zeng, H.; Cao, J.; Wang, C. MicroRNA-9 Inhibits NLRP3 Inflammasome Activation in Human Atherosclerosis Inflammation Cell Models through the JAK1/STAT Signaling Pathway. Cell. Physiol. Biochem. 2017, 41, 1555–1571. [Google Scholar] [CrossRef]
- Peng, Q.; Yin, R.; Zhu, X.; Jin, L.; Wang, J.; Pan, X.; Ma, A. MiR-155 Activates the NLRP3 Inflammasome by Regulating the MEK/ERK/NF-ΚB Pathway in Carotid Atherosclerotic Plaques in ApoE−/−Mice. J. Physiol. Biochem. 2022, 78, 365–375. [Google Scholar] [CrossRef]
- Martínez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 Inflammasome and the Emerging Role of Colchicine to Inhibit Atherosclerosis-Associated Inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Thompson, P.L. Targeting Cholesterol Crystal-Induced Inflammation for the Secondary Prevention of Cardiovascular Disease. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 45–52. [Google Scholar] [CrossRef]
- Otani, K.; Watanabe, T.; Shimada, S.; Takeda, S.; Itani, S.; Higashimori, A.; Nadatani, Y.; Nagami, Y.; Tanaka, F.; Kamata, N.; et al. Colchicine Prevents NSAID-Induced Small Intestinal Injury by Inhibiting Activation of the NLRP3 Inflammasome. Sci. Rep. 2016, 6, 32587. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ren, M.; Adhikari, B.K.; Wang, H.; He, Y. The NLRP3 Inflammasome as a Novel Therapeutic Target for Cardiac Fibrosis. J. Inflamm. Res. 2022, 15, 3847–3858. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Ding, S.; Deng, H.; Wang, J.; Yi, W.; Wang, L.; Zhu, S.; Gu, L.; Xiong, X. Probenecid Protects against Oxygen-Glucose Deprivation Injury in Primary Astrocytes by Regulating Inflammasome Activity. Brain Res. 2016, 1643, 123–129. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, Z.; Wang, Y.; Hou, Y.; Li, L.; Zhao, J. Resveratrol Alleviates Cerebral Ischemia/Reperfusion Injury in Rats by Inhibiting NLRP3 Inflammasome Activation through Sirt1-Dependent Autophagy Induction. Int. Immunopharmacol. 2017, 50, 208–215. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, M.; Zhang, J.; Cai, Q.; Lu, D.; Li, Y.; Dong, Y.; Zhao, T.; Chen, H. The Neuroprotection of Sinomenine against Ischemic Stroke in Mice by Suppressing NLRP3 Inflammasome via AMPK Signaling. Int. Immunopharmacol. 2016, 40, 492–500. [Google Scholar] [CrossRef]
- Pluta, R.; Furmaga-Jabłońska, W.; Januszewski, S.; Czuczwar, S.J. Post-Ischemic Brain Neurodegeneration in the Form of Alzheimer’s Disease Proteinopathy: Possible Therapeutic Role of Curcumin. Nutrients 2022, 14, 248. [Google Scholar] [CrossRef]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s Tyrosine Kinase Is Essential for NLRP3 Inflammasome Activation and Contributes to Ischaemic Brain Injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef]
- PubMd. Minocycline Suppresses NLRP3 Inflammasome Activation in Experimental Ischemic Stroke. Available online: https://pubmed.ncbi.nlm.nih.gov/27846628/ (accessed on 15 March 2022).
- Li, C.; Wang, J.; Fang, Y.; Liu, Y.; Chen, T.; Sun, H.; Zhou, X.-F.; Liao, H. Nafamostat Mesilate Improves Function Recovery after Stroke by Inhibiting Neuroinflammation in Rats. Brain Behav. Immun. 2016, 56, 230–245. [Google Scholar] [CrossRef]
- Zhou, K.; Shi, L.; Wang, Z.; Zhou, J.; Manaenko, A.; Reis, C.; Chen, S.; Zhang, J. RIP1-RIP3-DRP1 Pathway Regulates NLRP3 Inflammasome Activation following Subarachnoid Hemorrhage. Exp. Neurol. 2017, 295, 116–124. [Google Scholar] [CrossRef]
- Feng, L.; Chen, Y.; Ding, R.; Fu, Z.; Yang, S.; Deng, X.; Zeng, J. P2X7R Blockade Prevents NLRP3 Inflammasome Activation and Brain Injury in a Rat Model of Intracerebral Hemorrhage: Involvement of Peroxynitrite. J. Neuroinflamm. 2015, 12, 190. [Google Scholar] [CrossRef]
- Van Putten, M.J.A.M.; Fahlke, C.; Kafitz, K.W.; Hofmeijer, J.; Rose, C.R. Dysregulation of Astrocyte Ion Homeostasis and Its Relevance for Stroke-Induced Brain Damage. Int. J. Mol. Sci. 2021, 22, 5679. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gong, P.; Zhang, X.; Li, S.; Lu, X.; Zhao, C.; Yu, Q.; Wei, Z.; Yang, Y.; Liu, Q.; et al. NLRP3 Inflammasome Participates in Host Response to Neospora caninum Infection. Front. Immunol. 2018, 9, 1791. [Google Scholar] [CrossRef] [PubMed]
- Sheth, K.N.; Petersen, N.H.; Cheung, K.; Elm, J.J.; Hinson, H.E.; Molyneaux, B.J.; Beslow, L.A.; Sze, G.K.; Simard, J.M.; Kimberly, W.T. Long-Term Outcomes in Patients Aged ≤70 Years with Intravenous Glyburide from the Phase II GAMES-RP Study of Large Hemispheric Infarction: An Exploratory Analysis. Stroke 2018, 49, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Carlos, D.; Costa, F.R.C.; Pereira, C.A.; Rocha, F.A.; Yaochite, J.N.U.; Oliveira, G.G.; Carneiro, F.S.; Tostes, R.C.; Ramos, S.G.; Zamboni, D.S.; et al. Mitochondrial DNA Activates the NLRP3 Inflammasome and Predisposes to Type 1 Diabetes in Murine Model. Front. Immunol. 2017, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Bauernfried, S.; Hornung, V. Human NLRP1: From the Shadows to Center Stage. J. Exp. Med. 2022, 219, e20211405. [Google Scholar] [CrossRef]
- Bruey, J.-M.; Bruey-Sedano, N.; Luciano, F.; Zhai, D.; Balpai, R.; Xu, C.; Kress, C.L.; Bailly-Maitre, B.; Li, X.; Osterman, A.; et al. Bcl-2 and Bcl-XL Regulate Proinflammatory Caspase-1 Activation by Interaction with NALP1. Cell 2007, 129, 45–56. [Google Scholar] [CrossRef]
- Lu, J.; Liu, Q.-H.; Wang, F.; Tan, J.-J.; Deng, Y.-Q.; Peng, X.-H.; Liu, X.; Zhang, B.; Xu, X.; Li, X.-P. Exosomal MiR-9 Inhibits Angiogenesis by Targeting MDK and Regulating PDK/AKT Pathway in Nasopharyngeal Carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 147. [Google Scholar] [CrossRef]
- Rostamian Delavar, M.; Baghi, M.; Safaeinejad, Z.; Kiani-Esfahani, A.; Ghaedi, K.; Nasr-Esfahani, M.H. Differential Expression of MiR-34a, MiR-141, and MiR-9 in MPP+-Treated Differentiated PC12 Cells as a Model of Parkinson’s Disease. Gene 2018, 662, 54–65. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, H.; Lu, X.; Wang, J.; Zhang, X.; Sun, S.; Bao, Z.; Tian, W.; Ning, S.; Wang, L.; et al. Overexpression of MicroRNA-9a-5p Ameliorates NLRP1 Inflammasome-Mediated Ischemic Injury in Rats Following Ischemic Stroke. Neuroscience 2020, 444, 106–117. [Google Scholar] [CrossRef]
- Sun, X.; Song, X.; Zhang, L.; Sun, J.; Wei, X.; Meng, L.; An, J. NLRP2 Is Highly Expressed in a Mouse Model of Ischemic Stroke. Biochem. Biophys. Res. Commun. 2016, 479, 656–662. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Kim, E.J.; Kim, S.Y.; Kim, J.M.; Kam, E.H.; Park, J.-K.; Koo, B.-N. Apoptosis Signal-Regulating Kinase 1 Silencing on Astroglial Inflammasomes in an Experimental Model of Ischemic Stroke. Neuroscience 2018, 390, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P.-Y. NLR Members NLRC4 and NLRP3 Mediate Sterile Inflammasome Activation in Microglia and Astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, S.; Malato, A.; Saccullo, G.; Iorio, A.; Di Ianni, M.; Caracciolo, C.; Coco, L.L.; Raso, S.; Santoro, M.; Guarneri, F.P.; et al. Residual vein thrombosis for assessing duration of anticoagulation after unprovoked deep vein thrombosis of the lower limbs: The extended DACUS study. Am. J. Hematol. 2011, 86, 914–917. [Google Scholar] [CrossRef]
- Pinto, A.; Di Raimondo, D.; Tuttolomondo, A.; Fernandez, P.; Arnao, V.; Licata, G. Twenty-four hour ambulatory blood pressure monitoring to evaluate effects on blood pressure of physical activity in hypertensive patients. Clin. J. Sport Med. 2006, 16, 238–243. [Google Scholar] [CrossRef]
- Basili, S.; Raparelli, V.; Napoleone, L.; Talerico, G.; Corazza, G.R.; Perticone, F.; Sacerdoti, D.; Andriulli, A.; Licata, A.; Pietrangelo, A.; et al. Platelet Count Does Not Predict Bleeding in Cirrhotic Patients: Results from the PRO-LIVER Study. Am. J. Gastroenterol. 2018, 113, 368–375. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Maida, C.; Arnao, V.; Della Corte, V.; Simonetta, I.; Corpora, F.; Di Bona, D.; Maugeri, R.; et al. Early High-dosage Atorvastatin Treatment Improved Serum Immune-inflammatory Markers and Functional Outcome in Acute Ischemic Strokes Classified as Large Artery Atherosclerotic Stroke: A Randomized Trial. Medicine 2016, 95, e3186. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Casuccio, A.; Di Bona, D.; Aiello, A.; Accardi, G.; Arnao, V.; Clemente, G.; Corte, V.D.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute ischemic stroke. J. Neuroinflamm. 2019, 16, 88. [Google Scholar] [CrossRef]
- Zanoli, L.; Boutouyrie, P.; Fatuzzo, P.; Granata, A.; Lentini, P.; Oztürk, K.; Cappello, M.; Theocharidou, E.; Tuttolomondo, A.; Pinto, A.; et al. Inflammation and Aortic Stiffness: An Individual Participant Data Meta-Analysis in Patients With Inflammatory Bowel Disease. J. Am. Heart Assoc. 2017, 6, e007003. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Categories | Drugs or Molecules | References |
|---|---|---|
| Acting on gene expression products | MCC950, Bay 11-7082, NRF2, sinomenine, curcumin, minocycline | [92] |
| Acting on the process of gene expression | IVIG, IFN-β, ketone metabolite hydroxybutyrate, probenecid, nafamostat mesilate | [92] |
| Acting on gene expression processes and gene expression products | miR-223, miR-155, resveratrol | [92] |
| Drug | Characteristic Features | Therapeutical Actions | References |
|---|---|---|---|
| Small molecules SB 203580 Bay-11-7082 U-0126 JNK Inhibitor V | →P38-MAPK inhibitor →NF-κB inhibitor →ERK inhibitor →JNK inhibitor | Neuroprotection during induced cerebral ischemia in mouse tMCAO model and primary neuron OGD reduced expression and activation of the inflammasome and decreased release of cytokines IL-18 and IL 1β | [34] |
| Glyburide | NLRP3 oligomerization inhibitor | In PC12 cell OGD, anti-inflammatory and anti-oxidative stress action | [121] |
| MCC950 | NLRP3 oligomerization inhibitor | In photothrombotic ischemia mice and primary neuron OGD, hampered platelet activation/aggregation and thrombogenesis in vitro Alleviated neuronal cell apoptosis, reduced area size of cerebral ischemia, and neurological disability | [63,94,95] |
| β-hydroxybutyrate (BHB) | Kefflux inhibitor and ASC oligomerization inhibitor | Inhibited NLRP3 inflammasome priming process | [25] |
| Nuclear factor erythroid-2 related factor 2 (NRF2) | Redox-sensitive transcription factor | Suppressed ROS- and NF-kB, modulated TXNIP complex | [96] |
| Nitric oxide (NO) | Gas molecule | Suppressed ASC pyroptosome formation, caspase-1 and IL-1b release | [99] |
| IFN-α and IFN-β | Nonspecific NLRP3 inflammasome | Promoting phosphorylation of STAT1, inducting IL-10 production | [25] |
| Micro RNAs | Non-protein-coding RNA | Inhibited NLRP3 protein expression | [107] |
| Colchicine | Alkaloid | Prevents P2X7-induced pore formation and inhibits caspase-1 | [108] |
| Probenecid | Pannexin 1 inhibitor | In primary astrocyte OGD, reduced expression levels of NLRP3 and caspase-1 and prevented the extracellular release of IL-1β death of astrocytes and increased production of ROS | [112] |
| Sinomenine | Natural alkaloid compound | In mouse tMCAO model and primary mixed glial cell OGD inhibited the release of NLRP3, ASC, cleaved caspase-1, and pro-inflammatory cytokines attenuation of cerebral oedema, neurological deficit, apoptosis of neurons and reduction of infarction activation of the AMPK pathway-mitigated activation of microglia and astrocytes following ischemic damage | [114] |
| Paeoniflorin | Natural bioactive monoterpene glucoside | In hippocampal slices, OGD diminished expression levels of NLRP3 and its downstream proteins safeguarded neuronal cell death | [122] |
| Resveratrol | Natural polyphenolic compound | In mouse endothelin-1-induced MCAO model, counteracted the activation of NLRP3 and the release of IL-1β and prevented the expression of TXNIP, promoting the reduction of cerebral oedema and the size of the infarcted area | [113,115] |
| Curcumin | Polyphenolic compound | Inhibited endoplasmic reticulum stress, suppressed TXNIP/ NLRP3 inflammasome stimulation | [115] |
| Ibrutinib (PCI-32765) | Bruton’s tyrosine kinase inhibitors | Decreased levels of IL-1β IL-6, IL-23A and infiltrating microglia | [116] |
| Minocycline | Antibiotic immunosuppressor | In mouse tMCAO model and BV2 cell OGD inhibited activation of microglia and signals 1 and 2 of NLRP3 inflammasome activation | [117] |
| Nafamostat mesilate | Synthetic serine protease inhibitor | In mouse tMCAO model and primary microglial culture, OGD altered expression profiles of inflammation mediators and induced expression of anti-inflammatory mediators | [118] |
| Necrostatin-1 | Inhibitor of RIP1 kinase | inhibits inflammasome activation in murine models | [119] |
| Brilliant Blue G | P2X7 receptor antagonist | Attenuated caspase-3 dependent neuronal apoptosis | [120] |
| Drug | Characteristic Features | Therapeutical Actions | References |
|---|---|---|---|
| NLRP1 SB 203580 Bay-11-7082 U-0126 JNK Inhibitor V and SP600125 IVIg | →P38-MAPK inhibitor →NF-κB inhibitor →ERK inhibitor →JNK inhibitor Intravenous immune globuline | Reduced expression levels of cleaved XIAP, cleaved caspase-1, and caspase-11 and maturation of IL-1β and IL-18 | [34] |
| Mir-9a-5p | Non coding RNA | In OGD cells and in MCAO rats the overexpression of mir-9a-5p downregulates NLRP1 inflammasome | [130] |
| NLRP2 ASK-1 (Apoptosis signal-regulating kinase 1) | Immune-regulator and early activator of apoptosis | Silencing or inhibition of ASK-1 determines downexpression of NLRP2 levels | [132] |
| NLRC4 Fendrr | Long non-coding RNA | Fendrr knockdown in (H/R)-induced microglia reduced NLRC4 levels associated with pyroptosis | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puleo, M.G.; Miceli, S.; Di Chiara, T.; Pizzo, G.M.; Della Corte, V.; Simonetta, I.; Pinto, A.; Tuttolomondo, A. Molecular Mechanisms of Inflammasome in Ischemic Stroke Pathogenesis. Pharmaceuticals 2022, 15, 1168. https://doi.org/10.3390/ph15101168
Puleo MG, Miceli S, Di Chiara T, Pizzo GM, Della Corte V, Simonetta I, Pinto A, Tuttolomondo A. Molecular Mechanisms of Inflammasome in Ischemic Stroke Pathogenesis. Pharmaceuticals. 2022; 15(10):1168. https://doi.org/10.3390/ph15101168
Chicago/Turabian StylePuleo, Maria Grazia, Salvatore Miceli, Tiziana Di Chiara, Giuseppina Maria Pizzo, Vittoriano Della Corte, Irene Simonetta, Antonio Pinto, and Antonino Tuttolomondo. 2022. "Molecular Mechanisms of Inflammasome in Ischemic Stroke Pathogenesis" Pharmaceuticals 15, no. 10: 1168. https://doi.org/10.3390/ph15101168
APA StylePuleo, M. G., Miceli, S., Di Chiara, T., Pizzo, G. M., Della Corte, V., Simonetta, I., Pinto, A., & Tuttolomondo, A. (2022). Molecular Mechanisms of Inflammasome in Ischemic Stroke Pathogenesis. Pharmaceuticals, 15(10), 1168. https://doi.org/10.3390/ph15101168