
Recent Advances in Structure, Function, and Pharmacology of Class A Lipid GPCRs: Opportunities and Challenges for Drug Discovery

 , and
, and
Abstract
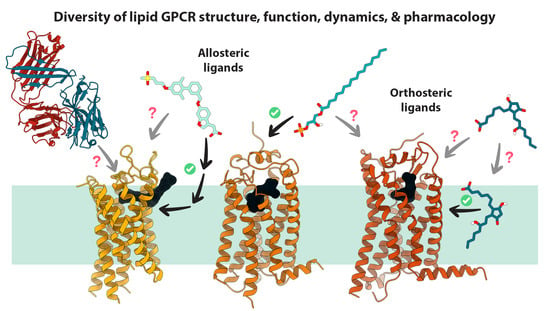
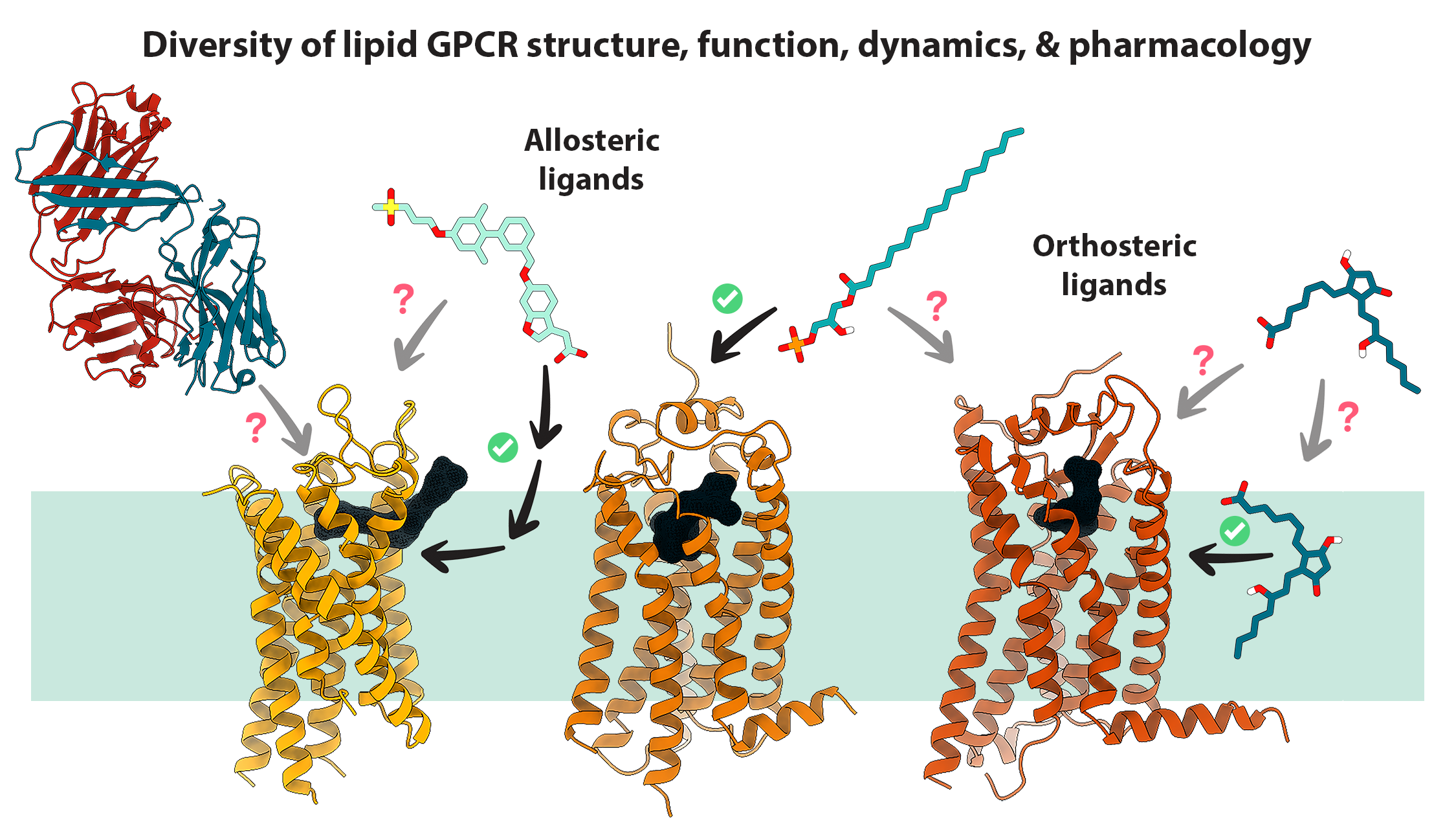
1. Introduction
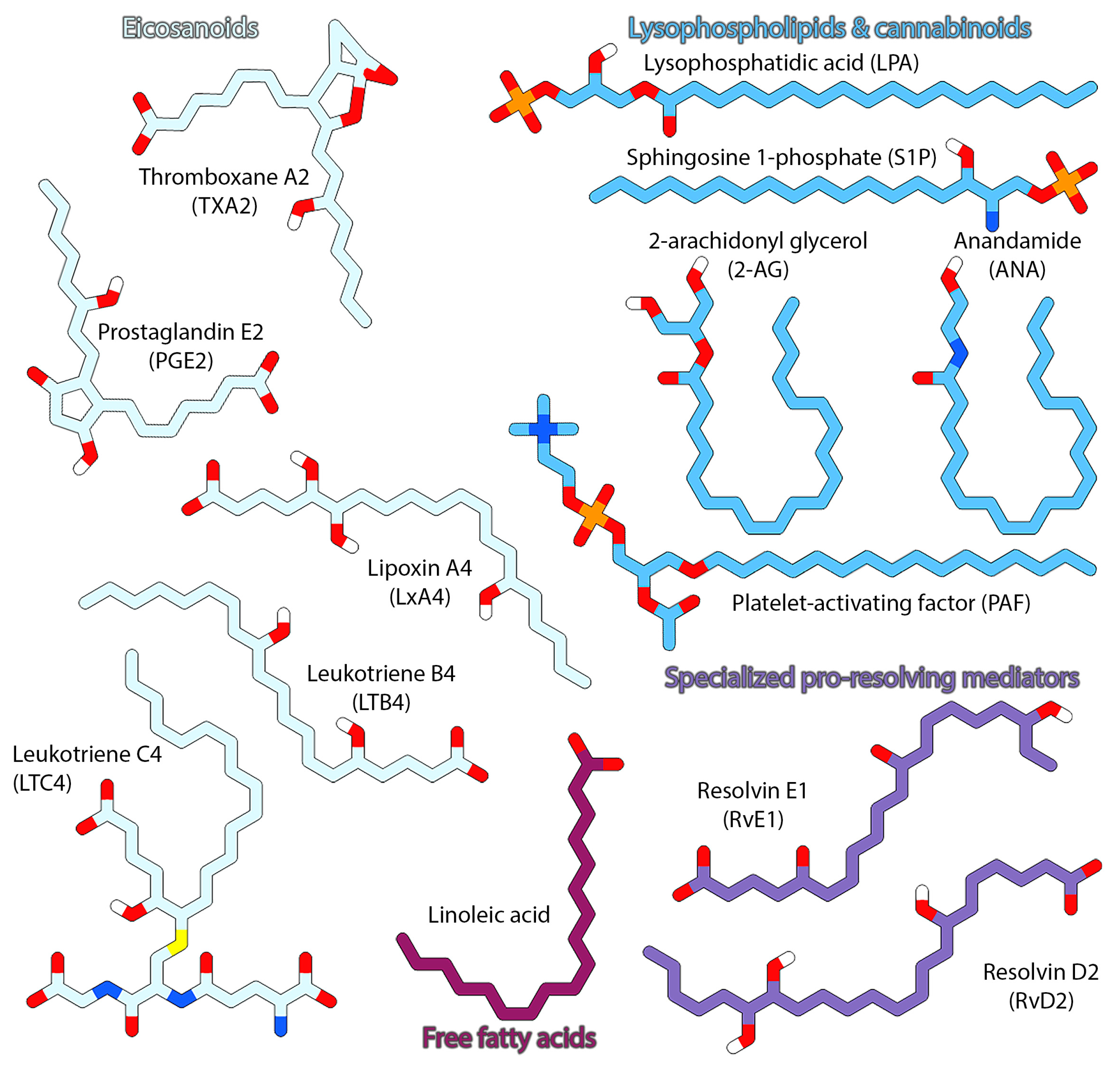
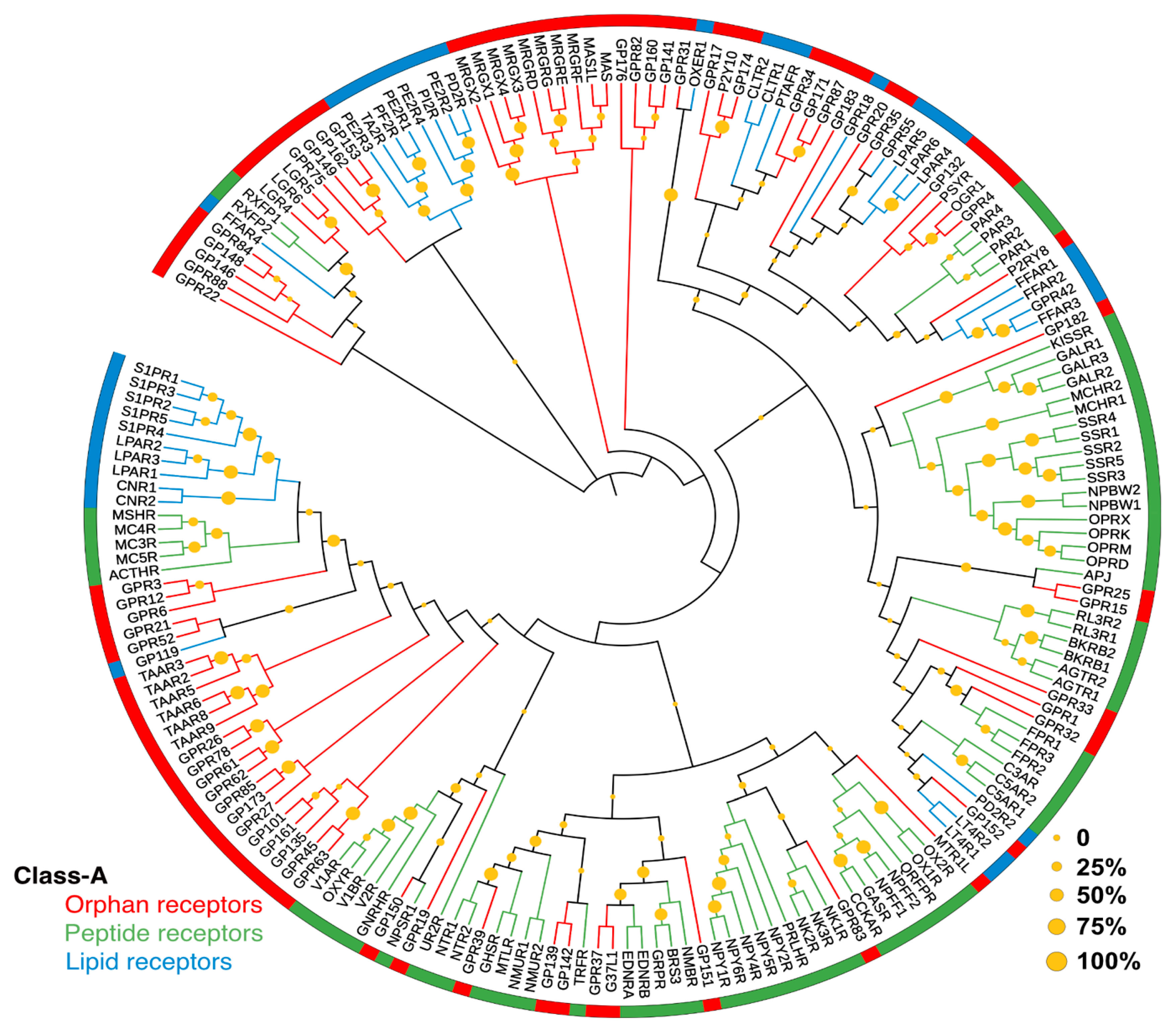
2. GPCRs Liganded by Lipid Mediators
2.1. A Brief Primer on GPCRs
2.2. Lipid GPCRs and Associated Drugs
3. Progress in Structural Biology of Lipid GPCRs
The First Lipid GPCR Structure
4. Structural and Functional Features of Lipid GPCRs
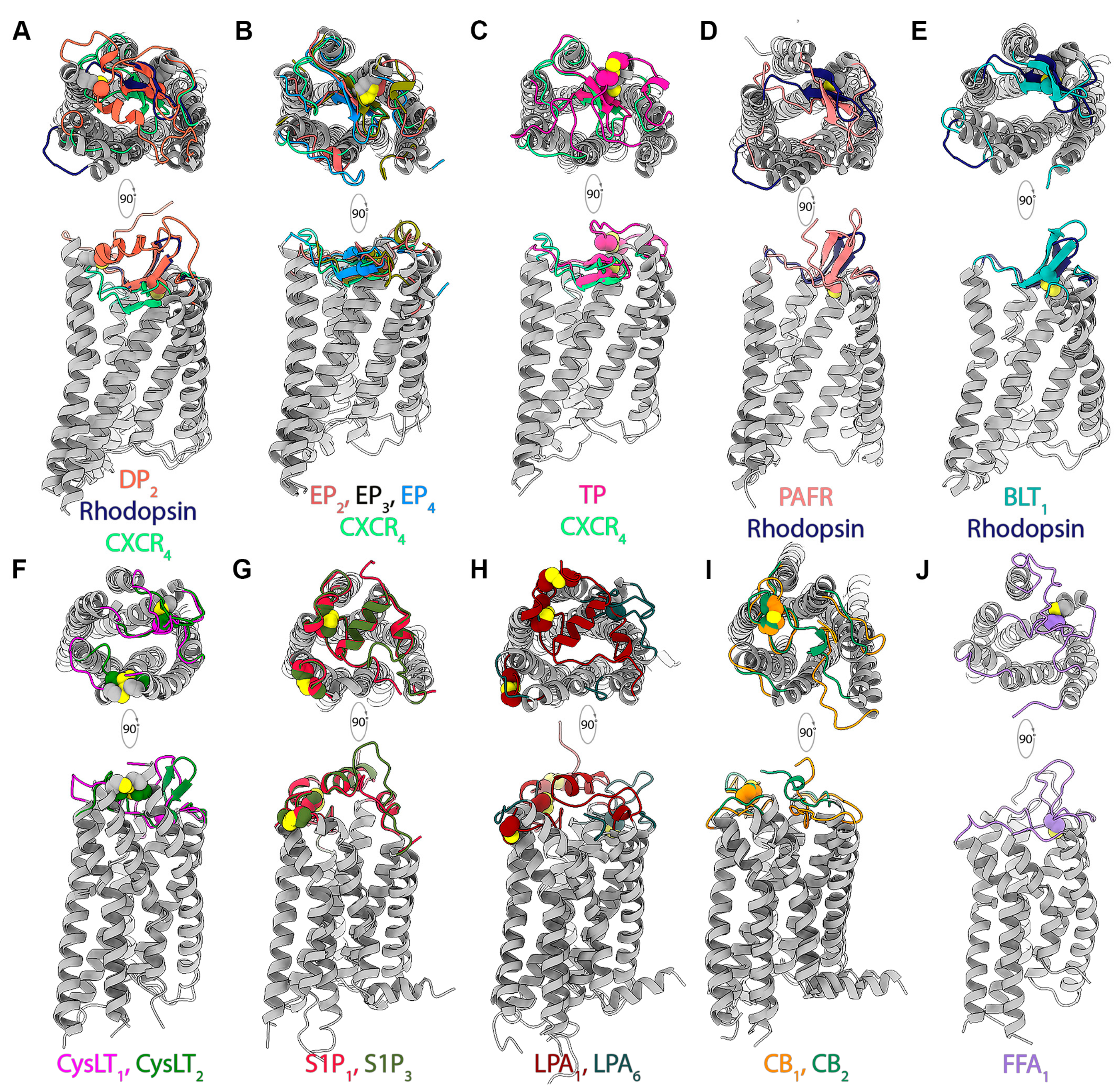
4.1. Organization of Extracellular Domains in Lipid GPCRs
4.2. Mode and Dynamics of Lipid Ligand Access
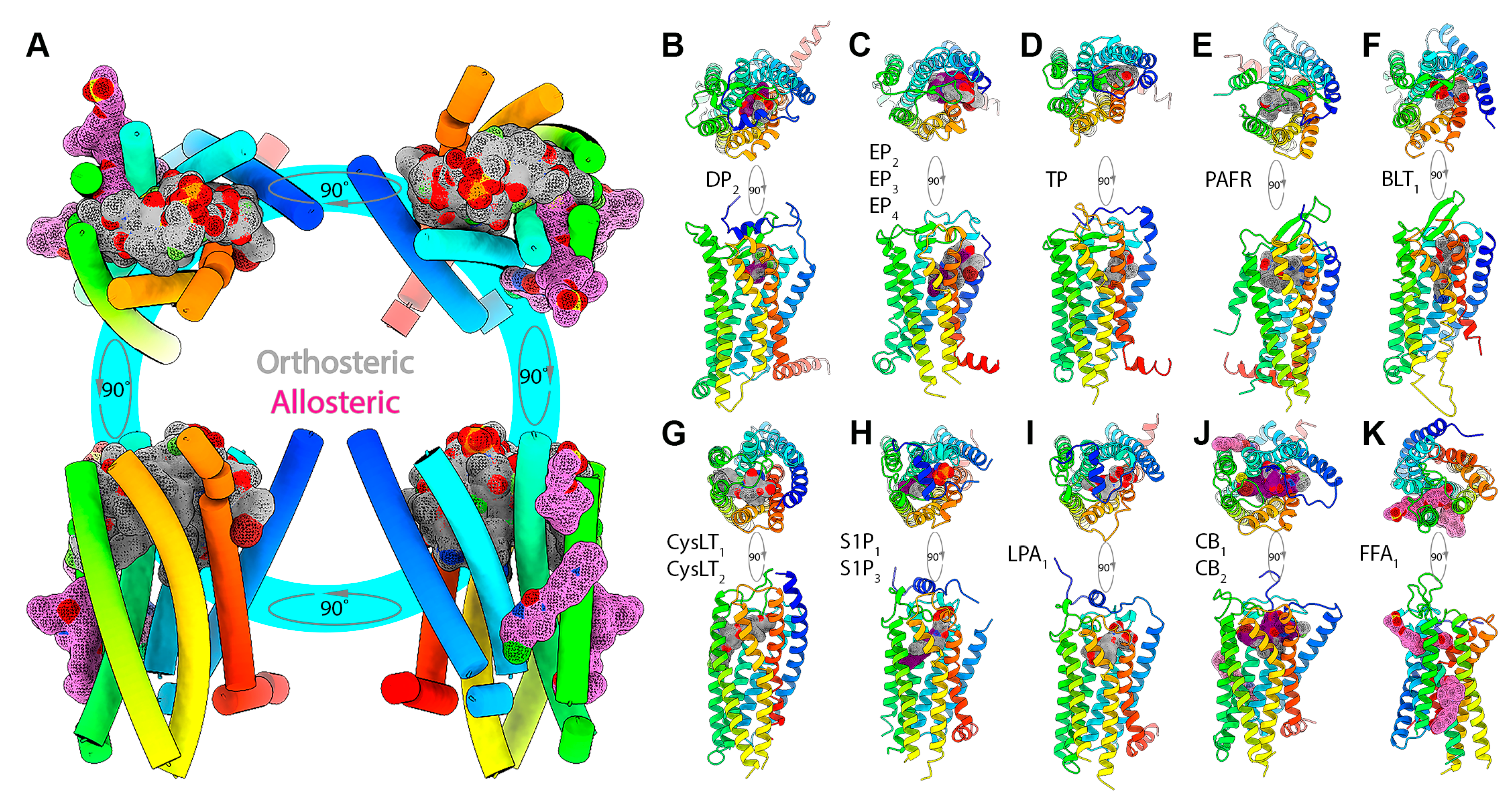
4.3. Canonical and Non-Canonical Ligand Binding in Lipid Receptors
4.4. Non-Canonical or Allosteric Sites
5. The Challenges and Future Perspectives on the Development of GPCR-Centric Therapeutics
5.1. Beyond Simple Agonism and Antagonism
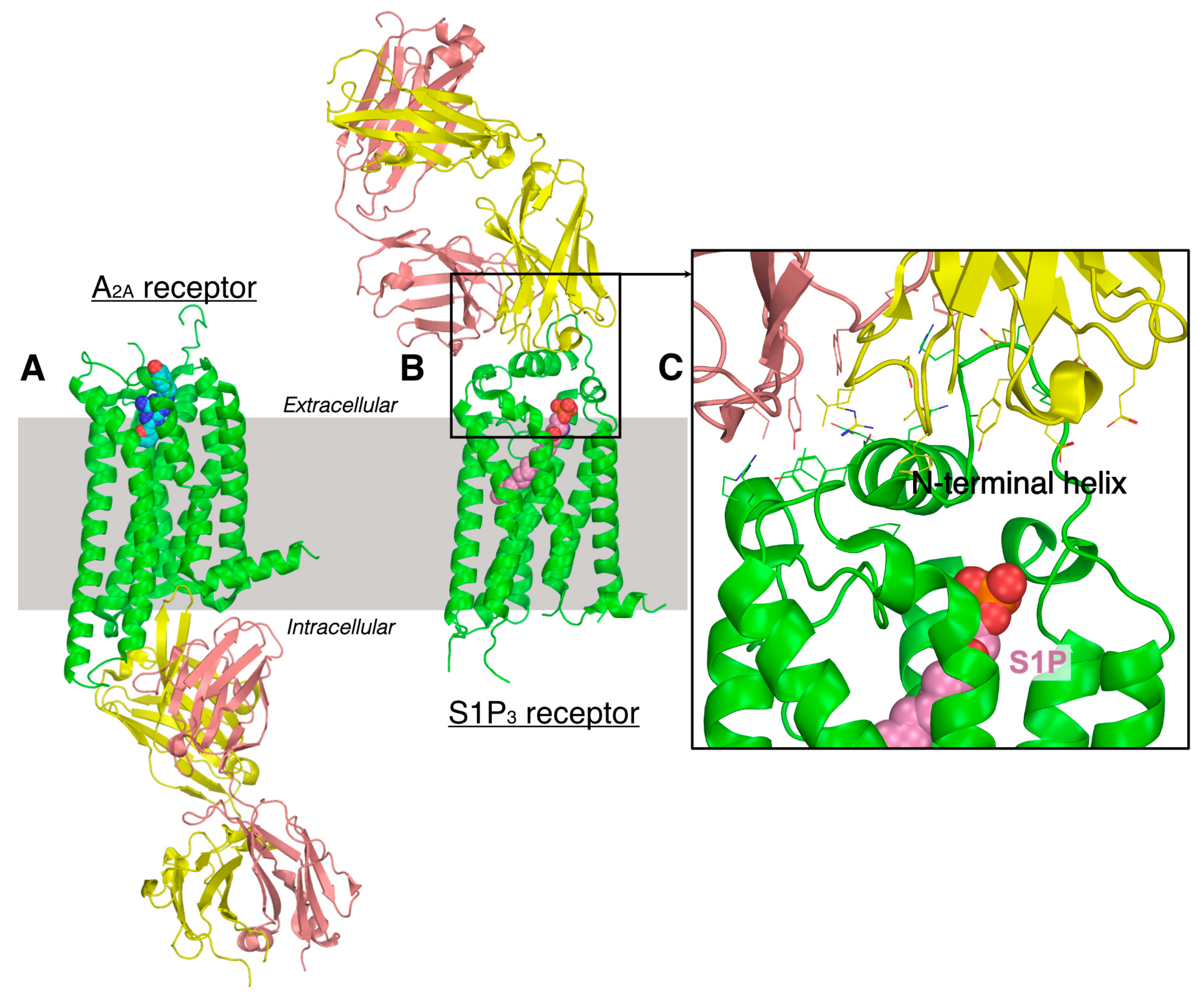
5.2. Antibody-Based Therapeutics
5.3. Advances in Computational Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandis, A.Z.; Wenk, M.R. Membrane lipids as signaling molecules. Curr. Opin. Lipidol. 2007, 18, 121–128. [Google Scholar] [CrossRef]
- Murakami, M. Lipid Mediators in Life Science. Exp. Anim. 2011, 60, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Foord, S.M.; Bonner, T.I.; Neubig, R.R.; Rosser, E.M.; Pin, J.-P.; Davenport, A.P.; Spedding, M.; Harmar, A. International Union of Pharmacology. XLVI. G Protein-Coupled Receptor List. Pharmacol. Rev. 2005, 57, 279–288. [Google Scholar]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, S.G. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Author response: Common activation mechanism of class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef]
- Attwood, T.K.; Findlay, J.B.C. Fingerprinting G-protein-coupled receptors. Protein Eng. Des. Sel. 1994, 7, 195–203. [Google Scholar] [CrossRef]
- Kolakowski, L.F., Jr. GCRDb: A G-protein-coupled receptor database. Recept. Channels 1994, 2, 1–7. [Google Scholar]
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gareri, C.; Rockman, H.A. G-protein–coupled receptors in heart disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef]
- Luo, J.; Sun, P.; Siwko, S.; Liu, M.; Xiao, J. The role of GPCRs in bone diseases and dysfunctions. Bone Res. 2019, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, R.; Anderman, M.F.; Shepard, B.D. GPCRs get fatty: The role of G protein-coupled receptor signaling in the development and progression of nonalcoholic fatty liver disease. Am. J. Physiol. Liver Physiol. 2021, 320, G304–G318. [Google Scholar] [CrossRef]
- Schöneberg, T.; Liebscher, I. Mutations in G Protein–Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol. Rev. 2021, 73, 89–119. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsøe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2016, 44, D356–D364. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef] [PubMed]
- Van Jaarsveld, M.T.M.; Houthuijzen, J.M.; Voest, E. Molecular mechanisms of target recognition by lipid GPCRs: Relevance for cancer. Oncogene 2016, 35, 4021–4035. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Kihara, Y. Druggable Lipid GPCRs: Past, Present, and Prospects. Druggable Lipid Signal. Pathw. 2020, 1274, 223–258. [Google Scholar]
- Zhao, Q.; Wu, B.-L. Ice breaking in GPCR structural biology. Acta Pharmacol. Sin. 2012, 33, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Vuckovic, Z.; Draper-Joyce, C.J.; Liang, Y.-L.; Glukhova, A.; Christopoulos, A.; Sexton, P.M. Recent advances in the determination of G protein-coupled receptor structures. Curr. Opin. Struct. Biol. 2018, 51, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Xu, H.; Hanson, M.; Liu, W. GPCR crystallization using lipidic cubic phase technique. Curr. Pharm. Biotechnol. 2014, 15, 971–979. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Cryo-Electron Microscopy: Moving Beyond X-ray Crystal Structures for Drug Receptors and Drug Development. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 51–71. [Google Scholar] [CrossRef]
- Wu, M.; Lander, G.C. Present and Emerging Methodologies in Cryo-EM Single-Particle Analysis. Biophys. J. 2020, 119, 1281–1289. [Google Scholar] [CrossRef]
- Lee, A.G. Interfacial Binding Sites for Cholesterol on G Protein-Coupled Receptors. Biophys. J. 2019, 116, 1586–1597. [Google Scholar] [CrossRef]
- Congreve, M.; Marshall, F. The impact of GPCR structures on pharmacology and structure-based drug design. Br. J. Pharmacol. 2009, 159, 986–996. [Google Scholar] [CrossRef]
- Lyumkis, D. Challenges and opportunities in cryo-EM single-particle analysis. J. Biol. Chem. 2019, 294, 5181–5197. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yao, D.; Deepak, R.K.; Liu, H.; Xiao, Q.; Fan, H.; Gong, W.; Wei, Z.; Zhang, C. Structures of the Human PGD2 Receptor CRTH2 Reveal Novel Mechanisms for Ligand Recognition. Mol. Cell 2018, 72, 48–59.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Deepak, R.N.V.K.; Shiriaeva, A.; Gati, C.; Batyuk, A.; Hu, H.; Weierstall, U.; Liu, W.; Wang, L.; Cherezov, V.; et al. Molecular basis for lipid recognition by the prostaglandin D2 receptor CRTH2. Proc. Natl. Acad. Sci. USA 2021, 118, e2102813118. [Google Scholar] [CrossRef]
- Qu, C.; Mao, C.; Xiao, P.; Shen, Q.; Zhong, Y.-N.; Yang, F.; Shen, D.-D.; Tao, X.; Zhang, H.; Yan, X.; et al. Ligand recognition, unconventional activation, and G protein coupling of the prostaglandin E2 receptor EP2 subtype. Sci. Adv. 2021, 7, eabf1268. [Google Scholar] [CrossRef]
- Morimoto, K.; Suno, R.; Hotta, Y.; Yamashita, K.; Hirata, K.; Yamamoto, M.; Narumiya, S.; Iwata, S.; Kobayashi, T. Crystal structure of the endogenous agonist-bound prostanoid receptor EP3. Nat. Chem. Biol. 2019, 15, 8–10. [Google Scholar] [CrossRef]
- Audet, M.; White, K.L.; Breton, B.; Zarzycka, B.; Han, G.W.; Lu, Y.; Gati, C.; Batyuk, A.; Popov, P.; Velasquez, J.; et al. Crystal structure of misoprostol bound to the labor inducer prostaglandin E2 receptor. Nat. Chem. Biol. 2019, 15, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Morimoto, K.; Suno, R.; Horita, S.; Yamashita, K.; Hirata, K.; Sekiguchi, Y.; Yasuda, S.; Shiroishi, M.; Shimizu, T.; et al. Ligand binding to human prostaglandin E receptor EP4 at the lipid-bilayer interface. Nat. Chem. Biol. 2019, 15, 18–26. [Google Scholar] [CrossRef]
- Nojima, S.; Fujita, Y.; Kimura, K.T.; Nomura, N.; Suno, R.; Morimoto, K.; Yamamoto, M.; Noda, T.; Iwata, S.; Shigematsu, H.; et al. Cryo-EM Structure of the Prostaglandin E Receptor EP4 Coupled to G Protein. Structure 2021, 29, 252–260.e6. [Google Scholar] [CrossRef]
- Fan, H.; Chen, S.; Yuan, X.; Han, S.; Zhang, H.; Xia, W.; Xu, Y.; Zhao, Q.; Wu, B. Structural basis for ligand recognition of the human thromboxane A2 receptor. Nat. Chem. Biol. 2019, 15, 27–33. [Google Scholar] [CrossRef]
- Cao, C.; Tan, Q.; Xu, C.; He, L.; Yang, L.; Zhou, Y.; Zhou, Y.; Qiao, A.; Lu, M.; Yi, C.; et al. Structural basis for signal recognition and transduction by platelet-activating-factor receptor. Nat. Struct. Mol. Biol. 2018, 25, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Roth, C.B.; Jo, E.; Griffith, M.T.; Scott, F.L.; Reinhart, G.; Desale, H.; Clemons, B.; Cahalan, S.M.; Schuerer, S.C.; et al. Crystal Structure of a Lipid G Protein–Coupled Receptor. Science 2012, 335, 851–855. [Google Scholar] [CrossRef]
- Maeda, S.; Shiimura, Y.; Asada, H.; Hirata, K.; Luo, F.; Nango, E.; Tanaka, N.; Toyomoto, M.; Inoue, A.; Aoki, J.; et al. Endogenous agonist–bound S1PR3 structure reveals determinants of G protein–subtype bias. Sci. Adv. 2021, 7, eabf5325. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Okuno, T.; Hirata, K.; Yamashita, K.; Kawano, Y.; Yamamoto, M.; Hato, M.; Nakamura, M.; Shimizu, T.; Yokomizo, T.; et al. Na+-mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat. Chem. Biol. 2018, 14, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Michaelian, N.; Sadybekov, A.; Besserer-Offroy, É.; Han, G.W.; Krishnamurthy, H.; Zamlynny, B.A.; Fradera, X.; Siliphaivanh, P.; Presland, J.; Spencer, K.B.; et al. Structural insights on ligand recognition at the human leukotriene B4 receptor 1. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiriaeva, A.; Besserer-Offroy, É.; Longpré, J.-M.; Lyapina, E.; et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 2019, 5, eaax2518. [Google Scholar] [CrossRef]
- Gusach, A.; Luginina, A.; Marin, E.; Brouillette, R.; Besserer-Offroy, É.; Longpré, J.-M.; Ishchenko, A.; Popov, P.; Patel, N.; Fujimoto, T.; et al. Structural basis of ligand selectivity and disease mutations in cysteinyl leukotriene receptors. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Srivastava, A.; Yano, J.; Hirozane, Y.; Kefala, G.; Gruswitz, F.; Snell, G.; Lane, W.; Ivetac, A.; Aertgeerts, K.; Nguyen, J.; et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nat. Cell Biol. 2014, 513, 124–127. [Google Scholar] [CrossRef]
- Lu, J.; Byrne, N.; Wang, J.; Bricogne, G.; Brown, F.K.; Chobanian, H.R.; Colletti, S.L.; Di Salvo, J.; Thomas-Fowlkes, B.; Guo, Y.; et al. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat. Struct. Mol. Biol. 2017, 24, 570–577. [Google Scholar] [CrossRef]
- Ho, J.; Chau, B.; Rodgers, L.; Lu, F.; Wilbur, K.L.; Otto, K.A.; Chen, Y.; Song, M.; Riley, J.P.; Yang, H.-C.; et al. Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; LaPrairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nat. Cell Biol. 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Kumar, K.K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e12. [Google Scholar] [CrossRef]
- Shao, Z.; Yan, W.; Chapman, K.; Ramesh, K.; Ferrell, A.J.; Yin, J.; Wang, X.; Xu, Q.; Rosenbaum, D.M. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat. Chem. Biol. 2019, 15, 1199–1205. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef]
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef] [PubMed]
- Audet, M.; Stevens, R.C. Emerging structural biology of lipid G protein-coupled receptors. Protein Sci. 2019, 28, 292–304. [Google Scholar] [CrossRef]
- Parrill, A.L.; Wang, D.-A.; Bautista, D.L.; Van Brocklyn, J.R.; Lorincz, Z.; Fischer, D.J.; Baker, D.L.; Liliom, K.; Spiegel, S.; Tigyi, G. Identification of Edg1 Receptor Residues that Recognize Sphingosine 1-Phosphate. J. Biol. Chem. 2000, 275, 39379–39384. [Google Scholar] [CrossRef]
- Wheatley, M.; Wootten, D.; Conner, M.T.; Simms, J.; Kendrick, R.; Logan, R.T.; Poyner, D.R.; Barwell, J. Lifting the lid on GPCRs: The role of extracellular loops. Br. J. Pharmacol. 2012, 165, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nat. Cell Biol. 2015, 523, 561–567. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Lone, A.M.; Taskén, K. Proinflammatory and Immunoregulatory Roles of Eicosanoids in T Cells. Front. Immunol. 2013, 4, 130. [Google Scholar] [CrossRef]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, S.A.; Leah, E.J.; Zhang, Q.; Bright, N.A.; Oxley, D.; Bootman, M.; Rudge, S.A.; Wakelam, M.J.O. Exosomes bind autotaxin and act as a physiological delivery mechanism to stimulate LPA receptor signalling in cells. J. Cell Sci. 2016, 129, 3948–3957. [Google Scholar] [CrossRef] [PubMed]
- Schädel, S.A.; Heck, M.; Maretzki, D.; Filipek, S.; Teller, D.C.; Palczewski, K.; Hofmann, K.P. Ligand channeling within a G-protein-coupled receptor: The entry and exit of retinals in native opsin. J. Biol. Chem. 2003, 278, 24896–24903. [Google Scholar] [CrossRef]
- Heck, M.; Schädel, S.A.; Maretzki, D.; Hofmann, K.P. Secondary binding sites of retinoids in opsin: Characterization and role in regeneration. Vis. Res. 2003, 43, 3003–3010. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; Yamamoto, M.; et al. Crystal structure of rhodopsin: AG protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef]
- Okada, T.; Sugihara, M.; Bondar, A.-N.; Elstner, M.; Entel, P.; Buss, V. The Retinal Conformation and its Environment in Rhodopsin in Light of a New 2.2Å Crystal Structure. J. Mol. Biol. 2004, 342, 571–583. [Google Scholar]
- Wang, T.; Duan, Y. Chromophore Channeling in the G-Protein Coupled Receptor Rhodopsin. J. Am. Chem. Soc. 2007, 129, 6970–6971. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hildebrand, P.W.; Scheerer, P.; Park, J.H.; Choe, H.-W.; Piechnick, R.; Ernst, O.P.; Hofmann, K.P.; Heck, M. A Ligand Channel through the G Protein Coupled Receptor Opsin. PLoS ONE 2009, 4, e4382. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Morizumi, T.; Li, Y.; Hong, J.E.; Pai, E.F.; Hofmann, K.P.; Choe, H.-W.; Ernst, O.P. Opsin, a Structural Model for Olfactory Receptors? Angew. Chem. 2013, 125, 11227–11230. [Google Scholar] [CrossRef]
- Guixà-González, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Martí-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martín, M.; et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Mercier, R.W.; Anday, J.K.; Thakur, G.A.; Zvonok, A.M.; Hurst, D.; Reggio, P.H.; Janero, D.R.; Makriyannis, A. Ligand-Binding Architecture of Human CB2 Cannabinoid Receptor: Evidence for Receptor Subtype-Specific Binding Motif and Modeling GPCR Activation. Chem. Biol. 2008, 15, 1207–1219. [Google Scholar] [CrossRef]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-coupled Receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef]
- Caliman, A.D.; Miao, Y.; McCammon, J.A. Activation mechanisms of the first sphingosine-1-phosphate receptor. Protein Sci. 2017, 26, 1150–1160. [Google Scholar] [CrossRef]
- Stanley, N.; Pardo, L.; De Fabritiis, G. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016, 6, 22639. [Google Scholar] [CrossRef]
- Lin, Y.; Deepak, R.N.V.K.; Zheng, J.Z.; Fan, H.; Zheng, L. A dual substrate-accessing mechanism of a major facilitator superfamily protein facilitates lysophospholipid flipping across the cell membrane. J. Biol. Chem. 2018, 293, 19919–19931. [Google Scholar] [CrossRef]
- Yuan, S.; Wu, R.; Latek, D.; Trzaskowski, B.; Filipek, S. Lipid Receptor S1P1 Activation Scheme Concluded from Microsecond All-Atom Molecular Dynamics Simulations. PLoS Comput. Biol. 2013, 9, e1003261. [Google Scholar] [CrossRef] [PubMed]
- Wenthur, C.J.; Gentry, P.R.; Mathews, T.P.; Lindsley, C.W. Drugs for Allosteric Sites on Receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. [Google Scholar] [CrossRef]
- Wakefield, A.E.; Mason, J.S.; Vajda, S.; Keserű, G.M. Analysis of tractable allosteric sites in G protein-coupled receptors. Sci. Rep. 2019, 9, 6180. [Google Scholar] [CrossRef]
- Langmead, C.J.; Christopoulos, A. Allosteric agonists of 7TM receptors: Expanding the pharmacological toolbox. Trends Pharmacol. Sci. 2006, 27, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef]
- Foster, D.; Conn, P.J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef]
- Leander, M.; Yuan, Y.; Meger, A.; Cui, Q.; Raman, S. Functional Plasticity and Evolutionary Adaptation of Allosteric Regulation. Proc. Natl. Acad. Sci. USA 2020, 117, 25445–25454. [Google Scholar] [CrossRef] [PubMed]
- Prakash, P.; Hancock, J.; Gorfe, A.A. Binding hotspots on K-ras: Consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins Struct. Funct. Bioinform. 2015, 83, 898–909. [Google Scholar] [CrossRef]
- Lakkaraju, S.K.; Yu, W.; Raman, E.P.; Hershfeld, A.V.; Fang, L.; Deshpande, D.A.; MacKerell, J.A.D. Mapping Functional Group Free Energy Patterns at Protein Occluded Sites: Nuclear Receptors and G-Protein Coupled Receptors. J. Chem. Inf. Model. 2015, 55, 700–708. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Jo, S.; Lakkaraju, S.K.; Lind, C.; Yu, W. Identification and characterization of fragment binding sites for allosteric ligand design using the site identification by ligand competitive saturation hotspots approach (SILCS-Hotspots). Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129519. [Google Scholar] [CrossRef]
- Tan, Y.S.; Verma, C.S. Straightforward Incorporation of Multiple Ligand Types into Molecular Dynamics Simulations for Efficient Binding Site Detection and Characterization. J. Chem. Theory Comput. 2020, 16, 6633–6644. [Google Scholar] [CrossRef]
- Ciancetta, A.; Gill, A.K.; Ding, T.; Karlov, D.S.; Chalhoub, G.; McCormick, P.J.; Tikhonova, I.G. Probe Confined Dynamic Mapping for G Protein-Coupled Receptor Allosteric Site Prediction. ACS Cent. Sci. 2021, 7, 1847–1862. [Google Scholar] [CrossRef]
- Lim, J.Y.; Park, C.-K.; Hwang, S.W. Biological Roles of Resolvins and Related Substances in the Resolution of Pain. BioMed Res. Int. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 2015, 7, a016311. [Google Scholar] [CrossRef]
- Chan, H.S.; Li, Y.; Dahoun, T.; Vogel, H.; Yuan, S. New Binding Sites, New Opportunities for GPCR Drug Discovery. Trends Biochem. Sci. 2019, 44, 312–330. [Google Scholar] [CrossRef]
- Fujino, H. Why PGD 2 has different functions from PGE 2. BioEssays 2021, 43, e2000213. [Google Scholar] [CrossRef] [PubMed]
- Abramovitz, M.; Adam, M.; Boie, Y.; Carrière, M.-C.; Denis, D.; Godbout, C.; Lamontagne, S.; Rochette, C.; Sawyer, N.; Tremblay, N.M.; et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2000, 1483, 285–293. [Google Scholar] [CrossRef]
- Al-Zoubi, R.; Morales, P.; Reggio, P.H. Structural Insights into CB1 Receptor Biased Signaling. Int. J. Mol. Sci. 2019, 20, 1837. [Google Scholar] [CrossRef]
- Sensken, S.-C.; Stäubert, C.; Keul, P.; Levkau, B.; Schöneberg, T.; Gräler, M.H. Selective activation of G alpha i mediated signalling of S1P3 by FTY720-phosphate. Cell. Signal. 2008, 20, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Amato, G.; Khan, N.S.; Maitra, R. A patent update on cannabinoid receptor 1 antagonists (2015–2018). Expert Opin. Ther. Patents 2019, 29, 261–269. [Google Scholar] [CrossRef]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.-M.; Ravetch, J.V. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Yonekura, K.; Kusumoto, S.; Choi, I.; Nakano, N.; Ito, A.; Suehiro, Y.; Imaizumi, Y.; Yoshimitsu, M.; Nosaka, K.; Ohtsuka, E.; et al. Mogamulizumab for adult T-cell leukemia-lymphoma: A multicenter prospective observational study. Blood Adv. 2020, 4, 5133–5145. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.C.; Elmes, J.B.; Shibu, P.A.; Larck, C.; Park, S.I. Mogamulizumab: An Anti-CC Chemokine Receptor 4 Antibody for T-Cell Lymphomas. Ann. Pharmacother. 2019, 54, 371–379. [Google Scholar] [CrossRef]
- Cancilla, D.; Rettig, M.P.; DiPersio, J.F. Targeting CXCR4 in AML and ALL. Front. Oncol. 2020, 10, 1672. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Barreto, C.A.V.; Baptista, S.J.; Preto, A.J.; Matos-Filipe, P.; Mourao, J.; Melo, R.; Moreira, I. Chapter Four—Prediction and targeting of GPCR oligomer interfaces. In Progress in Molecular Biology and Translational Science; Giraldo, J., Ciruela, F., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 105–149. [Google Scholar]
- Sadybekov, A.A.; Sadybekov, A.V.; Liu, Y.; Iliopoulos-Tsoutsouvas, C.; Huang, X.-P.; Pickett, J.; Houser, B.; Patel, N.; Tran, N.K.; Tong, F.; et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nat. Cell Biol. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.S.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.-P.; Savych, O.; Moroz, Y.S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nat. Cell Biol. 2020, 579, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.R.; Karpiak, J.; Huang, X.P.; Sassano, M.F.; Lyu, J.; Roth, B.L.; Shoichet, B.K. Selectivity Challenges in Docking Screens for GPCR Targets and Antitargets. J. Med. Chem. 2018, 61, 6830–6845. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.; Singh, I.; Levit, A.; Moroz, Y.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nat. Cell Biol. 2019, 566, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Gunera, J.; Baker, J.G.; van Hilten, N.; Rosenbaum, D.M.; Kolb, P. Structure-Based Discovery of Novel Ligands for the Orexin 2 Receptor. J. Med. Chem. 2020, 63, 11045–11053. [Google Scholar] [CrossRef]
- Zhang, Y.; Dijkman, P.M.; Zou, R.; Zandl-Lang, M.; Sanchez, R.M.; Eckhardt-Strelau, L.; Köfeler, H.; Vogel, H.; Yuan, S.; Kudryashev, M. Asymmetric opening of the homopentameric 5-HT3A serotonin receptor in lipid bilayers. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.-J. Structural basis of CXC chemokine receptor 2 activation and signalling. Nat. Cell Biol. 2020, 585, 135–140. [Google Scholar] [CrossRef]
- Jakowiecki, J.; Orzeł, U.; Chawananon, S.; Miszta, P.; Filipek, S. The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations. Molecules 2020, 25, 1930. [Google Scholar] [CrossRef] [PubMed]
- Zuzic, L.; Marzinek, J.K.; Warwicker, J.; Bond, P.J. A Benzene-Mapping Approach for Uncovering Cryptic Pockets in Membrane-Bound Proteins. J. Chem. Theory Comput. 2020, 16, 5948–5959. [Google Scholar] [CrossRef]
- Tan, Z.W.; Guarnera, E.; Tee, W.-V.; Berezovsky, I.N. AlloSigMA 2: Paving the way to designing allosteric effectors and to exploring allosteric effects of mutations. Nucleic Acids Res. 2020, 48, W116–W124. [Google Scholar] [CrossRef]
- Guarnera, E.; Berezovsky, I.N. Allosteric drugs and mutations: Chances, challenges, and necessity. Curr. Opin. Struct. Biol. 2020, 62, 149–157. [Google Scholar] [CrossRef]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Remington, J.M.; May, V.; Li, J. Molecular Basis of Class B GPCR Selectivity for the Neuropeptides PACAP and VIP. Front. Mol. Biosci. 2021, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Lovera, S.; Cuzzolin, A.; Kelm, S.; De Fabritiis, G.; Sands, Z.A. Reconstruction of apo A2A receptor activation pathways reveal ligand-competent intermediates and state-dependent cholesterol hotspots. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Abramyan, A.M.; Adhikari, P.; Keen, A.C.; Lee, K.-H.; Sanchez, J.; Verma, R.K.; Lim, H.D.; Yano, H.; A Javitch, J.; et al. Author response: Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism. eLife 2020, 9, e52189. [Google Scholar] [CrossRef]
- Troupiotis-Tsaïlaki, A.; Zachmann, J.J.; González-Gil, I.I.; Gonzalez, A.; Ortega-Gutiérrez, S.; Lopez-Rodriguez, M.L.; Pardo, L.; Govaerts, C. Ligand chain length drives activation of lipid G protein-coupled receptors. Sci. Rep. 2017, 7, 1–13. [Google Scholar]
- Jiang, Z.; Zhang, H. Molecular Mechanism of S1P Binding and Activation of the S1P1 Receptor. J. Chem. Inf. Model. 2019, 59, 4402–4412. [Google Scholar] [CrossRef]
- Schlick, T.; Portillo-Ledesma, S.; Myers, C.G.; Beljak, L.; Chen, J.; Dakhel, S.; Darling, D.; Ghosh, S.; Hall, J.; Jan, M.; et al. Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field. Annu. Rev. Biophys. 2021, 50, 267–301. [Google Scholar] [CrossRef]
- Lim, V.J.Y.; Du, W.; Chen, Y.Z.; Fan, H. A benchmarking study on virtual ligand screening against homology models of human GPCRs. Proteins: Struct. Funct. Bioinform. 2018, 86, 978–989. [Google Scholar] [CrossRef]
- Jaiteh, M.; Rodríguez-Espigares, I.; Selent, J.; Carlsson, J. Performance of virtual screening against GPCR homology models: Impact of template selection and treatment of binding site plasticity. PLoS Comput. Biol. 2020, 16, e1007680. [Google Scholar] [CrossRef]
- Nagasawa, T.; Horitani, M.; Kawaguchi, S.; Higashiyama, S.; Hama, Y.; Mitsutake, S. The molecular mechanism of phytosphingosine binding to FFAR4/GPR120 differs from that of other fatty acids. FEBS Open Bio 2021, 11, 3081–3089. [Google Scholar] [CrossRef]
- Kapla, J.; Rodríguez-Espigares, I.; Ballante, F.; Selent, J.; Carlsson, J. Can molecular dynamics simulations improve the structural accuracy and virtual screening performance of GPCR models? PLoS Comput. Biol. 2021, 17, e1008936. [Google Scholar] [CrossRef] [PubMed]
- Matricon, P.; Vo, D.D.; Gao, Z.-G.; Kihlberg, J.; Jacobson, K.A.; Carlsson, J. Fragment-based design of selective GPCR ligands guided by free energy simulations. Chem. Commun. 2021, 57, 12305–12308. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, F.; Kelemen, Á.; Baker, J.G.; Aranyodi, V.A.; Balzer, F.; Kolb, P.; Keserű, G.M. Fragment evolution for GPCRs: The role of secondary binding sites in optimization. Chem. Commun. 2021, 57, 10516–10519. [Google Scholar] [CrossRef] [PubMed]
- Scharf, M.; Bunemann, M.; Baker, J.G.; Kolb, P. Comparative Docking to Distinct G Protein–Coupled Receptor Conformations Exclusively Yields Ligands with Agonist Efficacy. Mol. Pharmacol. 2019, 96, 851–861. [Google Scholar] [CrossRef]
- Li, H.; Sze, K.-H.; Lu, G.; Ballester, P.J. Machine-learning scoring functions for structure-based virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 11, e1478. [Google Scholar] [CrossRef]
- Yin, Y.; Hu, H.; Yang, Z.; Xu, H.; Wu, J. RealVS: Toward Enhancing the Precision of Top Hits in Ligand-Based Virtual Screening of Drug Leads from Large Compound Databases. J. Chem. Inf. Model. 2021, 61, 4924–4939. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S. Automated discovery of GPCR bioactive ligands. Curr. Opin. Struct. Biol. 2019, 55, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wu, M.; Huang, Z.; An, J. Trends in application of advancing computational approaches in GPCR ligand discovery. Exp. Biol. Med. 2021, 246, 1011–1024. [Google Scholar] [CrossRef]
- Noé, F.; Tkatchenko, A.; Müller, K.-R.; Clementi, C. Machine Learning for Molecular Simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar] [CrossRef]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef]
- Xu, X.; Kaindl, J.; Clark, M.J.; Hübner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding pathway determines norepinephrine selectivity for the human beta1AR over beta2AR. Cell Res. 2021, 31, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Strachan, R.T.; Manglik, A.; Pani, B.; Kahsai, A.W.; Kim, T.H.; Wingler, L.M.; Ahn, S.; Chatterjee, A.; Masoudi, A.; et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 2016, 535, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, M.P.; Zou, Y.; Rasmussen, S.G.F.; Liu, C.W.; Nygaard, R.; Rosenbaum, D.M.; Fung, J.J.; Choi, H.-J.; Thian, F.S.; Kobilka, T.S.; et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 2010, 463, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Choi, H.-J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.P.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef]
- Liu, X.; Masoudi, A.; Kahsai, A.W.; Huang, L.-Y.; Pani, B.; Staus, D.P.; Shim, P.J.; Hirata, K.; Simhal, R.K.; Schwalb, A.M.; et al. Mechanism of beta2AR regulation by an intracellular positive allosteric modulator. Science 2019, 364, 1283–1287. [Google Scholar] [CrossRef]
- Weichert, D.; Kruse, A.C.; Manglik, A.; Hiller, C.; Hiller, C.; Hübner, H.; Kobilka, B.K.; Gmeiner, P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA 2014, 111, 10744–10748. [Google Scholar] [CrossRef]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar]
- Rasmussen, S.G.F.; Choi, H.-J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef]
- Masureel, M.; Zou, Y.; Picard, L.-P.; Westhuizen, E.V.D.; Mahoney, J.P.; Rodrigues, J.P.G.L.M.; Mildorf, T.J.; Dror, R.O.; Shaw, D.E.; Bouvier, M.; et al. Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat. Chem. Biol. 2018, 14, 1059–1066. [Google Scholar] [CrossRef]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 2019, 176, 479–490.e12. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, M.-C.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Shiimura, Y.; Horita, S.; Hamamoto, A.; Asada, H.; Hirata, K.; Tanaka, M.; Mori, K.; Uemura, T.; Kobayashi, T.; Iwata, S.; et al. Structure of an antagonist-bound ghrelin receptor reveals possible ghrelin recognition mode. Nat. Commun. 2020, 11, 4160. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; English, J.; Krumm, B.E.; Kim, K.; Pardon, E.; Olsen, R.H.J.; Wang, S.; Zhang, S.; Diberto, J.F.; Sciaky, N.; et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat. Commun. 2020, 11, 1145. [Google Scholar] [CrossRef] [PubMed]
- Ishchenko, A.; Wacker, D.; Kapoor, M.; Zhang, A.; Han, G.W.; Basu, S.; Patel, N.; Messerschmidt, M.; Weierstall, U.; Liu, W.; et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc. Natl. Acad. Sci. USA 2017, 114, 8223–8228. [Google Scholar] [CrossRef]
- Asada, H.; Horita, S.; Hirata, K.; Shiroishi, M.; Shiimura, Y.; Iwanari, H.; Hamakubo, T.; Shimamura, T.; Nomura, N.; Kusano-Arai, O.; et al. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 2018, 25, 570–576. [Google Scholar] [CrossRef]
- Asada, H.; Inoue, A.; Kadji, F.M.N.; Hirata, K.; Shiimura, Y.; Im, D.; Shimamura, T.; Nomura, N.; Iwanari, H.; Hamakubo, T.; et al. The Crystal Structure of Angiotensin II Type 2 Receptor with Endogenous Peptide Hormone. Structure 2020, 28, 418–425.e4. [Google Scholar] [CrossRef]
- Im, D.; Inoue, A.; Fujiwara, T.; Nakane, T.; Yamanaka, Y.; Uemura, T.; Mori, C.; Shiimura, Y.; Kimura, K.T.; Asada, H.; et al. Structure of the dopamine D2 receptor in complex with the antipsychotic drug spiperone. Nat. Commun. 2020, 11, 6442. [Google Scholar] [CrossRef]
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.M.; Dumelin, C.E.; Doré, A.S.; et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112–115. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cognate Receptor Name; Gene Name and Uniprot ID | Protein Engineering/Modification | Methodology | Structures | |
|---|---|---|---|---|
| Prostanoids | Prostaglandin D2 receptor 2 (DP2) #; PTGDR2; Q9Y5Y4 | T4-lysozyme (mT4L) with 8-aa linker insert in ICL3. | X-ray crystallography (XRD) | Antagonists fevipiprant [6D26 Inactive; 2.80 Å], CAY10471 [6D27 Inactive; 2.74 Å] [31] |
| T4-lysozyme (mT4L) with 8-aa linker insert in ICL3. | Serial femtosecond crystallography (SFX) with X-ray free electron laser (XFEL) | Agonist 15m-PGD2 [7M8W Inactive; 2.61 A] [32] | ||
| Prostaglandin E2 receptor EP2 subtype; PTGER2; P43116 | No modifications. | Cryo-electron microscopy (cryo-EM) | Endogenous agonist PGE2 + G-protein Gs [7CX2 Active; 2.80 Å], synthetic agonists taprenepag + Gs [7CX3 Active; 2.80 Å], evatanepag (CP-533536) + Gs [7CX4 Active; 2.90 Å] [33] | |
| Prostaglandin E2 receptor EP3 subtype; PTGER3; P43115 | Thermostabilized apocytochrome b562RIL (bRIL) insert in ICL3; N- and C-terminal truncation; four thermostabilizing mutations | Lipidic cubic phase crystallization (LCP); XRD | Agonist PGE2 [6AK3 Active; 2.90 Å] [34] | |
| T4-lysozyme insertion in ICL3; C-terminal truncation. | LCP; XFEL | Agonist misoprostol [6M9T Active; 2.5 Å] [35] | ||
| Prostaglandin E2 receptor EP4 subtype; PTGER4; P35408 | Stabilizing anti-human EP4 antibody (IgG#001); removal of N-glycosylation site; ICL3, N- and C-terminal truncation; two thermostabilizing point mutations | LCP; XRD | Antagonists ONO-9990614 [5YHL Inactive; 4.20 Å], ONO-AE3-208 [5YWY Inactive; 3.20 Å] [36]; | |
| Gs-stabilizing nanobody Nb35 | Cryo-EM | Agonist PGE2 + Gs + nanobody Nb35 [7D7M Active; 3.30 Å] [37] | ||
| Thromboxane A2 receptor; TBXA2R; P21731 | Thermostabilized b562RIL (bRIL) insert in N-terminal [6IIV]; rubredoxin insert in ICL3 [6IIV]; C-terminus truncation; one thermostabilizing point mutation | LCP; XRD | Antagonists ramatroban [6IIU Intermediate; 2.50 Å] dalotroban [6IIV Intermediate, 3.00 Å] [38] | |
| Platelet-activating factor | Platelet-activating factor receptor; PTAFR; P25105 | Flavodoxin insert [5ZKP] and T4-lysozyme [5ZKQ] insert in ICL3; | LCP; XRD | Antagonists SR 27417 [5ZKP Other; 2.81 Å] and BT-491 [5ZKQ Intermediate; 2.90 Å] [39] |
| Lysophospholipids | Sphingosine 1-phosphate receptor 1; S1PR1; P21453 | T4-lysozyme insert in ICL3 | XRD [3V2W]; microdiffraction [3V2Y] | Antagonist ML056 [3V2W Inactive, 3.35 Å; 3V2Y Inactive, 2.80 Å] [40] |
| Sphingosine 1-phosphate receptor 3; S1PR3; Q99500 | C-terminus truncation; removal of N-glycosylation site; stabilizing Fab antibody fragment (Fab AS55) | LCP; XRD | Natural agonist d18:1 S1P + Fab AS55 [7C4S Active; 3.2 Å] [41] | |
| Lysophosphatidic acid receptor 1; LPAR1; Q92633 | bRIL insert in ICL3; C-terminus truncation; engineered disulfide bridges [4Z36]; stabilizing antagonists | XRD | Selective antagonists ONO-9780307 [4Z34 Inactive; 3.0 Å], ONO-9910539 [4Z35 Inactive; 2.90 Å], ONO-3080573 [4Z36 Inactive; 2.90 Å] [42] | |
| Lysophosphatidic acid receptor 6; drlpar6a; Q08BG4 | T4-lysozyme insert in ICL3 | LCP; XRD | Apo state [5XSZ Intermediate; 3.20 Å] [43] | |
| Leukotrienes | Leukotriene B4 receptor 1; cpLTB4R; Q9WTK1 | T4-lysozyme insert in ICL3; N-terminus truncation; thermostabilizing point mutations | LCP; XRD | Antagonist BIIL260 [5X33 Inactive; 3.70 Å] [44] |
| Leukotriene B4 receptor 1; LTB4R; Q15722 | Flavodoxin insert in ICL3; N- and C-termini truncation; thermostabilizing point mutations | LCP; XRD | Antagonist ML-D-046 [7K15 Inactive; 2.88 Å] [45] | |
| Cysteinyl leukotriene receptor 1; CYSLTR1; Q9Y271 | Thermostabilized b562RIL (bRIL) insert in ICL3; C-terminal truncation. | LCP; SFX with XFEL | Antagonists pranlukast [6RZ4 Intermediate; 2.70 Å] and zafirlukast [6RZ5 Intermediate; 2.53 Å] [46] | |
| Cysteinyl leukotriene receptor 2; CYSLTR2; Q9NS75 | Thermostabilized b562RIL (bRIL) insert in ICL3; stabilizing mutations; N- and C-termini truncation | LCP; XRD | Antagonists ONO-2570366 [6RZ6 Intermediate, 6RZ7 Intermediate; 2.43 Å], ONO-2080365 [6RZ8 Intermediate; 2.70 Å], and ONO-2770372 [6RZ9 Intermediate; 2.73 Å] [47] | |
| Free fatty acids | Free fatty acid receptor 1 (GRP40); O14842; FFAR1 | T4-lysozyme insert in ICL3 | LCP; XRD | Allosteric partial agonist TAK-875 [4PHU Intermediate; 2.30 Å] [48] |
| T4-lysozyme insert in ICL3; three thermostabilizing mutations; | LCP; XRD | AgoPAM AP8 and partial agonist MK-8666 [5TZY Inactive; 3.22 Å], MK-8666 [5TZR Intermediate; 2.20 Å] [49]; | ||
| T4-lysozyme insert in ICL3 | LCP; XRD | Full agonist “compound 1” [5KW2 Intermediate; 2.76 Å] [50] | ||
| Cannabinoids | Cannabinoid receptor 1 (CB1); CNR1; P21554 | Flavodoxin insert in ICL3; stabilizing antagonist; N- and C-termini truncation; four thermostabilizing mutations | LCP; XRD | Antagonist AM6538 [5TGZ Inactive; 2.80 Å] [51] |
| Thermostable P. abyssi glycogen synthase (PGS) domain insert in ICL3; N- and C-termini truncation; one thermostabilizing mutation | LCP; XRD | Inverse agonist taranabant [5U09 Inactive; 2.6 Å] [52] | ||
| Flavodoxin insert in ICL3; N- and C-termini truncation; four thermostabilizing mutations; stabilizing agonists | LCP; XRD | Agonists AM11542 [5XRA Active; 2.80 Å], AM841 [5XR8 Active; 2.95 Å] [53] | ||
| Stabilizing single-chain variable fragment scFv16 | Single-particle cryo-EM | MDMB-Fubinaca (FUB) + Gi + scFv16 [6N4B Active; 3.0 Å] [54]; | ||
| Five stabilizing mutations | LCP; XRD | NAM ORG27569 [6KQI Inactive; 3.245 Å] [55]; | ||
| BRIL insert in N-terminus; ; CB1-Gi stabilized by svFv16 | Single-particle cryo-EM | Agonist AM841 + Gi + svFc16 [6KPG Active; 3.00 Å] [56] | ||
| Cannabinoid receptor 2 (CB2); CNR2; P34972 | Rationally designed stabilizing antagonist; T4-lysozyme insert in ICL3 | LCP; XRD | Antagonist AM10257 [5ZTY Inactive; 2.80 Å] [57]; | |
| CB2-Gi stabilized by svFv16 | Cryo-EM | Agonist WIN 55,212-2 + Gi + svFv16 [6PT0 Active, 3.2 Å] [58] | ||
| BRIL insert in N-terminus; CB2-Gi stabilized by svFv16 | X-ray [6KPC]; Single-particle cryo-EM | Agonist AM12033 [6KPC Active; 3.20 Å], Agonist AM12033 + Gi + svFc16 [6KPF Inactive; 2.90 Å] [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishna Deepak, R.N.V.; Verma, R.K.; Hartono, Y.D.; Yew, W.S.; Fan, H. Recent Advances in Structure, Function, and Pharmacology of Class A Lipid GPCRs: Opportunities and Challenges for Drug Discovery. Pharmaceuticals 2022, 15, 12. https://doi.org/10.3390/ph15010012
Krishna Deepak RNV, Verma RK, Hartono YD, Yew WS, Fan H. Recent Advances in Structure, Function, and Pharmacology of Class A Lipid GPCRs: Opportunities and Challenges for Drug Discovery. Pharmaceuticals. 2022; 15(1):12. https://doi.org/10.3390/ph15010012
Chicago/Turabian StyleKrishna Deepak, R. N. V., Ravi Kumar Verma, Yossa Dwi Hartono, Wen Shan Yew, and Hao Fan. 2022. "Recent Advances in Structure, Function, and Pharmacology of Class A Lipid GPCRs: Opportunities and Challenges for Drug Discovery" Pharmaceuticals 15, no. 1: 12. https://doi.org/10.3390/ph15010012
APA StyleKrishna Deepak, R. N. V., Verma, R. K., Hartono, Y. D., Yew, W. S., & Fan, H. (2022). Recent Advances in Structure, Function, and Pharmacology of Class A Lipid GPCRs: Opportunities and Challenges for Drug Discovery. Pharmaceuticals, 15(1), 12. https://doi.org/10.3390/ph15010012