
Targeting the Bromodomain of BRG-1/BRM Subunit of the SWI/SNF Complex Increases the Anticancer Activity of Temozolomide in Glioblastoma

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
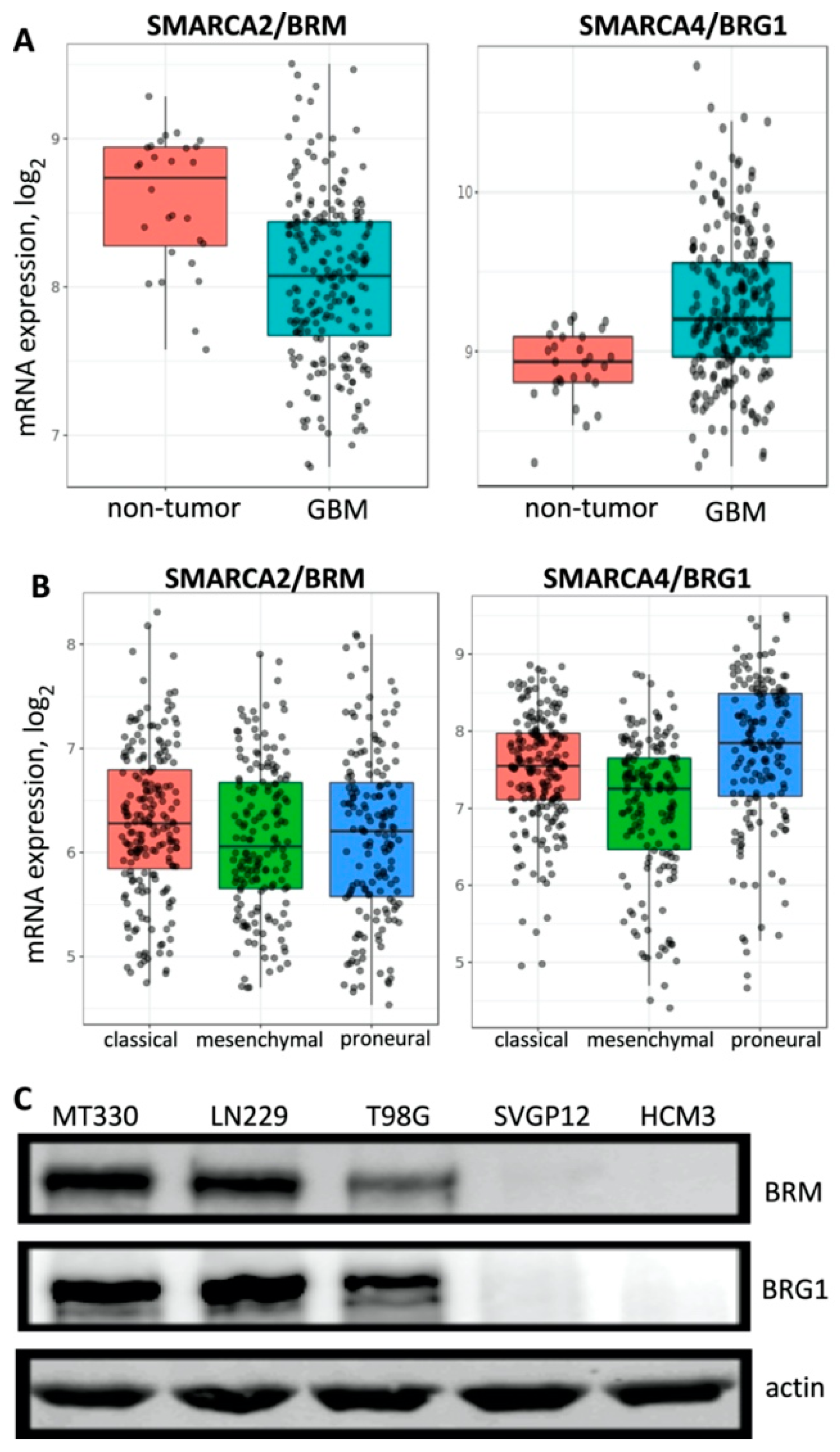
2.1. BRG1 Is Highly Expressed in GBM Tumor Tissue and in GBM Cells
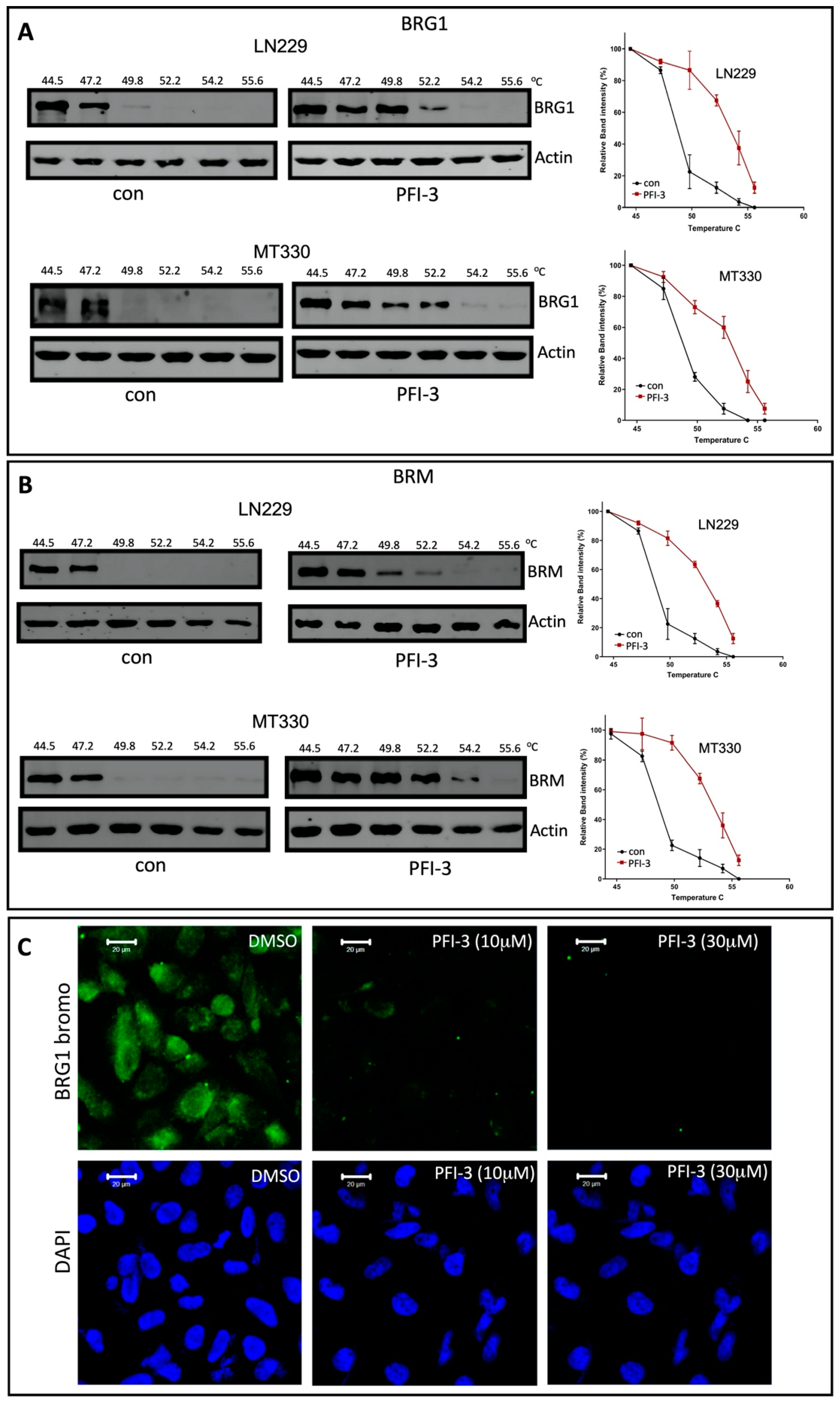
2.2. PFI-3 Binds to the Bromodomains of BRG1 and BRM
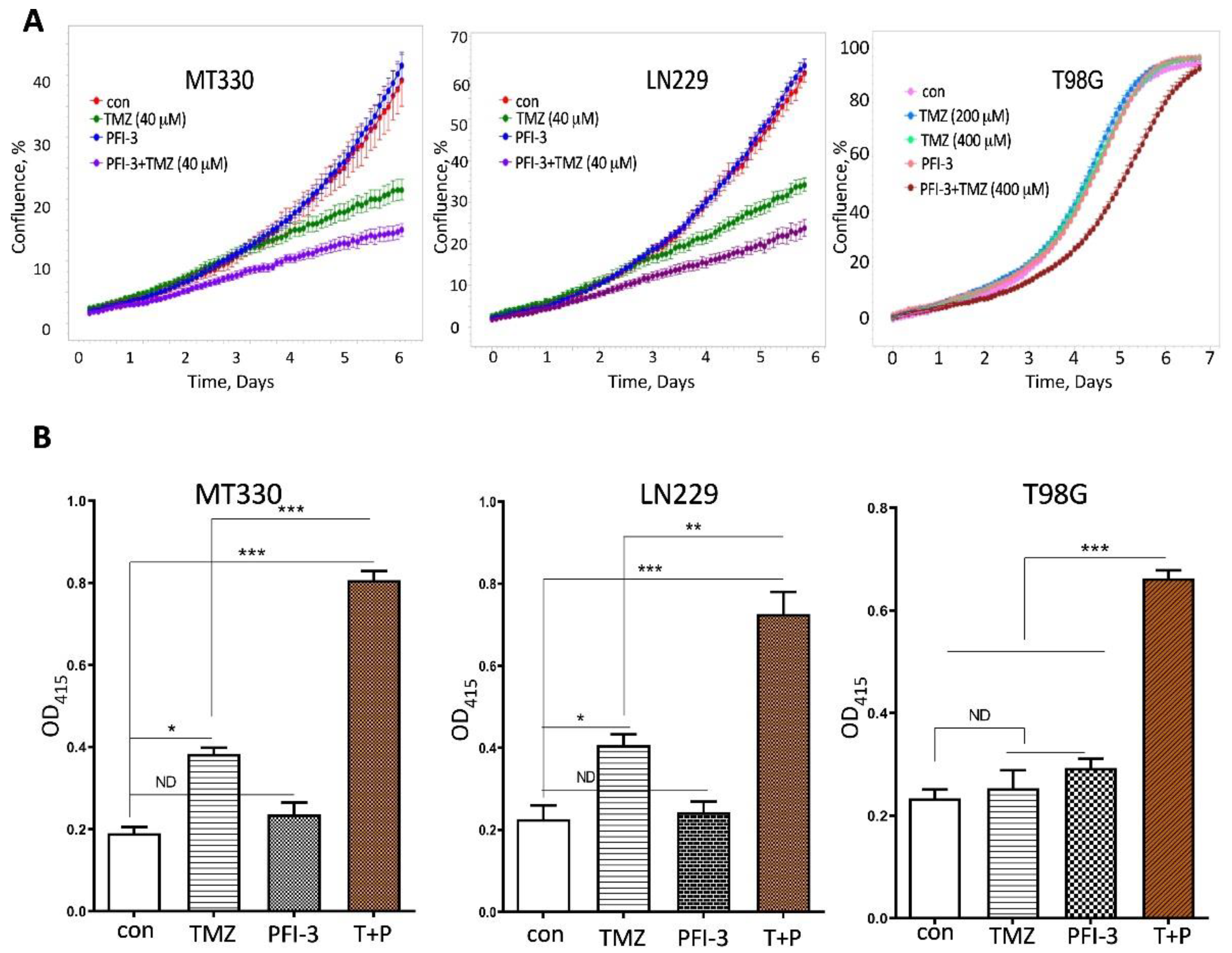
2.3. PFI-3 Treatment Increases TMZ-Sensitivity of GBM Cells
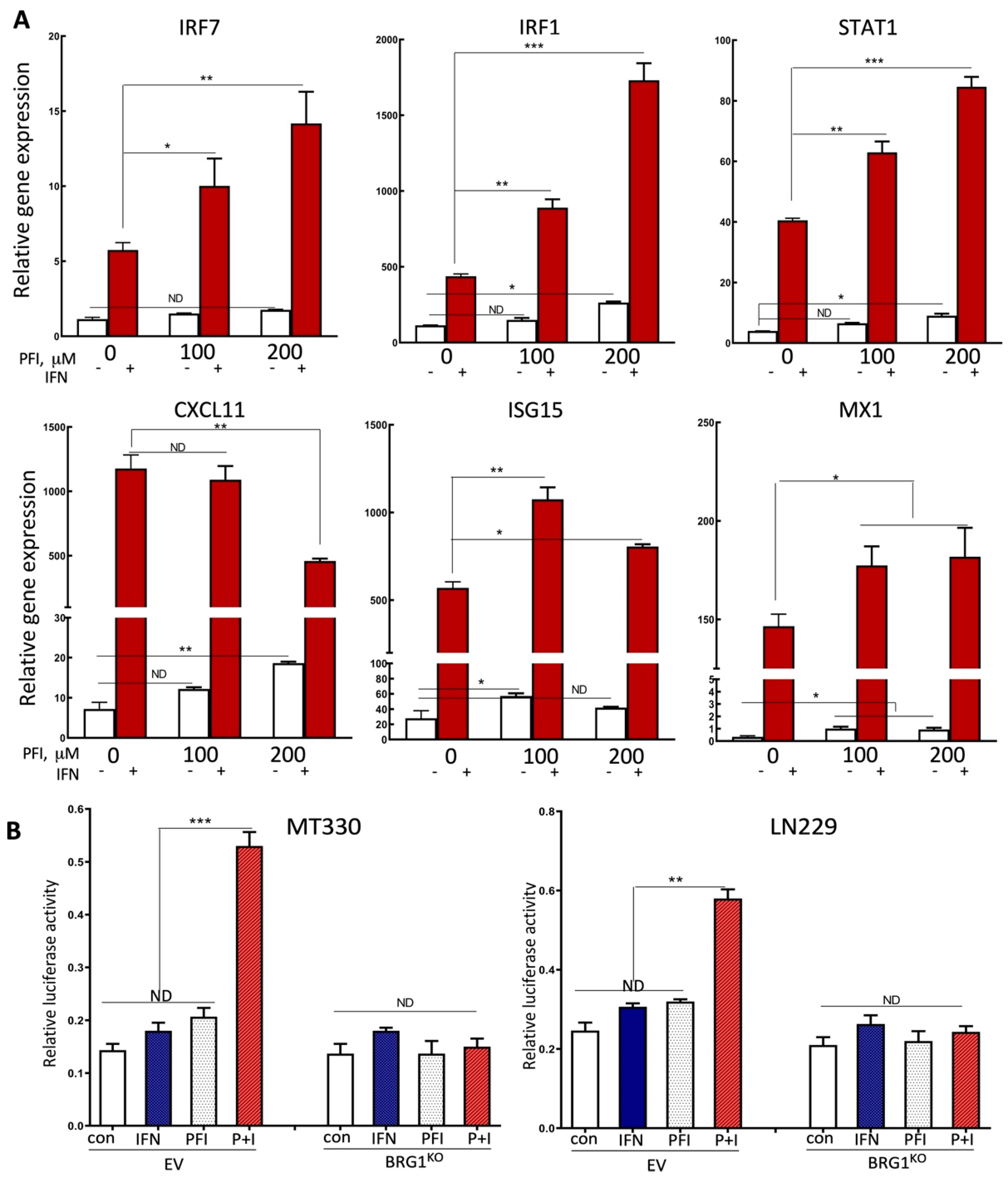
2.4. PFI-3 Regulates IFN-Induced Gene Expression in GBM Cells
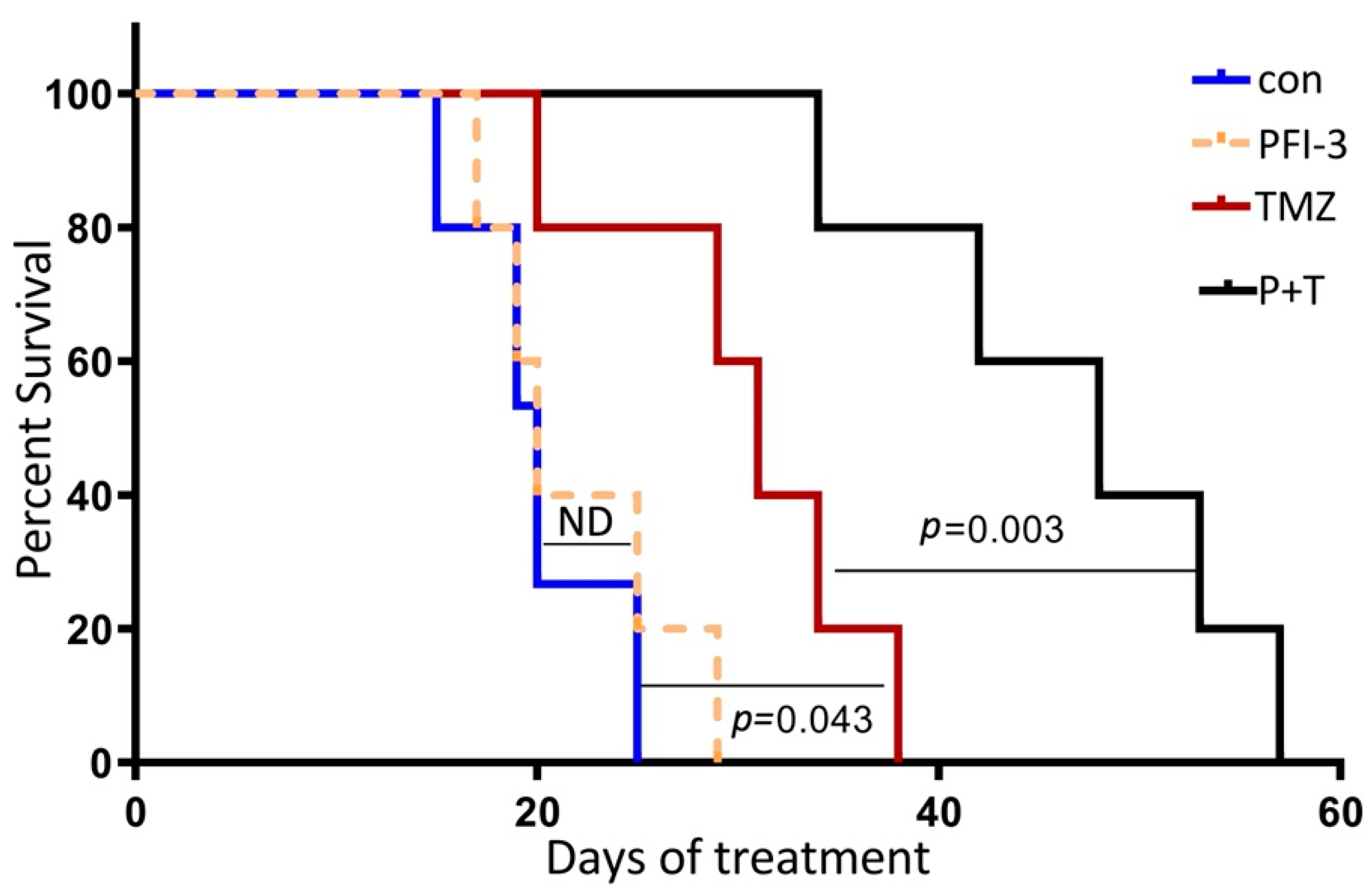
2.5. PFI-3 Enhances the Anticancer Activity of TMZ on GBM Tumorigenesis
3. Discussion
4. Materials and Methods
4.1. Biological and Chemical Reagents, and Cell Culture
4.2. Repository of Molecular Brain Neoplasia Data (REMBRANDT) Query
4.3. Immunoblot Analysis
4.4. Analysis of Binding to the Bromodomains of BRG1 and BRM
4.5. Cell Proliferation and Cell Death ELISA Assays
4.6. Gene Expression Analysis
4.7. IRF-1-Dependent Luciferase Reporter Assays
4.8. Tumor Xenograft
4.9. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Surawicz, T.S.; Davis, F.; Freels, S.; Laws, E.R., Jr.; Menck, H.R. Brain tumor survival: Results from the National Cancer Data Base. J. Neuro-Oncol. 1998, 40, 151–160. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Shain, A.H.; Pollack, J.R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef]
- Le Loarer, F.; Watson, S.; Pierron, G.; de Montpreville, V.T.; Ballet, S.; Firmin, N.; Auguste, A.; Pissaloux, D.; Boyault, S.; Paindavoine, S.; et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat. Genet. 2015, 47, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Hodges, H.C.; Stanton, B.Z.; Cermakova, K.; Chang, C.Y.; Miller, E.L.; Kirkland, J.G.; Ku, W.L.; Veverka, V.; Zhao, K.; Crabtree, G.R. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 2018, 25, 61–72. [Google Scholar] [CrossRef]
- Ganguly, D.; Sims, M.; Cai, C.; Fan, M.; Pfeffer, L.M. Chromatin Remodeling Factor BRG1 Regulates Stemness and Chemosensitivity of Glioma Initiating Cells. Stem Cells 2018, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yang, C.H.; Schultz, A.P.; Sims, M.M.; Miller, D.D.; Pfeffer, L.M. Brahma-Related Gene-1 (BRG1) promotes the malignant phenotype of glioblastoma cells. J. Cell. Mol. Med. 2021, 25, 2956–2966. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Castex, J.; Tallant, C.; Owen, D.R.; Martin, S.; Aldeghi, M.; Monteiro, O.; Filippakopoulos, P.; Picaud, S.; Trzupek, J.D.; et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci. Adv. 2015, 1, e1500723. [Google Scholar] [CrossRef] [Green Version]
- Wanior, M.; Preuss, F.; Ni, X.; Kramer, A.; Mathea, S.; Gobel, T.; Heidenreich, D.; Simonyi, S.; Kahnt, A.S.; Joerger, A.C.; et al. Pan-SMARCA/PB1 Bromodomain Inhibitors and Their Role in Regulating Adipogenesis. J. Med. Chem. 2020, 63, 14680–14699. [Google Scholar] [CrossRef]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Mobitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef] [PubMed]
- Rago, F.; Elliott, G.; Li, A.; Sprouffske, K.; Kerr, G.; Desplat, A.; Abramowski, D.; Chen, J.T.; Farsidjani, A.; Xiang, K.X.; et al. The Discovery of SWI/SNF Chromatin Remodeling Activity as a Novel and Targetable Dependency in Uveal Melanoma. Mol. Cancer Ther. 2020, 19, 2186–2195. [Google Scholar] [CrossRef]
- Vangamudi, B.; Paul, T.A.; Shah, P.K.; Kost-Alimova, M.; Nottebaum, L.; Shi, X.; Zhan, Y.; Leo, E.; Mahadeshwar, H.S.; Protopopov, A.; et al. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015, 75, 3865–3878. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundback, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Trotter, K.W.; Archer, T.K. The BRG1 transcriptional coregulator. Nucl. Recept Signal. 2008, 6, e004. [Google Scholar] [CrossRef] [Green Version]
- Tolstorukov, M.Y.; Sansam, C.G.; Lu, P.; Koellhoffer, E.C.; Helming, K.C.; Alver, B.H.; Tillman, E.J.; Evans, J.A.; Wilson, B.G.; Park, P.J.; et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. USA 2013, 110, 10165–10170. [Google Scholar] [CrossRef] [Green Version]
- Bourgo, R.J.; Siddiqui, H.; Fox, S.; Solomon, D.; Sansam, C.G.; Yaniv, M.; Muchardt, C.; Metzger, D.; Chambon, P.; Roberts, C.W.; et al. SWI/SNF deficiency results in aberrant chromatin organization, mitotic failure, and diminished proliferative capacity. Mol. Biol. Cell 2009, 20, 3192–3199. [Google Scholar] [CrossRef]
- Flanagan, J.F.; Peterson, C.L. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999, 27, 2022–2028. [Google Scholar] [CrossRef] [Green Version]
- Muchardt, C.; Yaniv, M. ATP-dependent chromatin remodelling: SWI/SNF and Co. are on the job. J. Mol. Biol. 1999, 293, 187–198. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Bultman, S.J.; Gebuhr, T.C.; Magnuson, T. A Brg1 mutation that uncouples ATPase activity from chromatin remodeling reveals an essential role for SWI/SNF-related complexes in beta-globin expression and erythroid development. Gene Dev. 2005, 19, 2849–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.; Ronan, J.L.; Wu, J.; Staahl, B.T.; Chen, L.; Kuo, A.; Lessard, J.; Nesvizhskii, A.I.; Ranish, J.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. USA 2009, 106, 5181–5186. [Google Scholar] [CrossRef] [Green Version]
- Gerstenberger, B.S.; Trzupek, J.D.; Tallant, C.; Fedorov, O.; Filippakopoulos, P.; Brennan, P.E.; Fedele, V.; Martin, S.; Picaud, S.; Rogers, C.; et al. Identification of a Chemical Probe for Family VIII Bromodomains through Optimization of a Fragment Hit. J. Med. Chem. 2016, 59, 4800–4811. [Google Scholar] [CrossRef] [Green Version]
- Garner, J.M.; Fan, M.; Yang, C.H.; Du, Z.; Sims, M.; Davidoff, A.M.; Pfeffer, L.M. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J. Biol. Chem. 2013, 288, 26167–26176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.H.; Yue, J.; Pfeffer, S.R.; Fan, M.; Paulus, E.; Hosni-Ahmed, A.; Sims, M.; Qayyum, S.; Davidoff, A.M.; Handorf, C.R.; et al. MicroRNA-21 promotes glioblastoma tumorigenesis by down-regulating insulin-like growth factor-binding protein-3 (IGFBP3). J. Biol. Chem. 2014, 289, 25079–25087. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Castro, S.; Brasier, A.R.; Jamaluddin, M.; Garofalo, R.P.; Casola, A. Reactive oxygen species mediate virus-induced STAT activation: Role of tyrosine phosphatases. J. Biol. Chem. 2004, 279, 2461–2469. [Google Scholar] [CrossRef] [Green Version]
- Garner, J.M.; Ellison, D.W.; Finkelstein, D.; Ganguly, D.; Du, Z.; Sims, M.; Yang, C.H.; Interiano, R.B.; Davidoff, A.M.; Pfeffer, L.M. Molecular heterogeneity in a patient-derived glioblastoma xenoline is regulated by different cancer stem cell populations. PLoS ONE 2015, 10, e0125838. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Sims, M.; Pfeffer, L.M. The curcumin analog EF24 targets NF-kappaB and miRNA-21, and has potent anticancer activity in vitro and in vivo. PLoS ONE 2013, 8, e71130. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Wang, Y.; Sims, M.M.; He, Y.; Miller, D.D.; Pfeffer, L.M. Targeting the Bromodomain of BRG-1/BRM Subunit of the SWI/SNF Complex Increases the Anticancer Activity of Temozolomide in Glioblastoma. Pharmaceuticals 2021, 14, 904. https://doi.org/10.3390/ph14090904
Yang C, Wang Y, Sims MM, He Y, Miller DD, Pfeffer LM. Targeting the Bromodomain of BRG-1/BRM Subunit of the SWI/SNF Complex Increases the Anticancer Activity of Temozolomide in Glioblastoma. Pharmaceuticals. 2021; 14(9):904. https://doi.org/10.3390/ph14090904
Chicago/Turabian StyleYang, Chuanhe, Yinan Wang, Michelle M. Sims, Yali He, Duane D. Miller, and Lawrence M. Pfeffer. 2021. "Targeting the Bromodomain of BRG-1/BRM Subunit of the SWI/SNF Complex Increases the Anticancer Activity of Temozolomide in Glioblastoma" Pharmaceuticals 14, no. 9: 904. https://doi.org/10.3390/ph14090904
APA StyleYang, C., Wang, Y., Sims, M. M., He, Y., Miller, D. D., & Pfeffer, L. M. (2021). Targeting the Bromodomain of BRG-1/BRM Subunit of the SWI/SNF Complex Increases the Anticancer Activity of Temozolomide in Glioblastoma. Pharmaceuticals, 14(9), 904. https://doi.org/10.3390/ph14090904