
Naproxen Based 1,3,4-Oxadiazole Derivatives as EGFR Inhibitors: Design, Synthesis, Anticancer, and Computational Studies

 , , ,
, , ,  , ,
, ,
Abstract
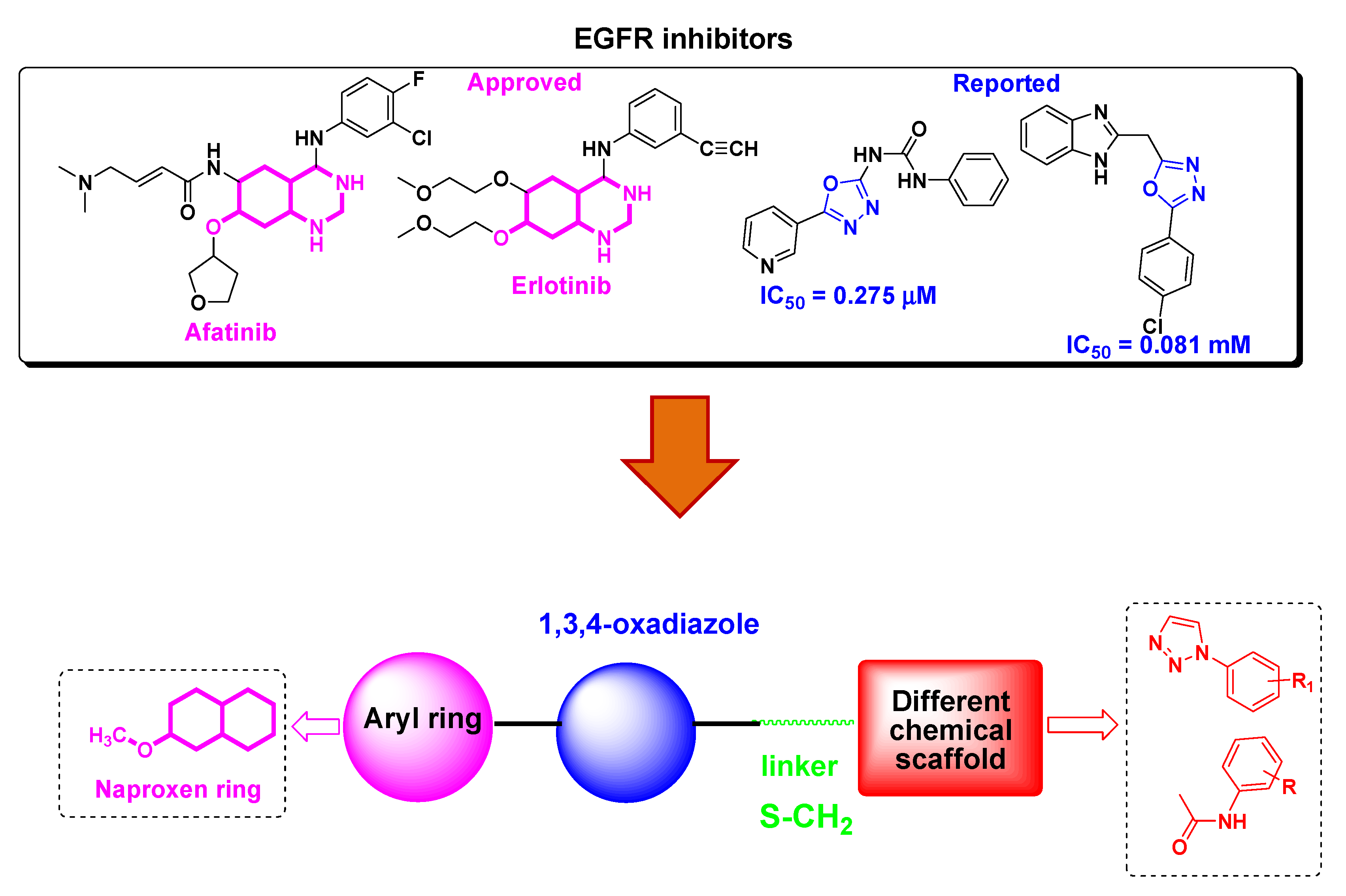
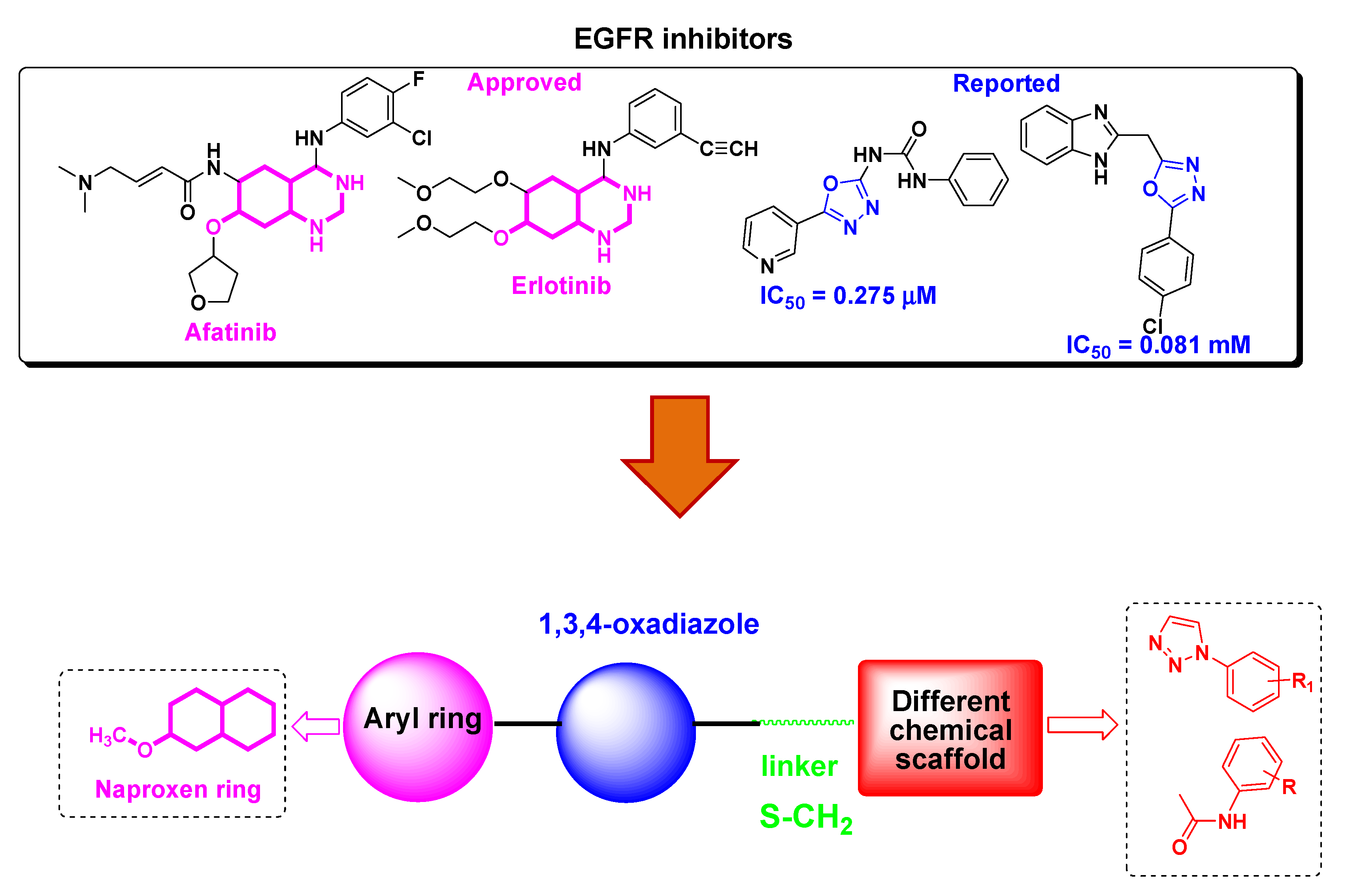
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General
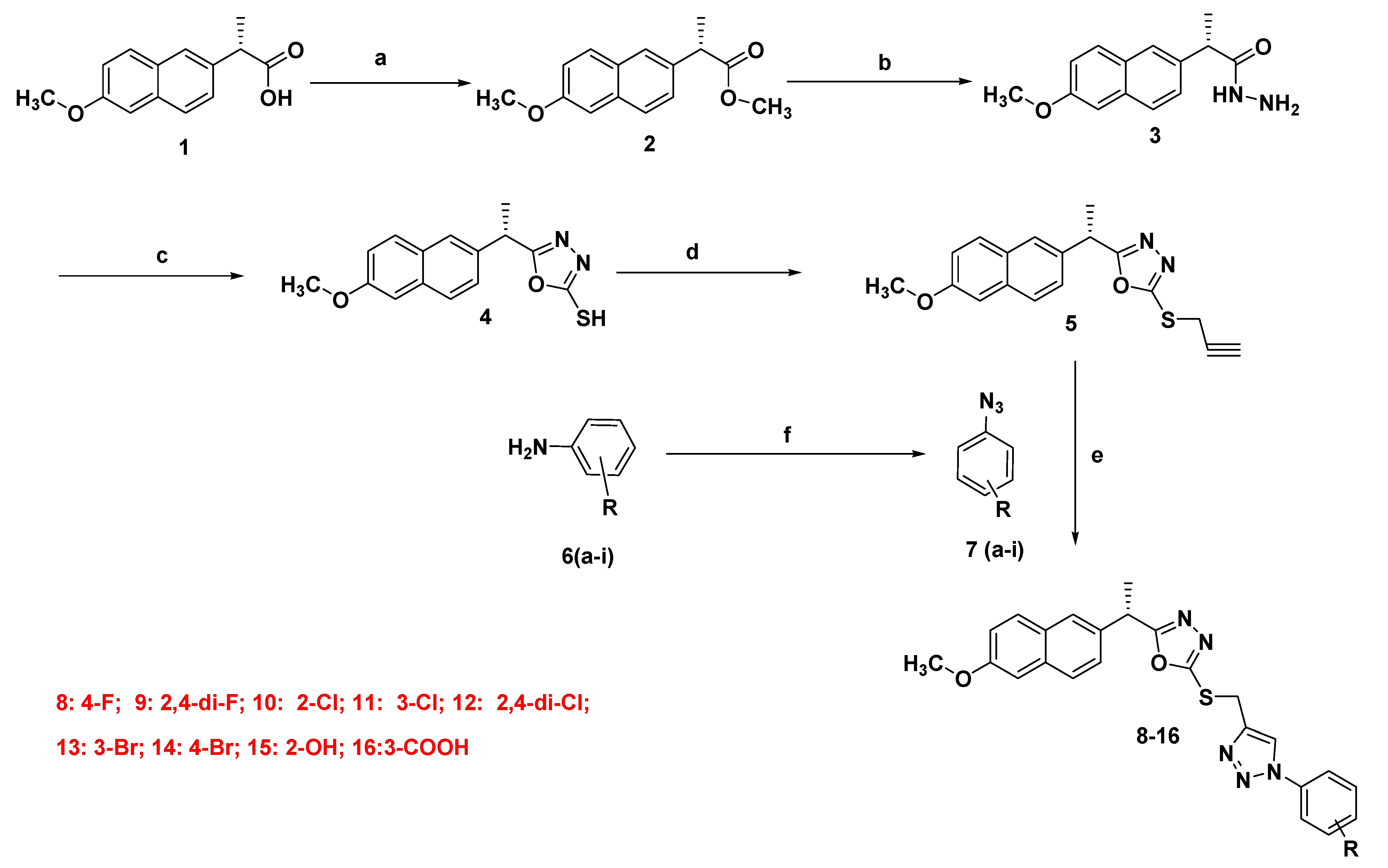
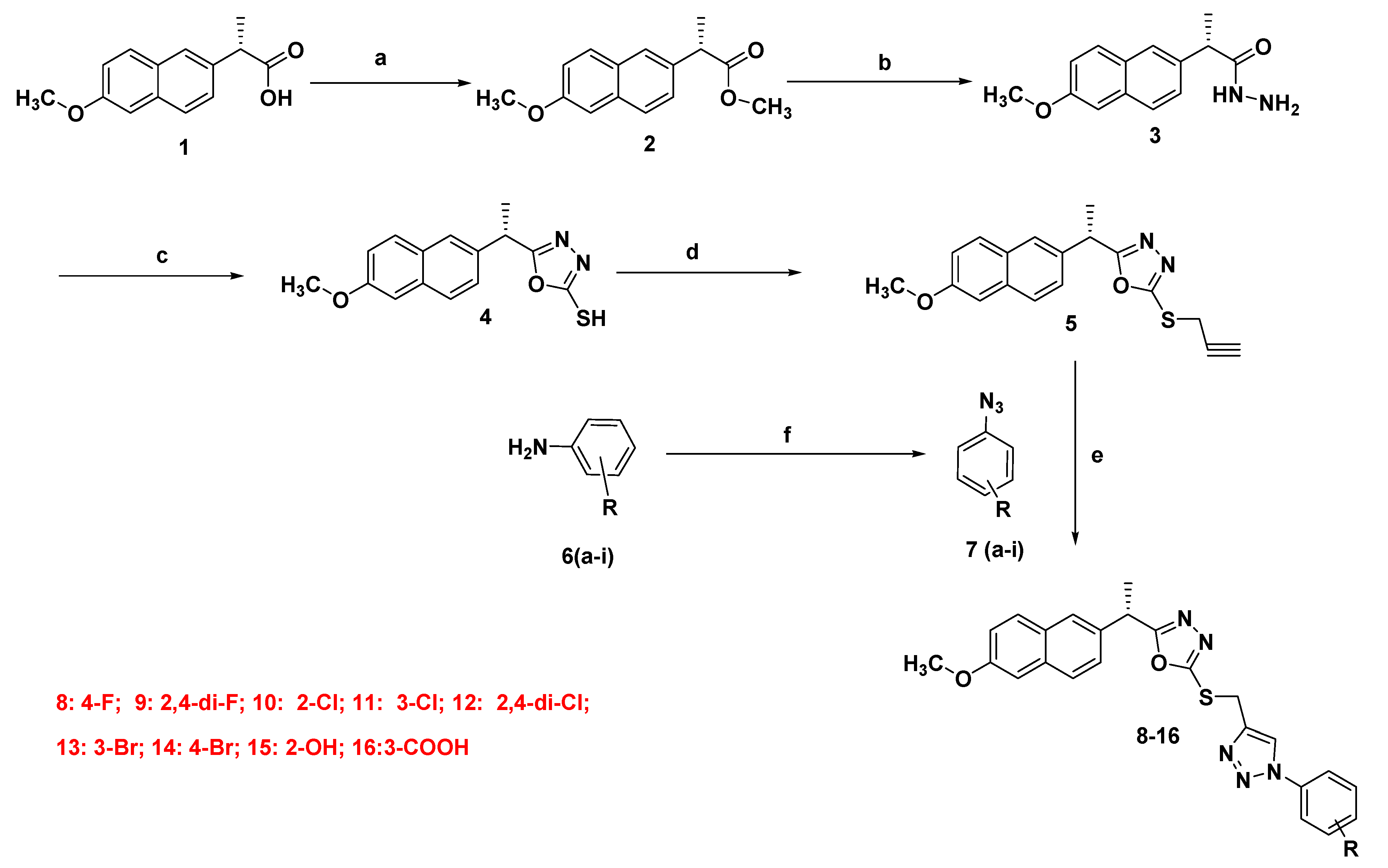
2.1.2. Synthesis of Final Compounds (8–16)
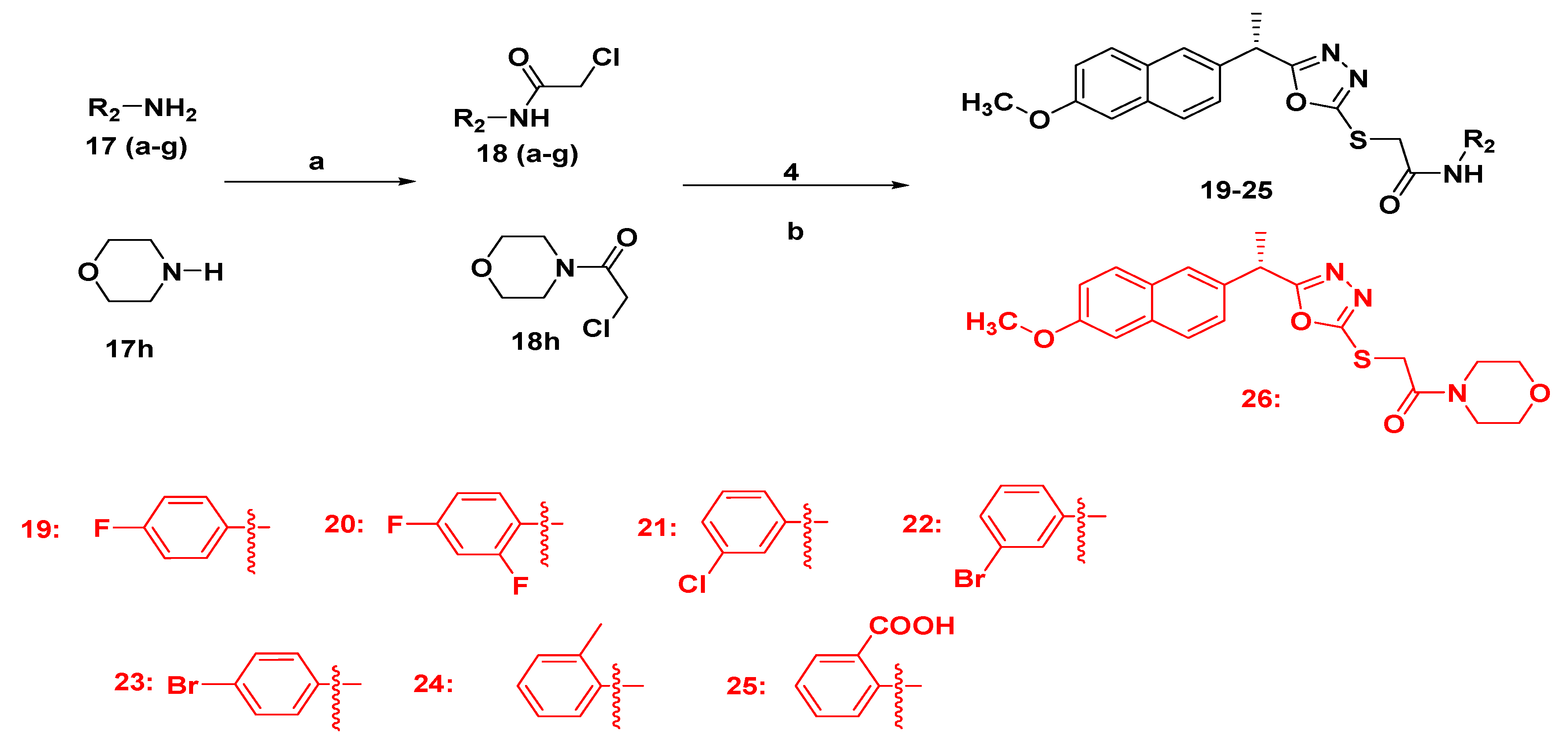
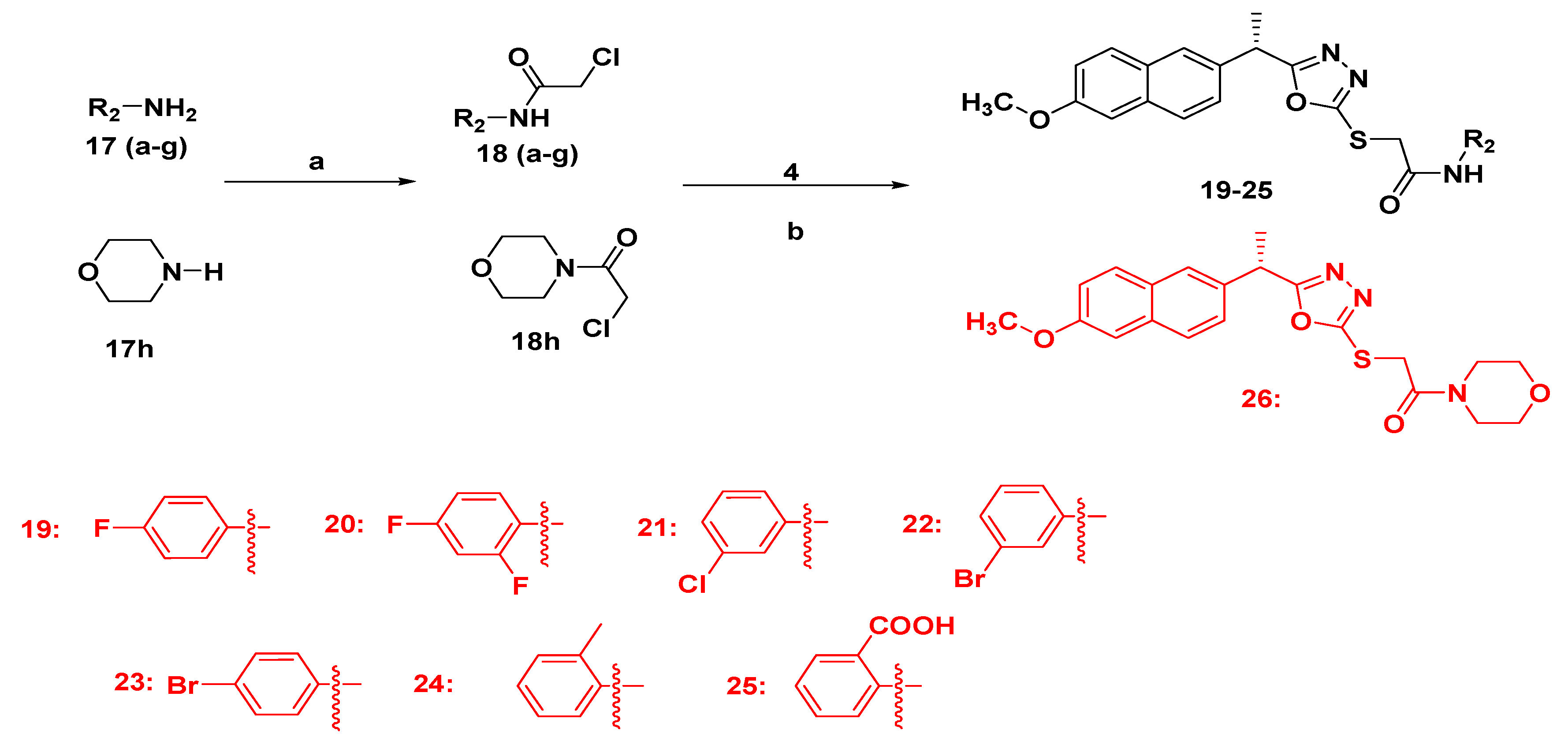
2.1.3. Synthesis of Final Compounds Bearing Acetamide (19–26)
2.1.4. Analytical Data
2.2. Biological Activities
2.2.1. Antiproliferative Activity
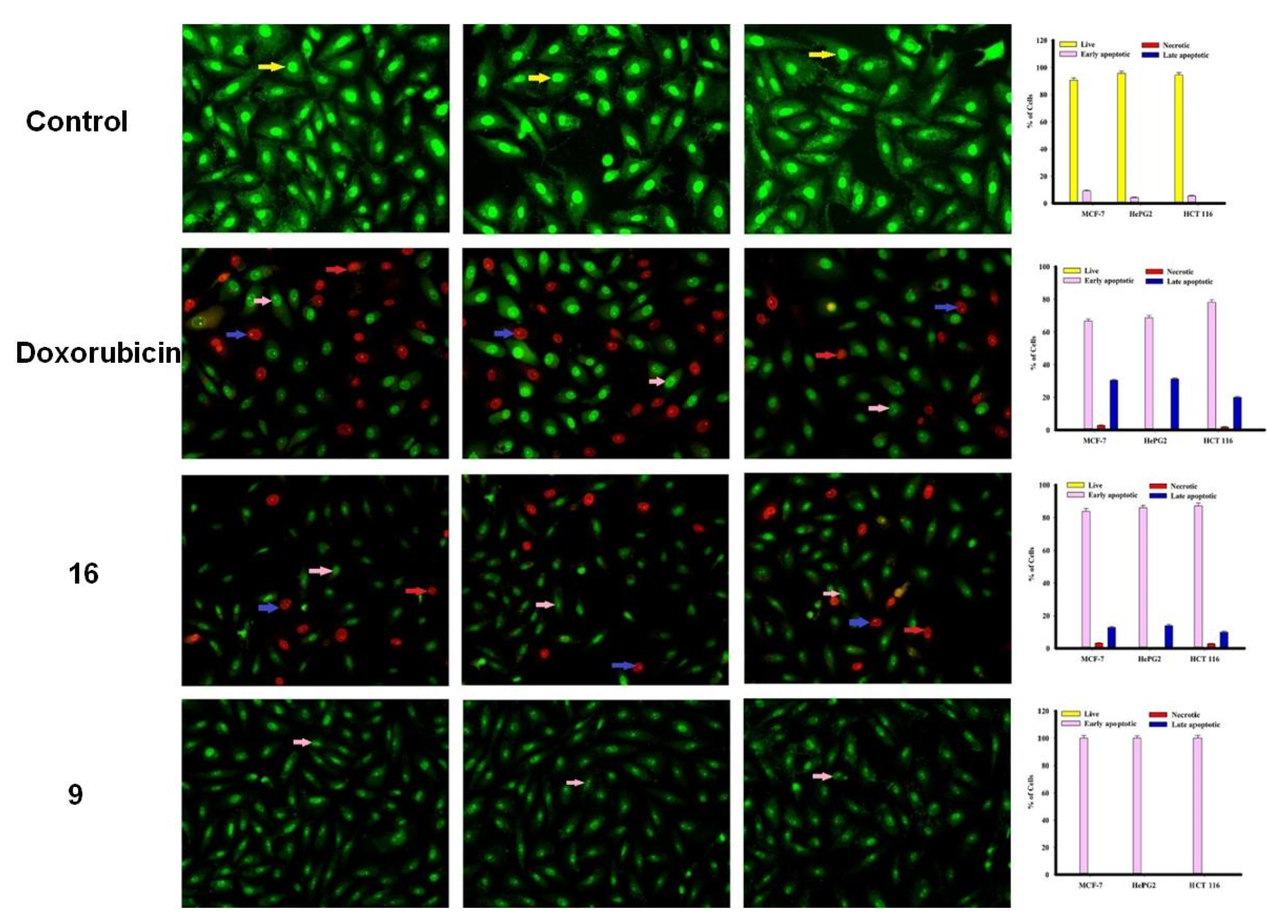
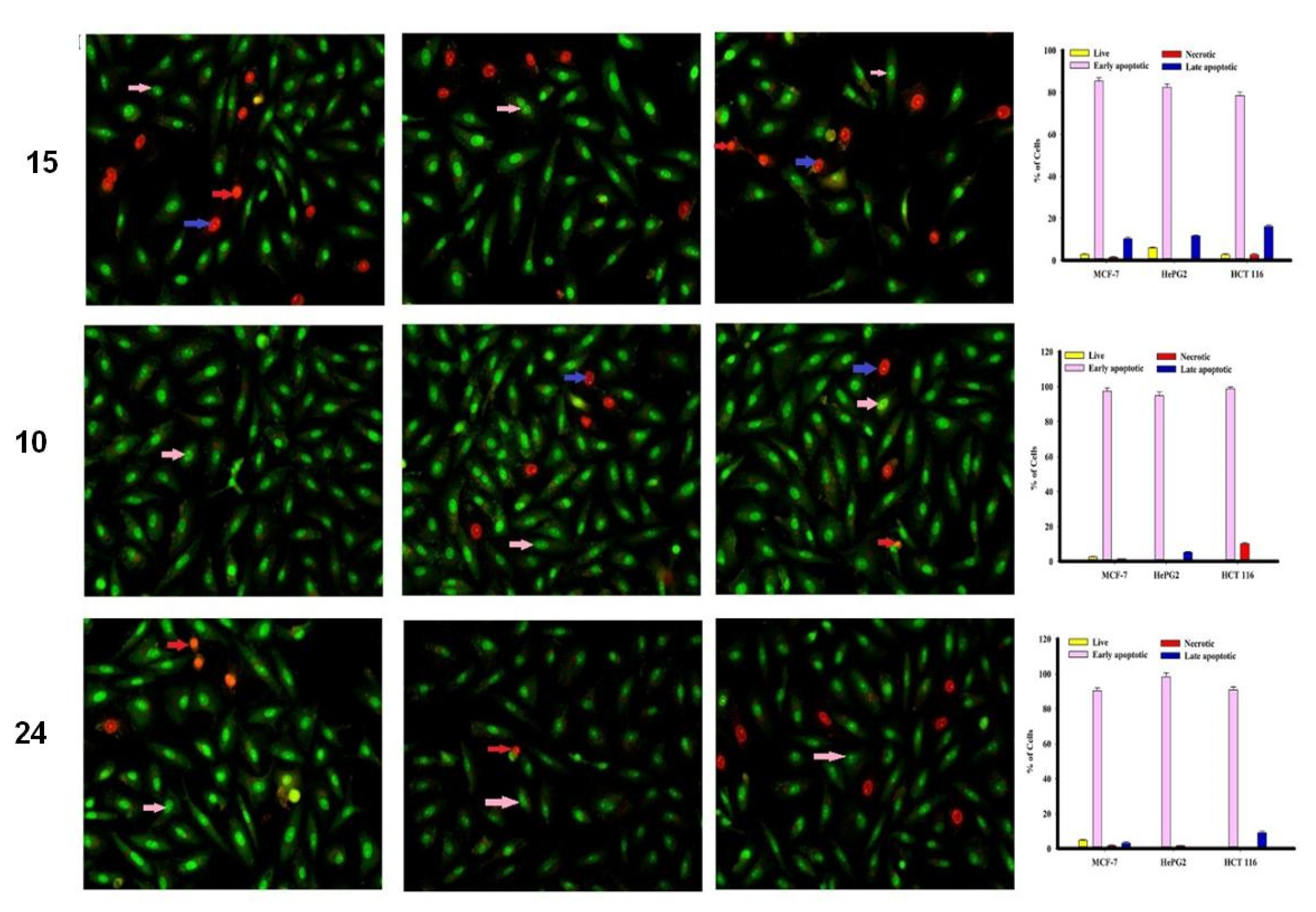
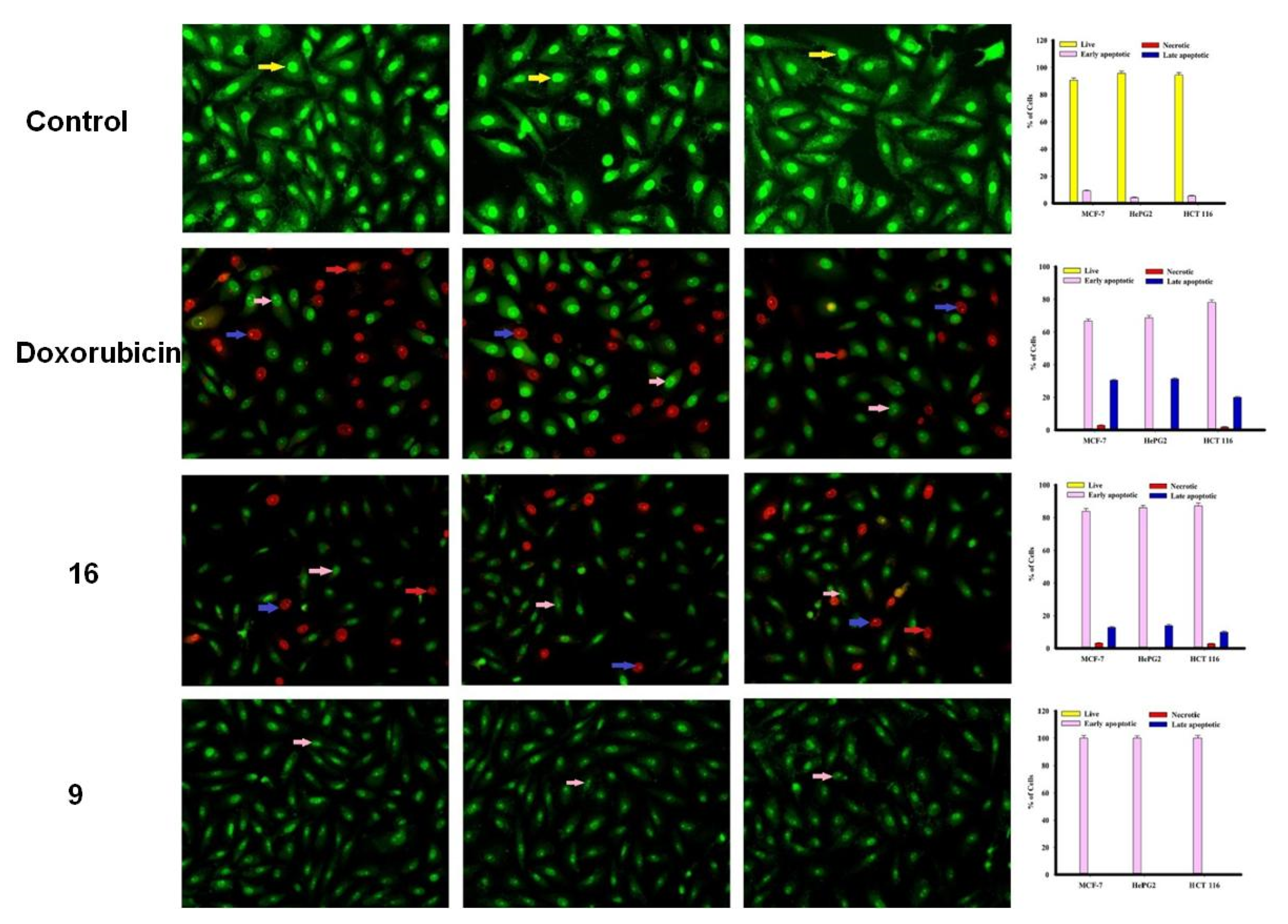
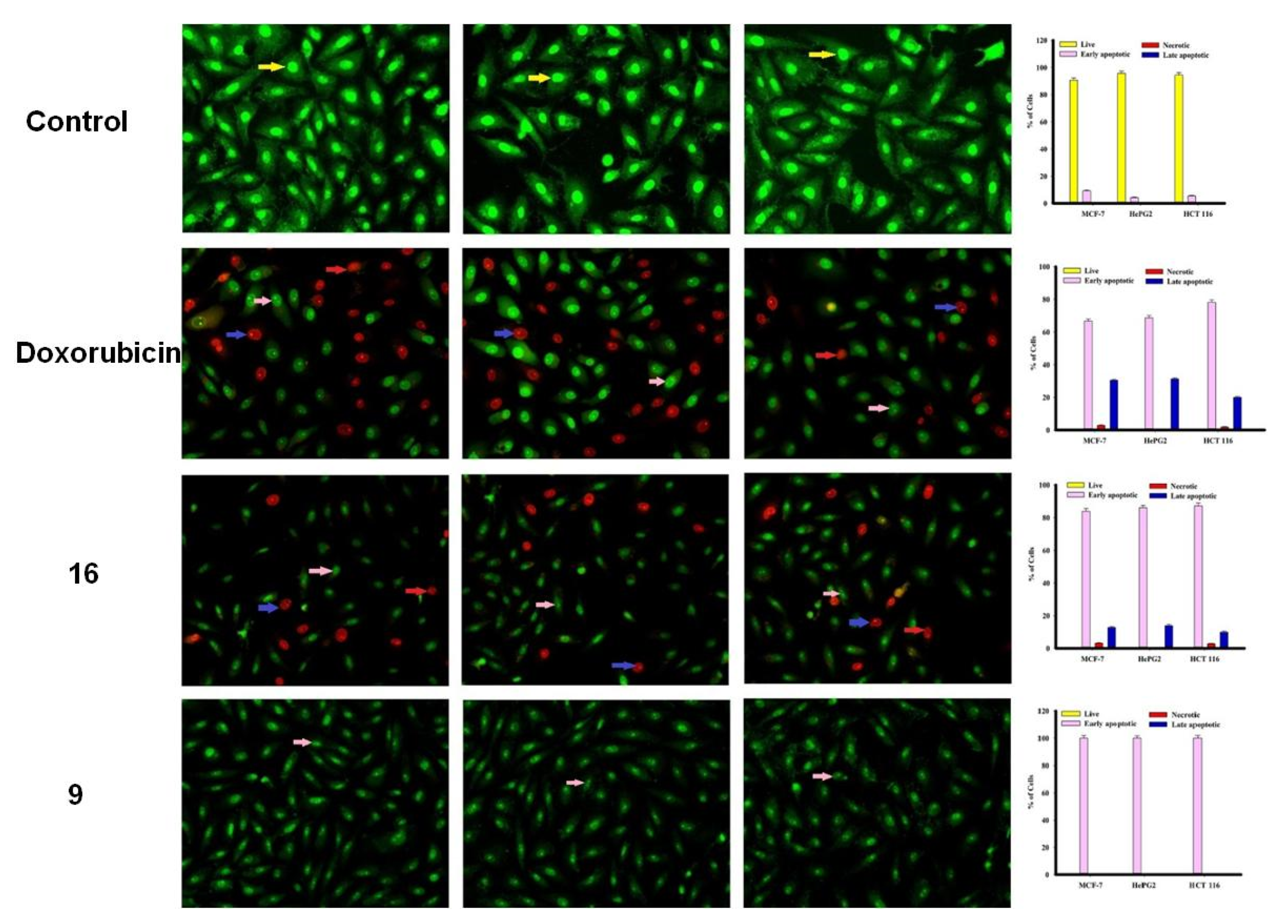
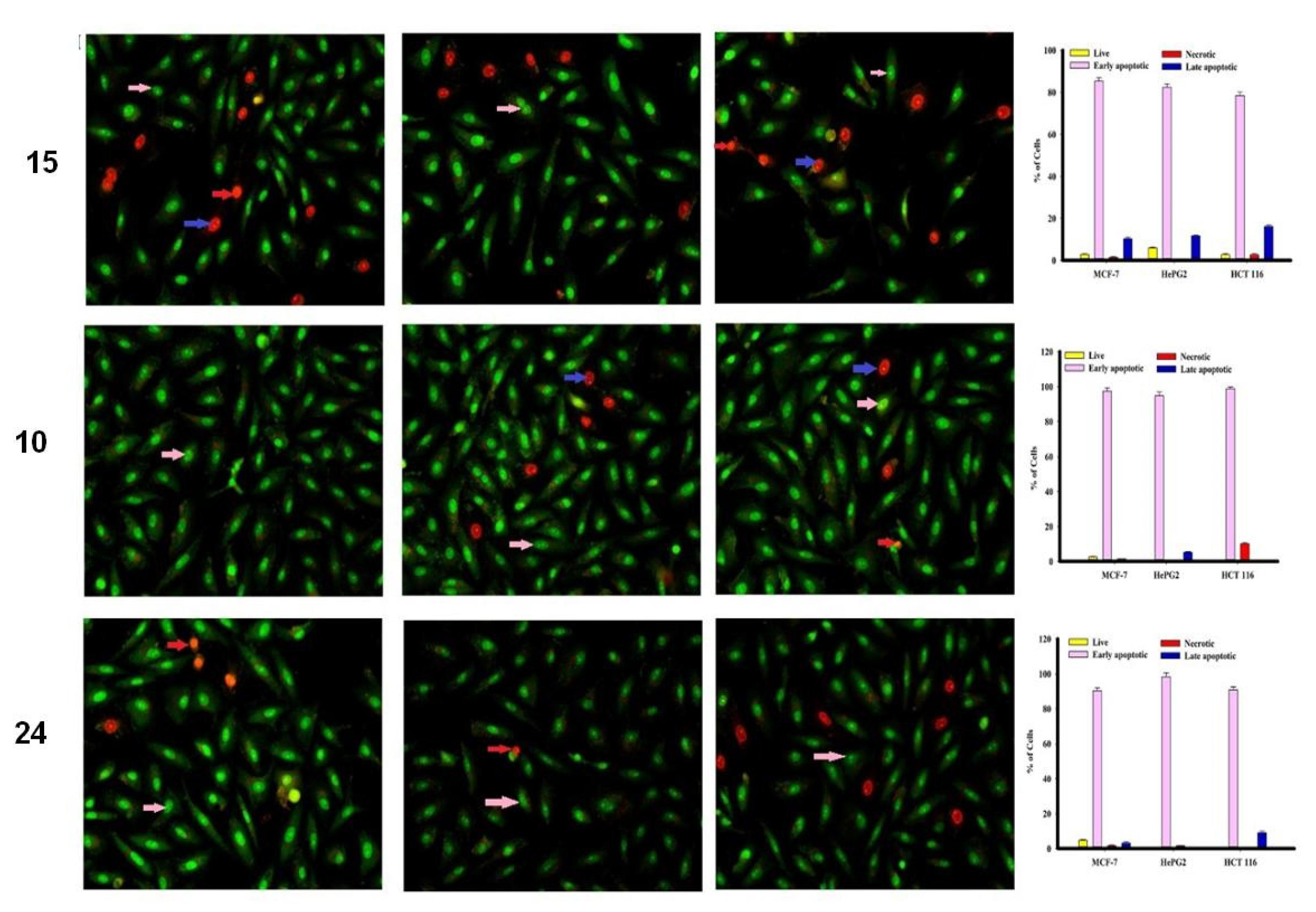
2.2.2. Apoptosis Using Acridine Orange/Ethidium Bromide Staining
2.2.3. EGFR Kinase Activity
2.2.4. Statistical Analysis
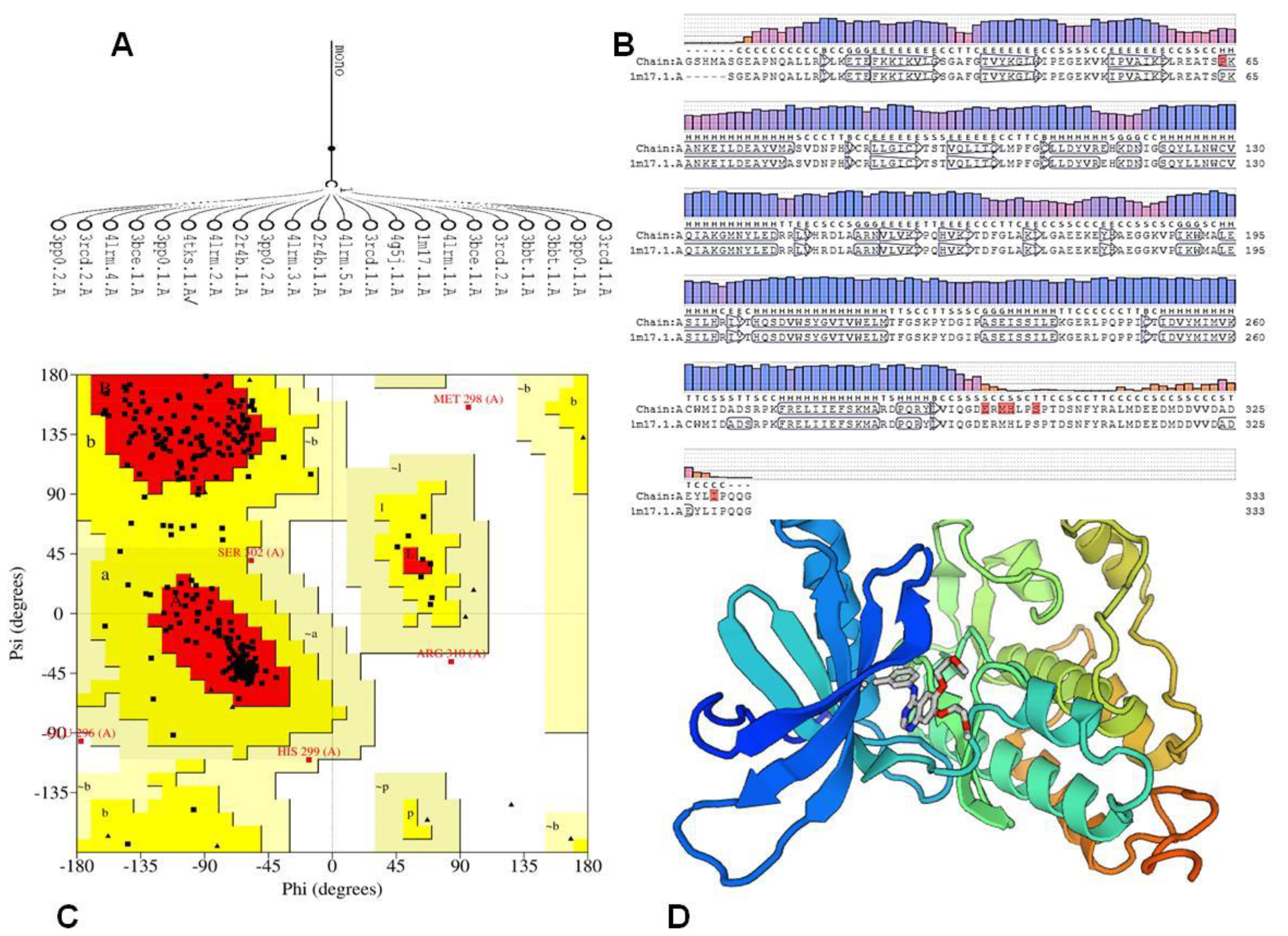
2.3. Computational Details and Molecular Docking
3. Results and Discussion
3.1. Chemistry
3.2. In Silico ADME/Pharmacokinetics Studies
3.3. Biological Activities
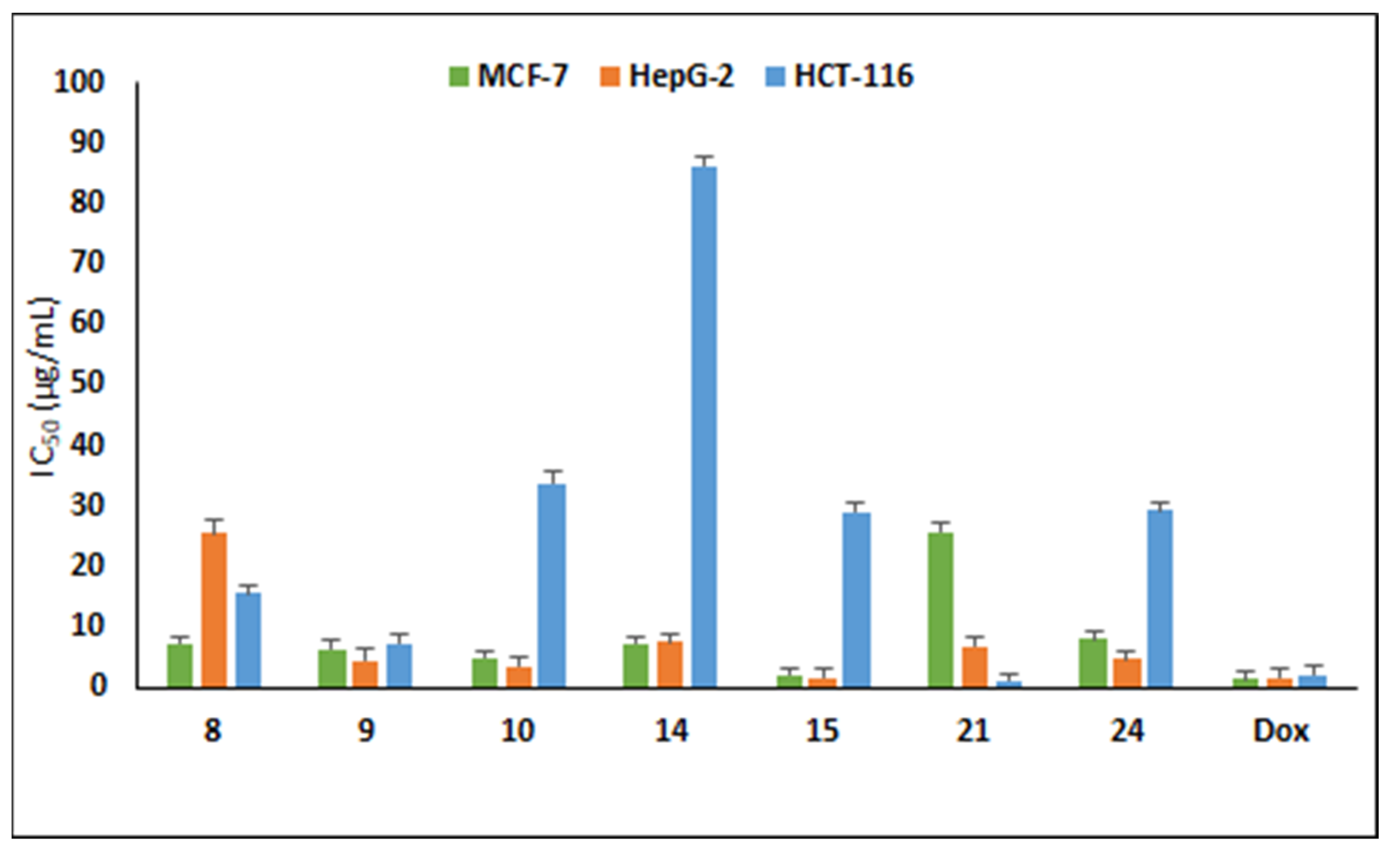
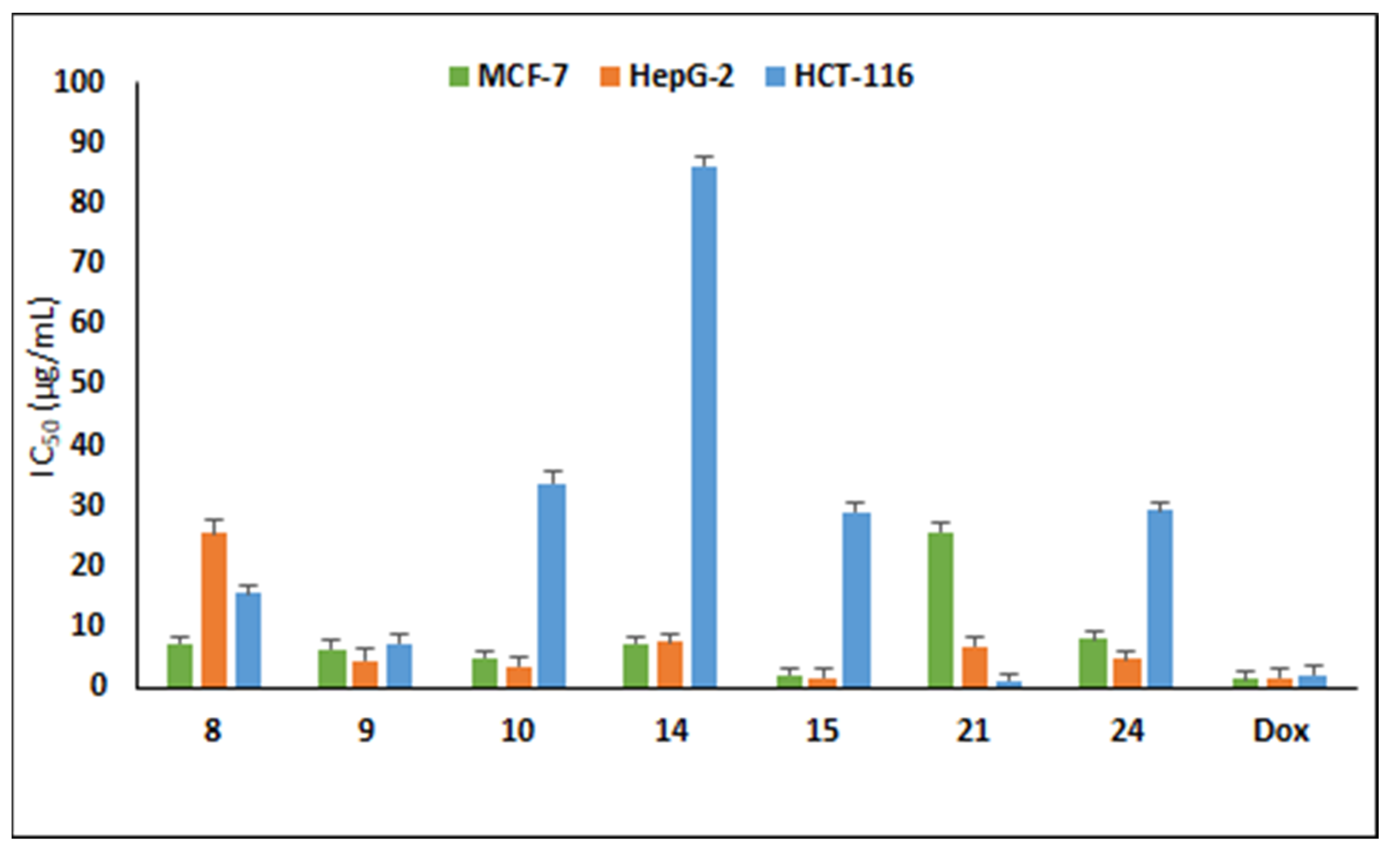
3.3.1. Antiproliferative Activity
3.3.2. In Vitro EGFR Activity
3.3.3. Apoptosis Studies
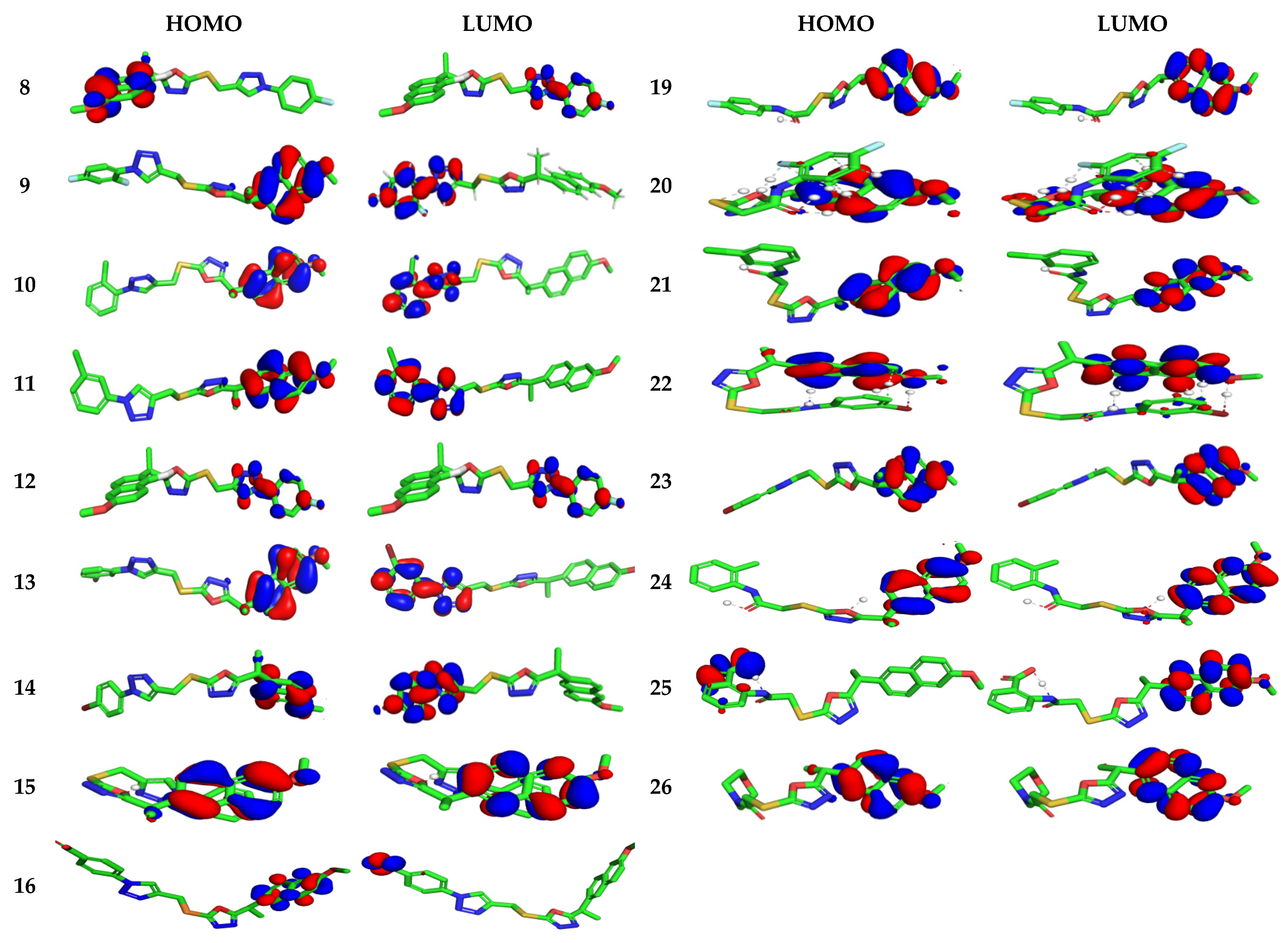
3.4. Computational Study
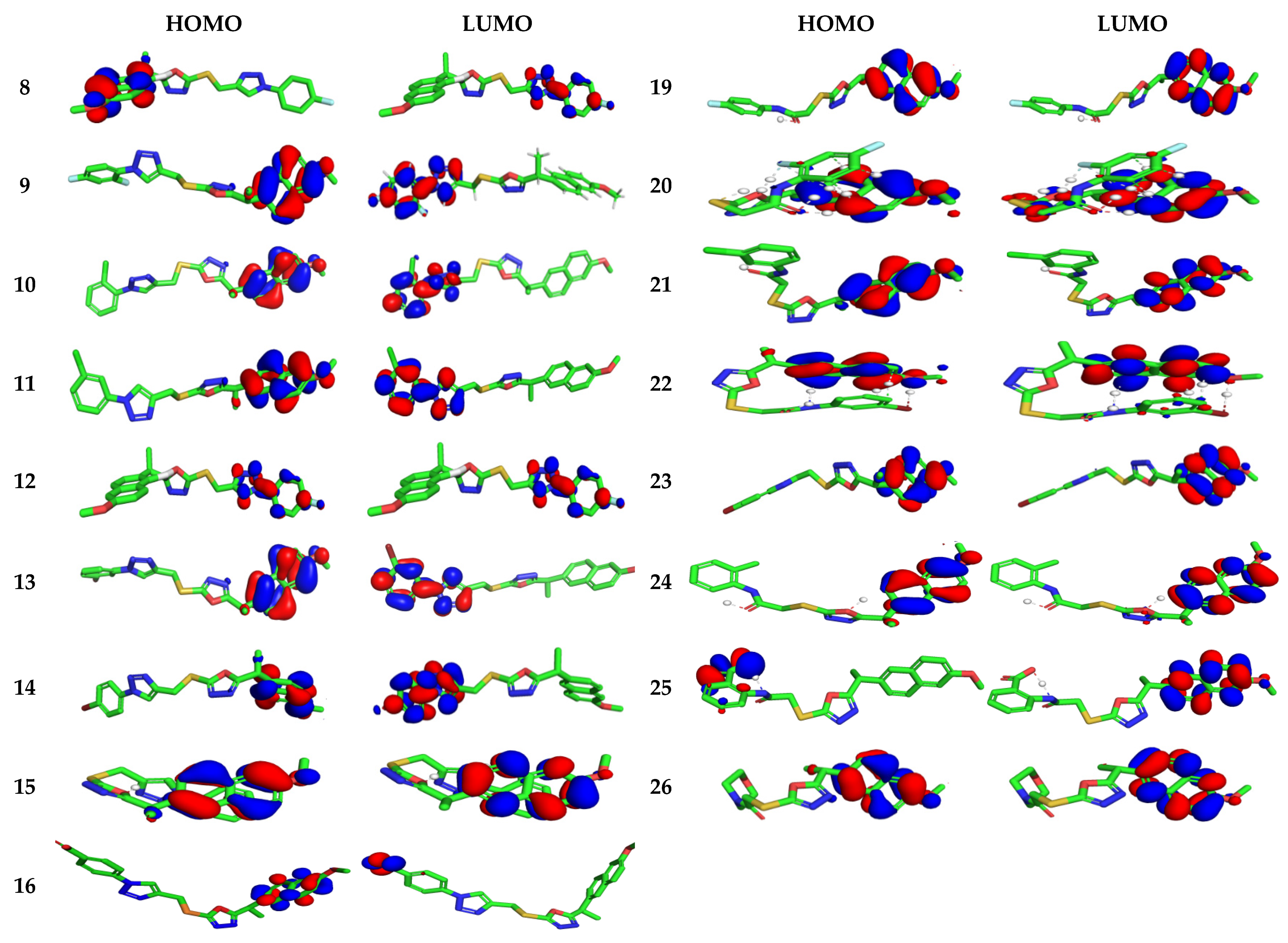
3.4.1. Electronic Properties
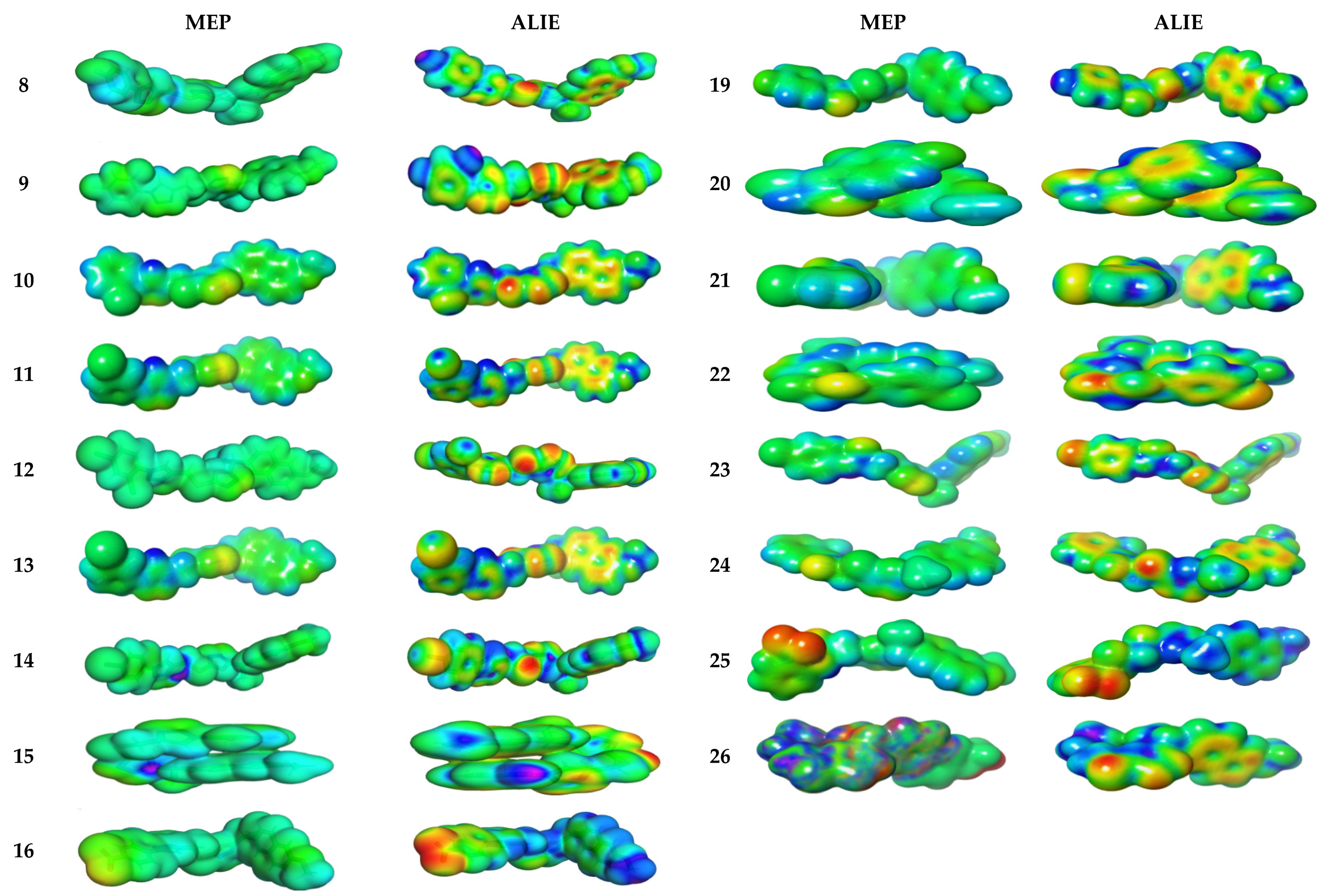
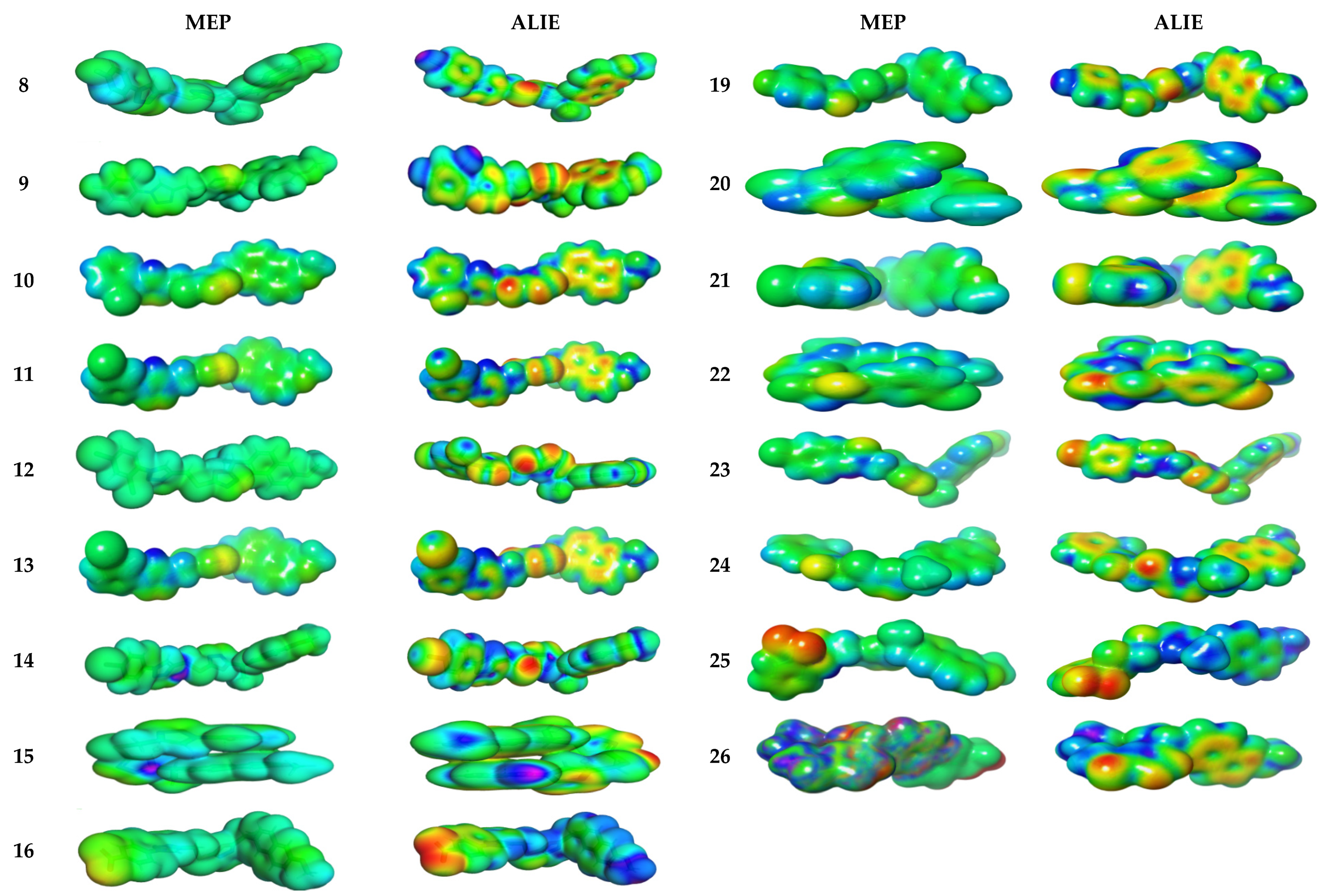
3.4.2. Molecular Electrostatic and Ionization Potential Maps
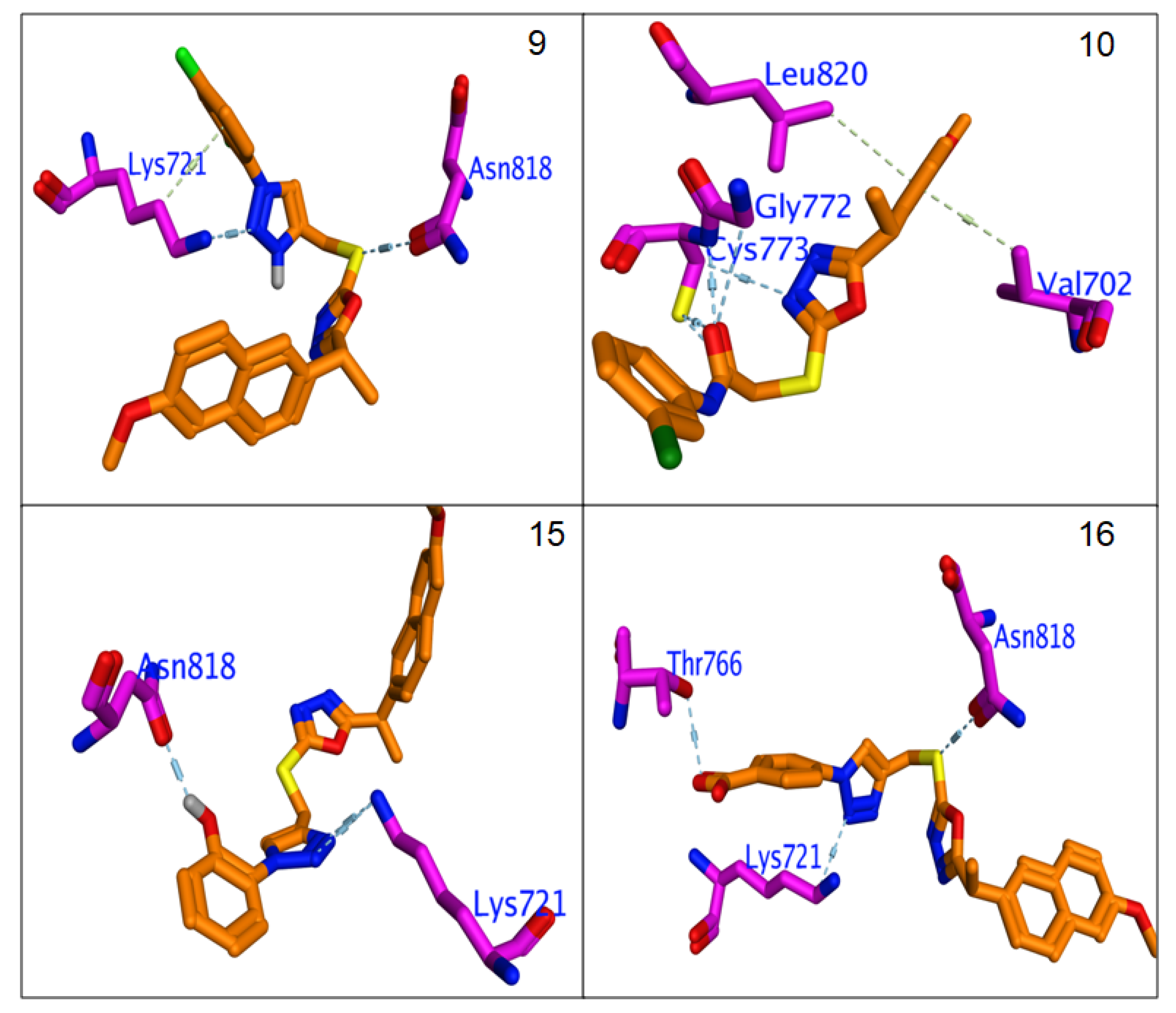
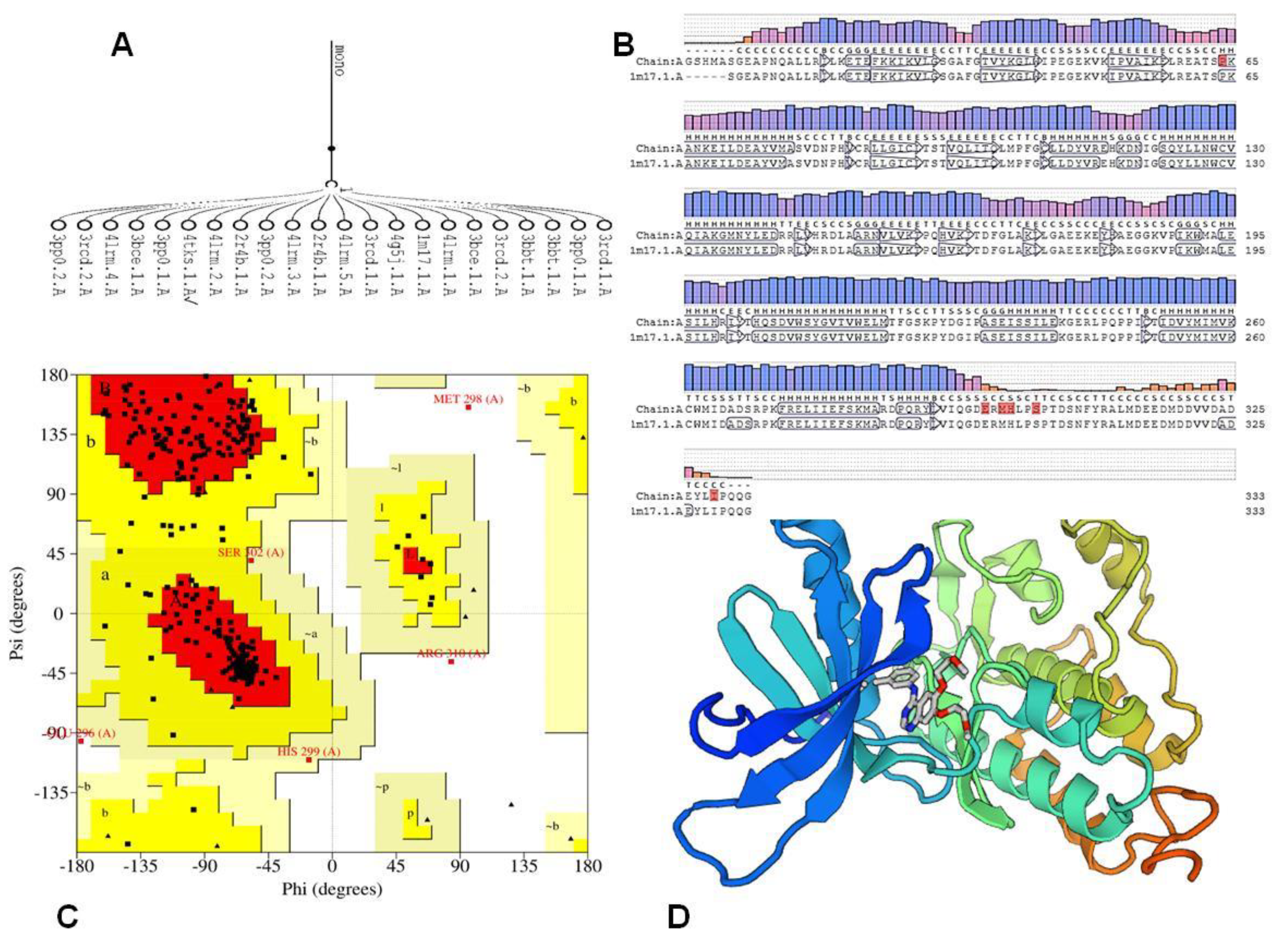
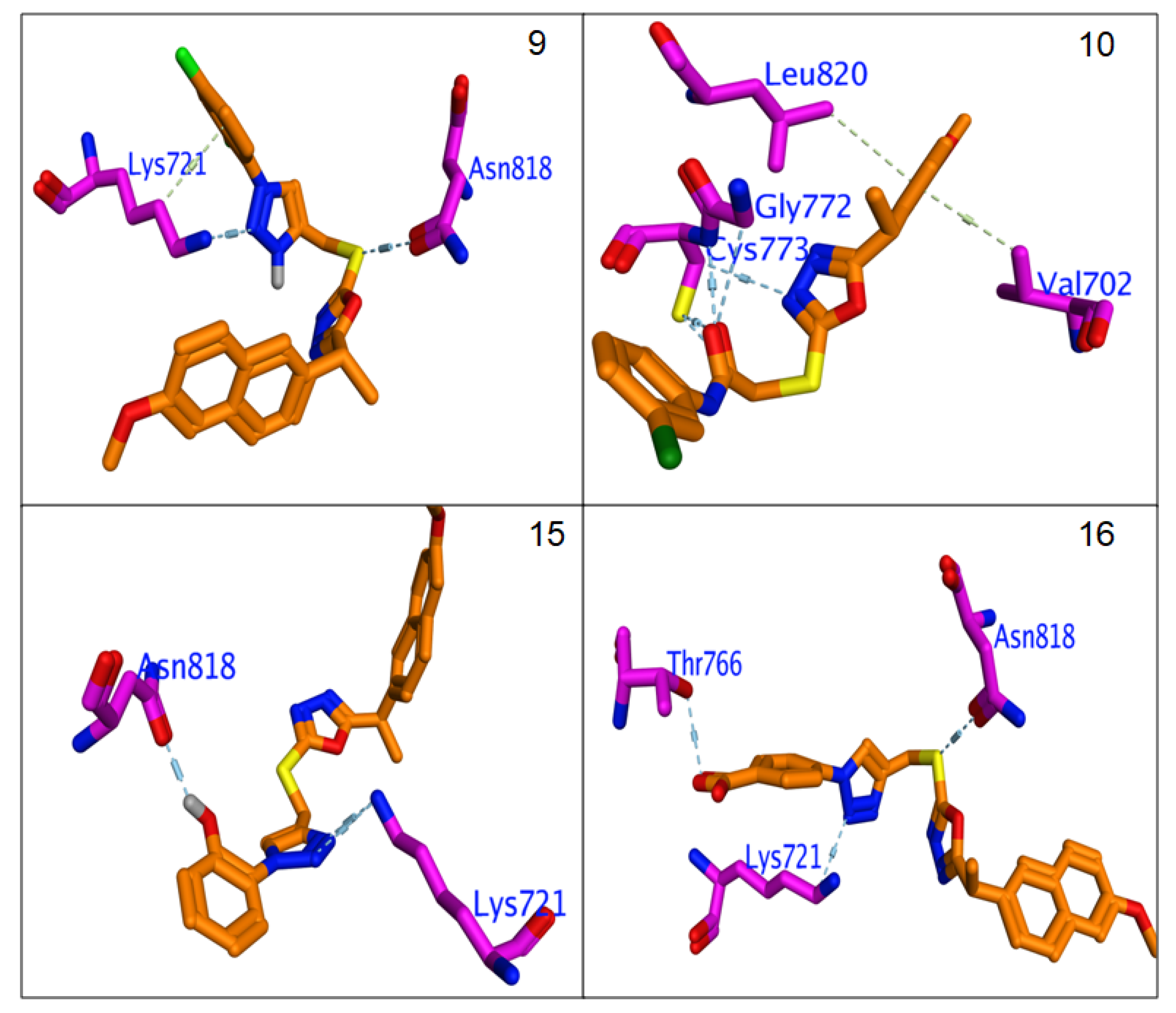
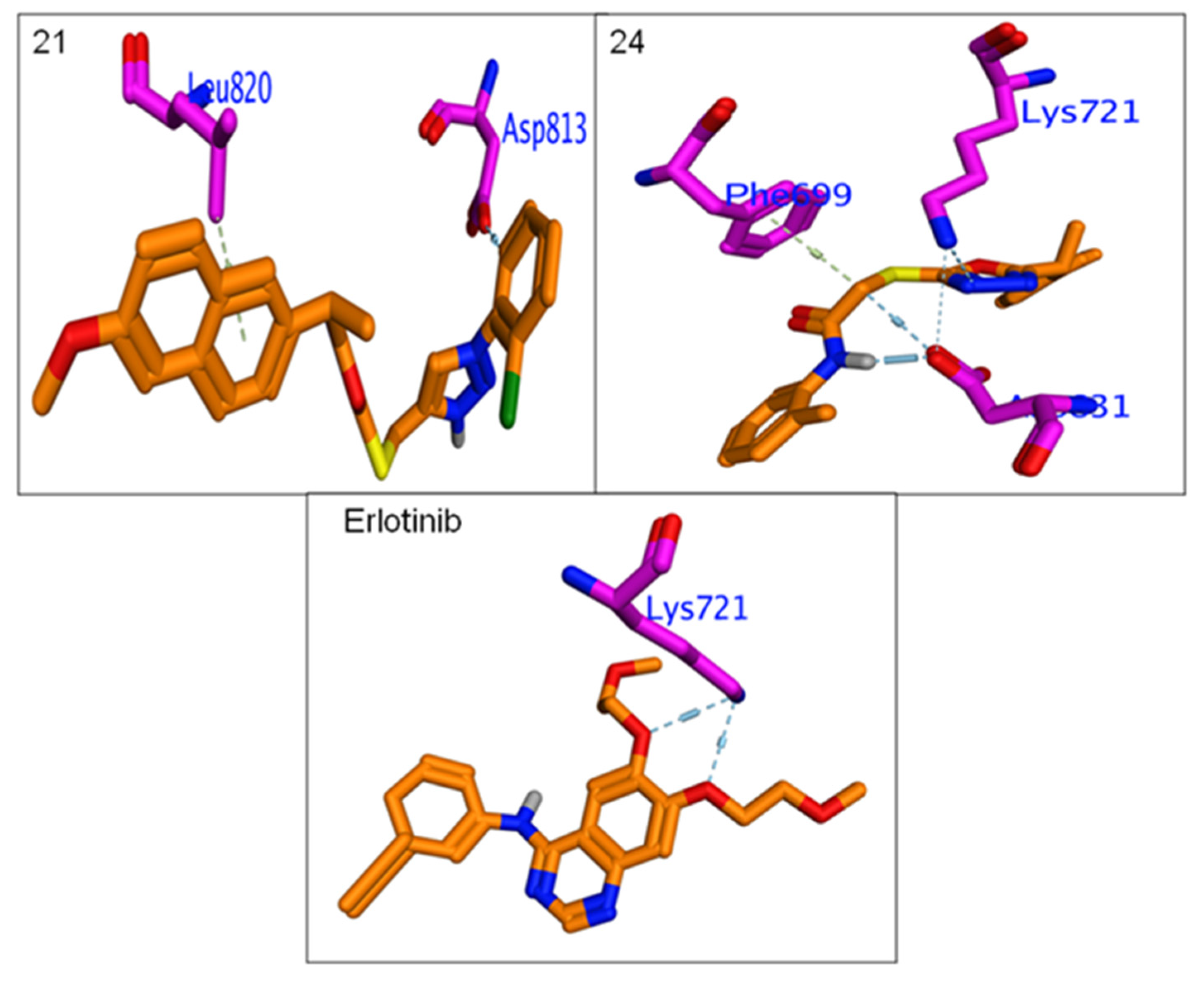
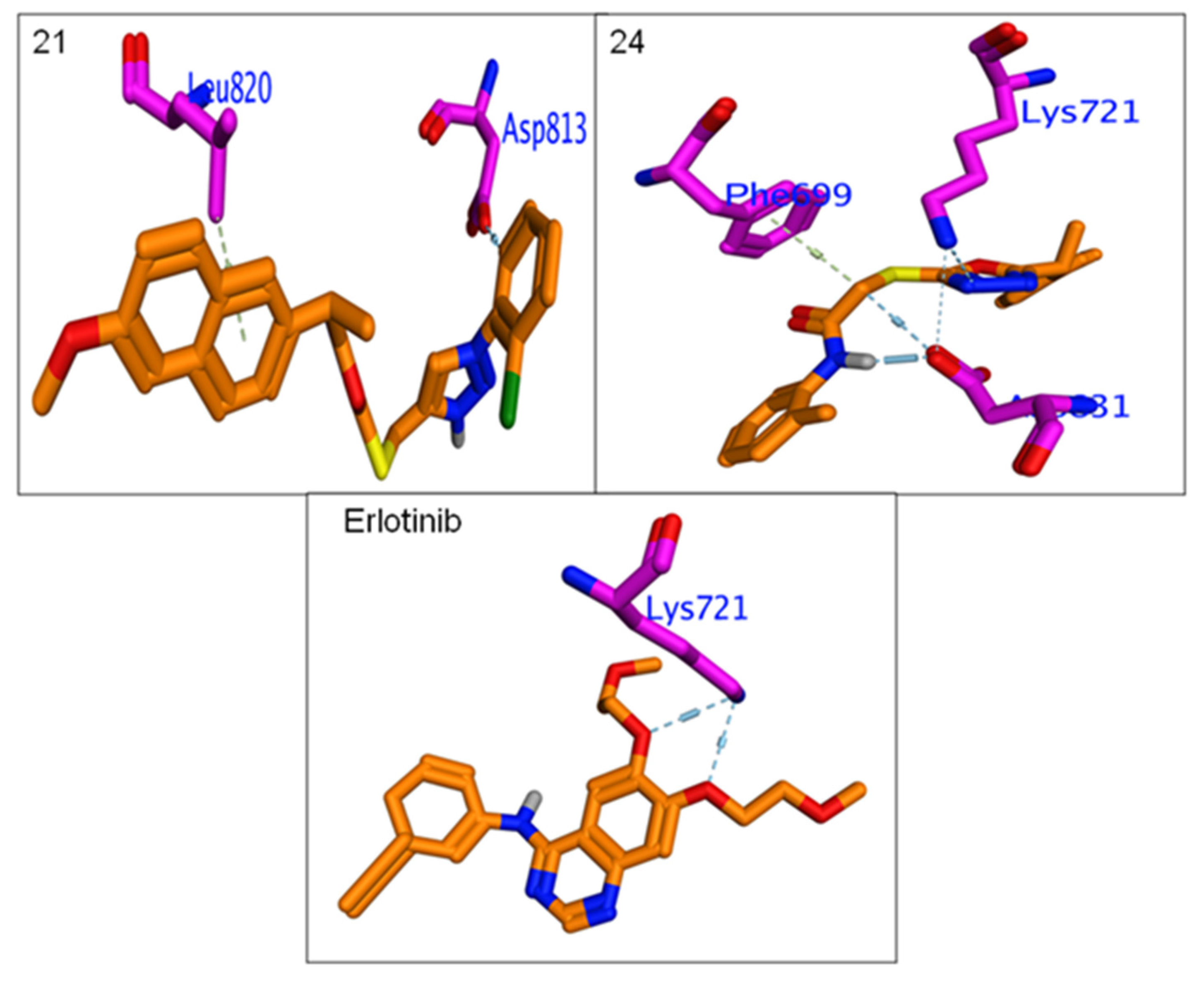
3.4.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Gohary, N.S.; Gabra, M.T.; Shaaban, M.I. Synthesis, molecular modeling and biological evaluation of new pyrazolo[3,4-b]pyridine analogs as potential antimicrobial, antiquorum-sensing and anticancer agents. Bioorg. Chem. 2019, 89, 102976. [Google Scholar] [CrossRef]
- Alghamdi, A.A.; Nazreen, S. Synthesis, characterization and cytotoxic study of 2-hydroxy benzothiazole incorporated 1,3,4-oxadiazole derivatives. Egypt. J. Chem. 2020, 63, 471–482. [Google Scholar] [CrossRef]
- Rajanarendar, E.; Reddy, M.N.; Krishna, S.R.; Reddy, K.G.; Reddy, Y.N.; Rajam, M.V. Design, synthesis, in vitro antimicrobial and anticancer activity of novel methylenebis-isoxazolo[4,5-b]azepines derivatives. Eur. J. Med. Chem. 2012, 50, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, N.; Bloom, D.; Bloom, R.; Stein, R. Breakaway: The Global Burden of Cancer: Challenges and Opportunities. Econ. Intell. Unit. 2009, 1–73. [Google Scholar]
- Bianco, R.; Gelardi, T.; Damiano, V.; Ciardiello, F.; Tortora, G. Rational bases for the development of EGFR inhibitors for cancer treatment. Int. J. Biochem. Cell Biol. 2007, 39, 1416–1431. [Google Scholar] [CrossRef] [PubMed]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Higgins, B.; Kolinsky, K.; Smith, M.; Beck, G.; Rashed, M.; Adames, V.; Linn, M.; Wheeldon, E.; Gand, L.; Birnboeck, H.; et al. Antitumor activity of erlotinib (OSI-774, Tarceva) alone or in combination in human non-small cell lung cancer tumor xenograft models. Anticancer Drugs 2014, 15, 503–512. [Google Scholar] [CrossRef]
- Celik, I.; Ayhan-Kılcıgil, G.; Guven, B.; Kara, Z.; Gurkan-Alp, S.; Karayel, A.; Onay-Besikci, A. Design, synthesis and docking studies of benzimidazole derivatives as potential EGFR inhibitors. Eur. J. Med. Chem. 2019, 173, 240–249. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.R.; Lanzarini, C.M.; Ricci-Junior, E. Preparation, in vitro characterization and in vivo release of naproxen loaded in poly-caprolactone nanoparticles. Pharm. Dev. Technol. 2011, 16, 12–21. [Google Scholar] [CrossRef]
- Elhenawy, A.A.; Al-Harbi, M.; Moustafa, G.O.; El-Gazzar, M.A.; Abdel-Rahman, R.F.; Salim, A.E. Synthesis, comparative docking, and pharmacological activity of naproxen amino acid derivatives as possible anti-inflammatory and analgesic agents. Drug Des. Dev. Ther. 2019, 13, 1773–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cıkla, P.; Ozsavcı, D.; Ozakpınar, O.B.; Sener, A.; Cevik, O.; Ozbas-Turan, S.; Akbuga, J.; Sahin, F.; Kucukguzel, S.G. Synthesis, Cytotoxicity, and Pro-Apoptosis Activity of Etodolac Hydrazide Derivatives as Anticancer Agents. Arch. Pharm. Chem. Life Sci. 2013, 346, 367–379. [Google Scholar] [CrossRef]
- Kucukguzel, S.G.; Koc, D.; Suzgun, P.C.; Ozsavci, D.; Ozakpinar, O.B.; Tiber, P.M.; Orun, O.; Erzincan, P.; Erdem, S.S.; Sahin, F. Synthesis of Tolmetin Hydrazide–Hydrazones and Discovery of a Potent Apoptosis Inducer in Colon Cancer Cells. Arch. Pharm. Chem. Life Sci. 2015, 348, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Han, M.I.; Bekci, H.; Uba, A.I.; Yildrim, Y.; Karasulu, E.; Cumaoglu, A.; Karasulu, H.Y.; Yelecksi, K.; Yilmaz, O.; Kucukguzel, S.G. Synthesis, molecular modeling, in vivo study, and anticancer activity of 1,2,4-triazole containing hydrazide-hydrazones derived from(S)-naproxen. Arch. Pharm. 2019, 352, 1800365. [Google Scholar] [CrossRef] [PubMed]
- Birgul, K.; Yıldırım, Y.; Karasulu, H.Y.; Karasulu, E.; Uba, A.I.; Yelekci, K.; Bekci, H.; Cumaoglu, A.; Kabasakal, L.; Yilmaz, O.; et al. Synthesis, molecular modeling, in vivo study and anticancer activity against prostate cancer of (+) (S)-naproxen derivatives. Eur. J. Med. Chem. 2020, 208, 112841. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, M.M.; Ismail, M.M.; Eissa, S.; Ammar, Y. Design and synthesis of some novel 6-methoxynaphthalene derivatives with Potential anticancer activity. Der Pharma. Chem. 2012, 4, 1552–1566. [Google Scholar]
- Lubet, R.A.; Steele, V.E.; Juliana, M.M.; Grubbs, C.J. Screening agents for preventive efficacy in a bladder cancer model: Study design, end points, and gefitinib and naproxen efficacy. J. Urol. 2010, 183, 1598. [Google Scholar] [CrossRef]
- Han, M.I.; Atalay, P.; Tunc, C.U.; Unal, G.; Dayan, S.; Aydın, O.; Kuçukguzel, S.G. Design and synthesis of novel (S)-Naproxen hydrazide-hydrazones as potent VEGFR-2 inhibitors and their evaluation in vitro/in vivo breast cancer models. Bioorg. Med. Chem. 2021, 37, 116097. [Google Scholar] [CrossRef]
- Chen, P.C.; Patil, V.; Guerrant, W.; Green, P.; Oyelere, A.K. Synthesis and structure-activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg. Med. Chem. 2008, 16, 4839–4853. [Google Scholar] [CrossRef]
- Dua, R.; Shrivastava, S.; Sonwane, S.K.; Srivastava, S.K. Pharmacological Significance of Synthetic Heterocycles Scaffold: A Review. Adv. Biol. Res. 2011, 5, 120–144. [Google Scholar]
- Nazreen, S. Design, synthesis, and molecular docking studies of thiazolidinediones as PPAR-γ agonists and thymidylate synthase inhibitors. Arch. Der Pharm. 2021, e2100021. [Google Scholar] [CrossRef]
- Chawla, G.; Naaz, B.; Siddiqui, A.A. Exploring 1,3,4-Oxadiazole Scaffold for Anti-inflammatory and Analgesic Activities: A Review of Literature From 2005–2016. Mini. Rev. Med. Chem. 2018, 18, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Alghmadi, A.A.; Alam, M.M.; Nazreen, S. In silico ADME predictions and in vitro antibacterial evaluation of 2-hydroxy benzothiazole-based 1,3,4-oxadiazole derivatives. Turk. J. Chem. 2020, 44, 1068–1084. [Google Scholar] [CrossRef] [PubMed]
- Albratty, M.; El-Sharkawy, K.A.; Alhazmi, H.A. Synthesis and evaluation of some new 1,3,4-oxadiazoles bearing thiophene, thiazole, coumarin, pyridine and pyridazine derivatives as antiviral agents. Acta. Pharm. 2019, 69, 261–276. [Google Scholar] [CrossRef] [Green Version]
- Abou-Seri, S.M. Synthesis and biological evaluation of novel 2,4′-bis substituted diphenylamines as anticancer agents and potential epidermal growth factor receptor tyrosine kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 4113–4121. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Siddiqui, A.A.; Khan, A.A.; Ali, Z.; Dewangan, R.P.; Pasha, S.; Yar, M.S. Design, synthesis, docking and QSAR study of substituted benzimidazole linked oxadiazole as cytotoxic agents, EGFR and erbB2 receptor inhibitors. Eur. J. Med. Chem. 2017, 126, 853–869. [Google Scholar] [CrossRef]
- El-Sayed, N.A.; Nour, M.S.; Alaraby Salem, M.; Arafa, R.K. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: Design, synthesis, cytotoxicity evaluation and in silico studies. Eur. J. Med. Chem. 2019, 183, 111693. [Google Scholar] [CrossRef] [PubMed]
- Fathi, M.A.A.; Abd-El-Hafeez, A.A.; Abdelhamid, D.; Abbas, S.H.; Montano, M.M.; Abdel-Aziz, M. 1,3,4-oxadiazole/chalcone hybrids: Design, synthesis, and inhibition of leukemia cell growth and EGFR, Src, IL-6 and STAT3 activities. Bioorg. Chem. 2019, 84, 150–163. [Google Scholar] [CrossRef]
- Alzhrani, Z.M.M.; Alam, M.M.; Neamatallah, T.; Nazreen, S. Design, synthesis and in vitro antiproliferative activity of new thiazolidinedione-1,3,4-oxadiazole hybrids as thymidylate synthase inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1116–1123. [Google Scholar] [CrossRef]
- Alam, M.M.; Almalki, A.S.A.; Neamatallah, T.; Ali, N.M.; Malebari, A.M.; Nazreen, S. Synthesis of New 1,3,4-Oxadiazole-Incorporated 1, 2, 3-Triazole Moieties as Potential Anticancer Agents Targeting Thymidylate Synthase and Their Docking Studies. Pharmaceuticals 2020, 13, 390. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Malebari, A.M.; Nazreen, S.; Neamatallah, T.; Almalki, A.S.A.; Elhenawy, A.A.; Obaid, R.J.; Alsherif, M.A. Design, synthesis and molecular docking studies of thymol based 1,2,3-triazole hybrids as thymidylate synthase inhibitors and apoptosis inducers against breast cancer cells. Bioorg. Med. Chem. 2021, 38, 116136. [Google Scholar] [CrossRef]
- Alarif, W.M.; Ghandourah, M.A.; Abdel-Lateff, A.; Bawakid, N.O.; Alotaibi, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Two new xeniolide diterpenes from the soft coral Xenia umbellata; displayed anti proliferative effects. Pharmacogn. Mag. 2020, 16, 774–779. [Google Scholar] [CrossRef]
- Althagbi, H.I.; Budiyanto, F.; Abdel-Lateff, A.; Al-Footy, K.O.; Bawakid, N.O.; Ghandourah, M.A.; Alfaifi, M.Y.; Elbehairi, S.E.I.; Alarif, W.M. Antiproliferative Isoprenoid Derivatives from the Red Sea Alcyonacean Xenia umbellata. Molecules 2021, 26, 1311. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Chen, E.; Shu, K.; Chen, W.; Zhang, G.; Yu, Y. 6-Oxooxazolidine–quinazolines as noncovalent inhibitors with the potential to target mutant forms of EGFR. Bioorg. Med. Chem. 2016, 24, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Freener, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Elgendy, A.; Nady, H.; El-Rabiei, M.M.; Elhenawy, A.A. Understanding the adsorption performance of two glycine derivatives as novel and environmentally safe anti-corrosion agents for copper in chloride solutions: Experimental, DFT, and MC studies. RSC Adv. 2019, 9, 42120–42131. [Google Scholar] [CrossRef] [Green Version]
- El Gaafary, M.; Syrovets, T.; Mohamed, H.M.; Elhenawy, A.A.; El-Agrody, A.M.; Amr, A.E.; Ghabbour, H.A.; Almehizia, A.A. Synthesis, Cytotoxic Activity, Crystal Structure, DFT Studies and Molecular Docking of 3-Amino-1-(2,5-dichlorophenyl)-8-methoxy-1H-benzo[f]chromene-2-carbonitrile. Crystals 2021, 11, 184. [Google Scholar] [CrossRef]
- Elhenawy, A.A.; Al-Harbi, L.M.; El-Gazzar, M.A.; Khowdiary, M.M.; Ouidate, A.; Alosaimi, A.M.; Salim, A.E. Naproxenylamino acid derivatives: Design, synthesis, docking, QSAR and anti-inflammatory and analgesic activity. Biomed. Pharmacother. 2019, 116, 109024. [Google Scholar] [CrossRef] [PubMed]
- Elhenawy, A.A.; Al-Harbi, L.M.; El-Gazzar, M.A.; Khowdiary, M.M.; Moustfa, A. Synthesis, molecular properties and comparative docking and QSAR of new 2-(7-hydroxy-2-oxo-2H-chromen-4-yl) acetic acid derivatives as possible anticancer agents. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 218, 248–262. [Google Scholar] [CrossRef]
- Abusaif, M.S.; Fathy, M.; Abu-Saied, M.A.; Elhenawy, A.A.; Kashyout, A.B.; Selim, M.R.; Ammar, Y.A. New carbazole-based organic dyes with different acceptors for dye-sensitized solar cells: Synthesis, characterization, dssc fabrications and density functional theory studies. J. Mol. Struct. 2021, 1225, 129297. [Google Scholar] [CrossRef]
- Naglah, A.M.; Moustafa, G.O.; Elhenawy, A.A.; Mounier, M.M.; El-Sayed, H.; Al-Omar, M.A.; Almehizia, A.A.; Bhat, M.A. Nα-1, 3-Benzenedicarbonyl-Bis-(Amino Acid) and Dipeptide Candidates: Synthesis, Cytotoxic, Antimicrobial and Molecular Docking Investigation. Drug Des. Devel. Ther. 2021, 15, 1315–1332. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. Chapter 21.4 PROCHECK: Validation of protein-structure coordinates. Crystallogr. Biol. Macromol. 2012, F, 722–725. [Google Scholar]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2018-1; 2018; Available online: https://www.mdpi.com/1420-3049/23/6/1313/htm (accessed on 27 August 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipinski Parameters | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MW a | HBA b | HBD c | LogP d | Violations | nROTB e | TPSA f | %ABS g | BBB h | GI ABS i | |
| 8 | 461.51 | 7 | 0 | 4.41 | 0 | 7 | 104.16 | 73.06 | No | Low |
| 9 | 479.5 | 8 | 0 | 4.18 | 1 | 7 | 104.16 | 73.06 | No | Low |
| 10 | 477.97 | 6 | 0 | 4.30 | 0 | 7 | 104.16 | 73.06 | No | Low |
| 11 | 477.97 | 6 | 0 | 4.47 | 0 | 7 | 104.16 | 73.06 | No | Low |
| 12 | 512.41 | 6 | 0 | 4.47 | 2 | 7 | 104.16 | 73.06 | No | Low |
| 13 | 522.42 | 6 | 0 | 4.59 | 2 | 7 | 104.16 | 73.06 | No | Low |
| 14 | 522.42 | 6 | 0 | 4.67 | 2 | 7 | 104.16 | 73.06 | No | Low |
| 15 | 459.52 | 7 | 1 | 4.21 | 0 | 7 | 124.39 | 66.08 | No | Low |
| 16 | 487.53 | 8 | 1 | 3.49 | 0 | 8 | 141.46 | 60.19 | No | Low |
| 19 | 437.49 | 6 | 1 | 3.63 | 0 | 8 | 102.55 | 73.62 | No | High |
| 20 | 455.48 | 7 | 1 | 4.02 | 0 | 8 | 102.55 | 73.62 | No | Low |
| 21 | 453.94 | 5 | 1 | 3.71 | 0 | 8 | 102.55 | 73.62 | No | High |
| 22 | 498.39 | 5 | 1 | 3.88 | 0 | 8 | 102.55 | 73.62 | No | Low |
| 23 | 498.39 | 5 | 1 | 3.87 | 0 | 8 | 102.55 | 73.62 | No | Low |
| 24 | 433.52 | 5 | 1 | 3.9 | 0 | 8 | 102.55 | 73.62 | No | High |
| 25 | 463.51 | 7 | 2 | 2.92 | 0 | 9 | 139.85 | 60.75 | No | Low |
| 26 | 413.49 | 6 | 0 | 3.81 | 0 | 7 | 102.99 | 73.46 | No | High |
| IC50(μg/mL) | |||
|---|---|---|---|
| Compounds | MCF-7 | HepG2 | HCT-116 |
| 8 | 7.38 | 25.49 | 15.66 |
| 9 | 6.47 | 4.52 | 7.46 |
| 10 | 5.05 | 3.53 | 33.91 |
| 11 | 60.45 | 37.88 | 83.92 |
| 12 | 45.7 | 26.37 | 28.15 |
| 13 | 15.84 | 7.07 | 29.28 |
| 14 | 7.50 | 7.63 | 86.1 |
| 15 | 2.13 | 1.63 | 29.1 |
| 16 | 10.44 | 10.67 | 8.36 |
| 19 | 387.46 | 829.66 | 1107.03 |
| 20 | 32.65 | 31.32 | 73.42 |
| 21 | 25.90 | 7.00 | 1.24 |
| 22 | 97.29 | 159.2 | 20.24 |
| 23 | 25.56 | 14.97 | 35.15 |
| 24 | 8.15 | 4.88 | 29.34 |
| 25 | 29.79 | 28.02 | 29.21 |
| 26 | 22.28 | 23.64 | 11.09 |
| Doxorubicin | 1.41 | 1.62 | 2.11 |
| Compounds | EGFR (IC50, μM) |
|---|---|
| 8 | 7.31 ± 0.25 |
| 9 | 0.71 ± 0.17 |
| 10 | 0.67 ± 0.33 |
| 13 | 3.42 ± 0.11 |
| 14 | 5.12 ± 0.21 |
| 15 | 0.41 ± 0.12 |
| 16 | 2.27 ± 0.17 |
| 21 | 0.82 ± 0.28 |
| 24 | 1.08 ± 0.31 |
| Erlotinib | 0.30 ± 0.09 |
| ΔG | Rmsd | E.vdw | E.Int | E.H.B | Eele | LE | |
|---|---|---|---|---|---|---|---|
| Erlotinib | −6.27 | 1.43 | −33.45 | −69.42 | −10.72 | −37.91 | −4.38 |
| 8 | −6.61 | 1.92 | 58.42 | −83.02 | −9.87 | −42.21 | −3.45 |
| 9 | −6.33 | 1.15 | 30.35 | −43.38 | −11.56 | −42.17 | −2.95 |
| 10 | −6.53 | 1.76 | 34.61 | −51.40 | −11.27 | −36.07 | −3.72 |
| 11 | −6.69 | 1.28 | 54.56 | −47.98 | −9.74 | −40.32 | −2.04 |
| 12 | −6.58 | 1.38 | 21.52 | −35.34 | −10.71 | −41.74 | −4.76 |
| 13 | −6.83 | 1.76 | 56.07 | −71.30 | −12.22 | −36.19 | −3.88 |
| 14 | −6.69 | 1.42 | 54.59 | −33.93 | −9.62 | −38.08 | −1.95 |
| 15 | −7.14 | 1.22 | 45.95 | −77.82 | −10.91 | −41.24 | −5.87 |
| 16 | −7.00 | 1.56 | −12.27 | −44.06 | −10.55 | −41.11 | −2.73 |
| 19 | −6.52 | 1.86 | 3.54 | −62.10 | −9.17 | −38.25 | −3.50 |
| 20 | −6.39 | 1.15 | 8.52 | −65.83 | −10.70 | −39.01 | −2.97 |
| 21 | −6.60 | 0.98 | −9.42 | −43.49 | −10.10 | −39.53 | −6.74 |
| 22 | −6.33 | 1.52 | −8.45 | −29.15 | −9.14 | −34.41 | −4.17 |
| 23 | −6.33 | 1.55 | −2.47 | −55.54 | −10.02 | −32.07 | −4.09 |
| 24 | −6.68 | 1.46 | −2.82 | −85.90 | −8.95 | −37.87 | −4.56 |
| 25 | −6.33 | 1.74 | −77.81 | −41.75 | −10.47 | −39.31 | −3.64 |
| 26 | −6.32 | 1.24 | 56.04 | −34.78 | −11.08 | −38.89 | −2.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, M.M.; Nazreen, S.; Almalki, A.S.A.; Elhenawy, A.A.; Alsenani, N.I.; Elbehairi, S.E.I.; Malebari, A.M.; Alfaifi, M.Y.; Alsharif, M.A.; Alfaifi, S.Y.M. Naproxen Based 1,3,4-Oxadiazole Derivatives as EGFR Inhibitors: Design, Synthesis, Anticancer, and Computational Studies. Pharmaceuticals 2021, 14, 870. https://doi.org/10.3390/ph14090870
Alam MM, Nazreen S, Almalki ASA, Elhenawy AA, Alsenani NI, Elbehairi SEI, Malebari AM, Alfaifi MY, Alsharif MA, Alfaifi SYM. Naproxen Based 1,3,4-Oxadiazole Derivatives as EGFR Inhibitors: Design, Synthesis, Anticancer, and Computational Studies. Pharmaceuticals. 2021; 14(9):870. https://doi.org/10.3390/ph14090870
Chicago/Turabian StyleAlam, Mohammad Mahboob, Syed Nazreen, Abdulraheem S. A. Almalki, Ahmed A. Elhenawy, Nawaf I. Alsenani, Serag Eldin I. Elbehairi, Azizah M. Malebari, Mohammad Y. Alfaifi, Meshari A. Alsharif, and Sulaiman Y. M. Alfaifi. 2021. "Naproxen Based 1,3,4-Oxadiazole Derivatives as EGFR Inhibitors: Design, Synthesis, Anticancer, and Computational Studies" Pharmaceuticals 14, no. 9: 870. https://doi.org/10.3390/ph14090870
APA StyleAlam, M. M., Nazreen, S., Almalki, A. S. A., Elhenawy, A. A., Alsenani, N. I., Elbehairi, S. E. I., Malebari, A. M., Alfaifi, M. Y., Alsharif, M. A., & Alfaifi, S. Y. M. (2021). Naproxen Based 1,3,4-Oxadiazole Derivatives as EGFR Inhibitors: Design, Synthesis, Anticancer, and Computational Studies. Pharmaceuticals, 14(9), 870. https://doi.org/10.3390/ph14090870