1. Introduction
To overcome issues arising from the poor water solubility of many newly discovered drugs, lipid-based colloidal dispersions are under investigation as a promising formulation approach for the parenteral administration of these substances [
1]. Such dispersions may, e.g., be liposomes, nanoemulsions, or may contain solid or liquid crystalline nanoparticles [
2,
3,
4]. Information on the release behavior (or drug retention properties, respectively) of the lipid carrier particles is crucial for quality control, as well as to predict in vivo behavior. Since there is no officially approved test, efforts are being made to design appropriate setups for release testing of nanoparticulate drug carriers [
5].
Challenges to face are, for example, related to the small size of the carrier particles. Only drugs with special properties, such as fluorescence, acidic/basic moieties, or electrochemically active groups, enable the detection of released drug with analytical methods that do not interfere with the dispersed phase particles [
6,
7,
8]. Hence, many methods described in the literature require a separation step, such as filtration or centrifugation, in order to perform quantitative analysis of the released drug [
9,
10]. Other approaches are based on membrane barrier techniques [
11,
12,
13] or continuous flow setups [
14,
15]. Depending on the method, the drug release behavior may be affected by the experimental conditions, e.g., due to high shear stress or long high-speed circulation times, insufficient time resolution, fluctuations in flow rates, or filter clogging [
12,
16,
17,
18,
19].
For dosage forms that are designed to deliver lipophilic drugs via the intravenous (i.v.) route, release testing should be performed in an appropriate medium that reflects the physiological environment. Lipophilic compounds display poor aqueous solubility, and their distribution into mainly aqueous release media, for instance, simple buffer solutions, is limited. As an example, (lipo)proteins or cell compartments represent lipophilic acceptors in the blood that may be available for drug binding. Studies addressing this issue, e.g., investigated the transfer of lipophilic fluorescent dyes or temoporfin as model drugs into the oily droplets of o/w emulsions as acceptor using a flow cytometric approach [
20], or focused on liposomes as acceptor, applying asymmetrical flow field-flow fractionation [
21].
As an even closer approach to physiological conditions, Roese and Bunjes investigated drug transfer into porcine serum and blood using a method based on differential scanning calorimetry (DSC) for detection [
22]. This method circumvents the necessity of separating the donor from the acceptor compartment, but may be limited in applicability to supercooled trimyristin donor nanoparticles and similar systems.
The highest proportion of proteins in human plasma can be attributed to albumin (~55%) [
23]. Upon entering the bloodstream after parenteral administration, many drugs do not only associate with albumin, but also with lipoproteins [
24]. Studies that analyzed the transfer properties of temoporfin from liposomes to some of the individual lipoprotein fractions and albumin in human plasma found significant differences regarding the distribution profiles of the drug [
25,
26]. After i.v. administration of liposomal amphotericin B, a large fraction of the drug was transferred to high density lipoproteins (HDL) [
27]. For α-tocopherol, in contrast, low density lipoproteins (LDL) seem to be the predominant transport vehicle, as this substance was found to considerably associate with LDL after incubation in human plasma [
28].
In order to predict the release performance in vivo, it is most desirable to investigate drug transfer into the original media relevant for administration. Unfortunately, the small size of the carrier particles on the one hand, as well as the complexity of the physiological environment present in vivo on the other hand, entails complications. During in vitro method development, it may thus be preferable to replace the very complex physiological media with simple and robust in vitro media that only contain the ingredients essential for the drug release process.
In a recent study, the transfer of lipophilic drugs from drug-loaded trimyristin emulsions was investigated using an unloaded trimyristin nanoemulsion incorporated into small calcium alginate hydrogel microbeads as a lipophilic acceptor [
29]. This setup combined the advantages of small acceptor particles (with a large corresponding interfacial area) with a simple, filtration-based separation procedure from the donor particles. However, trimyristin emulsion droplets alone may not be sufficiently representative as components of “model-blood”, since other lipophilic substances, such as albumin and lipoproteins, might also have an impact on the drug distribution process. For example, LDL, which represent a large proportion of the plasma lipoprotein fraction, consist, for the most part, of cholesteryl esters [
30,
31].
To achieve an even closer approximation to the lipophilic acceptors in the blood, the trimyristin emulsion employed in the previous study was supplemented in the present study by the additional incorporation of a cholesteryl nonanoate dispersion into the hydrogel particles. Cholesteryl nonanoate was chosen as model cholesteryl ester as it forms nanodispersions in which it remains physically stable in a supercooled liquid crystalline state over a long period of time [
3]. As another advantage, the saturated fatty acid chain of the cholesteryl nonanoate molecule is more resistant to chemical degradation, such as oxidation, in comparison to unsaturated derivates. It was an aim of this study to comprehensively characterize the trimyristin and cholesteryl nonanoate-containing hydrogel particles in order to perform transfer studies in a more advanced transfer medium. Using this approach, the contribution of the “model lipoprotein” acceptor hydrogel particles to the transfer of fenofibrate, retinyl acetate, and orlistat was investigated from trimyristin donor emulsions. Additionally, the transfer performance of these drugs was investigated from the same donor emulsions into albumin solution as acceptor by applying the DSC method for drug detection [
22].
3. Materials and Methods
3.1. Materials
The triglyceride trimyristin (Dynasan® 114) was donated by IOI Oleo, Witten, Germany, and the surfactant poloxamer 407 (Kolliphor® P127) by BASF AG, Ludwigshafen, Germany. Sodium alginate (Manugel® GMB) was a kind gift from FMC International, Wallingstown, Ireland. As estimated by the supplier, the molecular weight was ~124 kDa, the content of guluronic acid was 60–70%, and that of mannuronic acid was 30–40%. Cholesteryl nonanoate was purchased from TCI, Zwijndrecht, Belgium. Tetrahydrofuran (HPLC grade), acetonitrile (HPLC grade), bovine serum albumin (BSA, heat shock fraction, pH 7, ≥98%), and the drugs fenofibrate and retinyl acetate were obtained from Sigma-Aldrich, Steinheim, Germany. Orlistat was donated by Formosa Laboratories Inc., Taoyuan, Taiwan. Sodium azide, anhydrous glycerol, calcium chloride, acetonitrile (LC MS grade), and tetrahydrofuran (ultra LC MS grade) were obtained from Carl Roth, Karlsruhe, Germany. All materials were used as received. Water was purified by deionization and filtration (EASYpureTM LF, Barnstead, Dubuque, IA, USA) or was of bidistilled quality. The logP values of the drugs were obtained from DrugBank (calculated by ALOGPS).
3.2. Preparation of Donor and Acceptor Lipid Nanodispersions
The nanoemulsions consisted of 10% trimyristin (TM) as lipid phase, which was dispersed in an aqueous phase containing 5% poloxamer 407 as a stabilizer. The aqueous phase was isotonized with 2.25% anhydrous glycerol. Additionally, nanodispersions were prepared that contained 10% cholesteryl nonanoate (CN) as lipid phase. These dispersions were stabilized with 8% poloxamer 407. All lipid nanodispersions were preserved with 0.05% sodium azide. The concentrations are given related to the total weight of the dispersions (w/w).
The aqueous and lipid phases were preheated separately to 75 °C (TM nanoemulsions) or 95 °C (CN dispersions). After mixing, a pre-emulsion was formed using an Ultra-Turrax (T25 digital, IKA, Staufen, Germany) for four minutes at 11,000 rpm. Subsequently, the mixture was processed in the heat by high-pressure homogenization in 10 cycles at 700 bar (TM emulsions) or 900 bar (CN dispersions) using a Microfluidizer (M110-PS, interaction chamber type F12Y DIXC, Microfluidics, Newton, MA, USA).
After homogenization, all dispersions were filtered through a polyvinylidene fluoride (PVDF) filter with 0.45 μm pore size (Rotilabo
®, Karlsruhe, Germany) and stored in glass vials at 20 °C. Under these conditions, TM remained in a liquid state due to supercooling [
22], whereas the CN transformed into a liquid crystalline state [
3]. The trimyristin nanoemulsion and the cholesteryl nonanoate dispersion were mixed in a ratio of 9 CN + 1 TM (CNTM dispersion; lipid ratio based on quantification via HPLC), and served as the acceptor system to be incorporated in alginate beads (cf.
Section 3.4).
For the preparation of donor emulsions to be studied in transfer experiments, fenofibrate, retinyl acetate, or orlistat were dissolved in the melted trimyristin prior to emulsification. In order to exclude any possible influence of the drug loading on the transfer kinetics, all emulsions were loaded at the same concentration of ~3% related to trimyristin.
3.3. Lipid Quantification via High Performance Liquid Chromatography
A slight reduction in the lipid concentration may occur during dispersion production by dilution with process water remaining in the homogenization device. In order to achieve the lipid mixing ratio of 9 CN + 1 TM accurately, the lipid content of the unloaded dispersions was determined by HPLC after preparation. A Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) equipped with an LPG-3400SD pump, a WPS-3000TSL autosampler, and a Corona Veo Charged Aerosol detector was used to perform the analysis. The column (Thermo Fisher Scientific Hypersil Gold C18, 2.1 × 150 mm, 1.9 μm) was kept at 25 °C and the flow rate was set to 0.3 mL/min. The mobile phase consisted of acetonitrile/tetrahydrofuran 70/30 (v/v). Under these conditions, the retention time of both lipids was between 3 and 5 min, with cholesteryl nonanoate eluting from the column prior to trimyristin.
For sample preparation, dispersions were dissolved in tetrahydrofuran/acetonitrile 50/50 (v/v) and diluted to an appropriate detector response; 1 μL was injected and detected at a nebulizer temperature of 50 °C. Every sample was diluted twice and every dilution measured two times (n = 4). Lipid concentrations were calculated with the Chromeleon 7.2 software (Thermo Fisher Scientific, Waltham, MA, USA) using a calibration curve for trimyristin or cholesteryl nonanoate in different concentrations.
3.4. Preparation of Lipid-Containing Alginate Beads
Calcium alginate beads were produced with a spraying method as described earlier, with minor modifications [
29]. The lipid nanodispersion that was incorporated into the hydrogel beads was composed of cholesteryl nonanoate and trimyristin (lipid mixing ratio 9 + 1 as determined by HPLC). The drug-free lipid nanodispersion was mixed with the same volume of bidistilled water (final volume approximately 20–25 mL each per batch) and 2% (
w/w) sodium alginate was added to the dispersion. Under stirring at 200 rpm, the mixture was left to swell overnight. With the aid of a syringe pump (Fusion 200, Chemyx, Stafford, TX, USA), the resulting alginate-containing dispersion was fed (1 mL/min) into the two-fluid spray nozzle (diameter: 0.7 mm) of a BÜCHI Mini Spray Dryer B-191 (BÜCHI Labortechnik AG, Flawil, Switzerland) and sprayed under compressed air (650 L/h) into a continuously stirred 5% (
w/w) CaCl
2 solution (approximately 500 mL). The hydrogel particles were stored in the CaCl
2 solution overnight to ensure thorough cross-linking. After hardening, excess CaCl
2 was washed off with purified water via centrifugation (SIGMA
® 3–15, Sigma Laborzentrifugen GmbH, Osterode am Harz, Germany) three times at 3200 rpm. The microbeads were stored in water and used as acceptor particles. The resulting volume of each batch of dispersion was about 40–50 mL in total. The overall lipid concentration encapsulated in each batch of hydrogel bead dispersion was evaluated via DSC (cf.
Section 3.6). Plain microbeads used for the cryo-SEM and SAXS analysis were produced in the same way but without lipid dispersion.
3.5. Particle Size Analysis
The particle size of the lipid nanodispersions was measured by PCS using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). Prior to the measurement, samples were diluted with purified water to an appropriate scattering intensity (attenuator 5–7). After an equilibration time of 300 s, three consecutive measurements of 5 min each were performed at 25 °C using a laser wavelength of 633 nm at an angle of 173°. As an average of three runs, the z-Average and the PdI were calculated.
The particle sizes of the hydrogel beads were determined via laser diffraction (LD; Beckman Coulter LS 13 320, Beckman Coulter GmbH, Krefeld, Germany). The samples were diluted with water to an appropriate optical density in the measuring chamber. Three consecutive measurements of 90 s each were averaged and the volume distribution, mean particle size, and D10, D50, and D90 values were calculated using Fraunhofer approximation.
3.6. Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) measurements were carried out using a DSC 1 calorimeter (Mettler Toledo, Gießen, Germany) equipped with an FRS 5+ sensor that was calibrated with indium. The calibration was checked by measuring indium before a series of measurements. About 20 mg of the samples were accurately weighed into 40 µL aluminum pans (Mettler Toledo, Gießen, Germany), which were hermetically sealed by cold welding. An empty pan was used as reference and all measurements were performed under nitrogen purge.
To examine the trimyristin concentration in the hydrogel bead dispersion, as well as in the drug-loaded and unloaded trimyristin-containing dispersions, samples were heated from 20 °C to 70 °C (20 K/min) and subsequently cooled to −5 °C with a scan rate of 10 K/min. The crystallization enthalpies from the cooling curves were evaluated and the trimyristin content was calculated using a calibration curve obtained from measuring different amounts of bulk trimyristin under the same conditions. The overall lipid content in mixed CNTM dispersions (composed of 9 CN + 1 TM), as well as the lipid amount in the hydrogel particle dispersions, was calculated by multiplying the determined trimyristin amount by 10.
The onset value of the crystallization signal of trimyristin was determined as an indicator for the crystallization temperature (T
cryst.). Samples from transfer experiments were cooled from 25 °C to 0 °C with a scan rate of 2.5 K/min. The changes in T
cryst. of drug-loaded nanoemulsions were used to quantify the transferred amount of drug, as described in earlier studies; cf.
Section 3.9.
In order to obtain information on the liquid crystalline structure, the cholesteryl no-nanoate dispersion and the CNTM-containing nanodispersion (also enclosed in the hydrogel beads) were heated from 15 °C to 100 °C (5 K/min), and subsequently cooled to −10 °C (5 K/min). The phase transitions indicated in the resulting curves were examined to characterize the structure of the incorporated lipid particles in comparison to those of unencapsulated counterparts.
If necessary, baseline correction was performed using OriginLab 2018.
3.7. Cryo-SEM
To evaluate the inner appearance of the placebo and the lipid-containing hydrogel beads, cryo-scanning electron microscopy (cryo-SEM) was performed using a Helios G4 CX DualBeam system and a Through the Lens Detector (FEI, Hillsboro, OR, USA). For sample preparation, the hydrogel microbeads were frozen in a high-pressure freezer (Leica EM ICE, Leica Microsystems GmbH, Wetzlar, Germany) using liquid nitrogen, subsequently fractured in the cryo-chamber at −150 °C, and sputtered with a 4 nm platinum layer in a high-vacuum coater (Leica EM ACE600, Leica Microsystems GmbH, Wetzlar, Germany). Imaging was performed at a voltage of 3 kV at different magnifications. The samples were kept under cryo conditions throughout the entire workflow.
3.8. X-ray Scattering
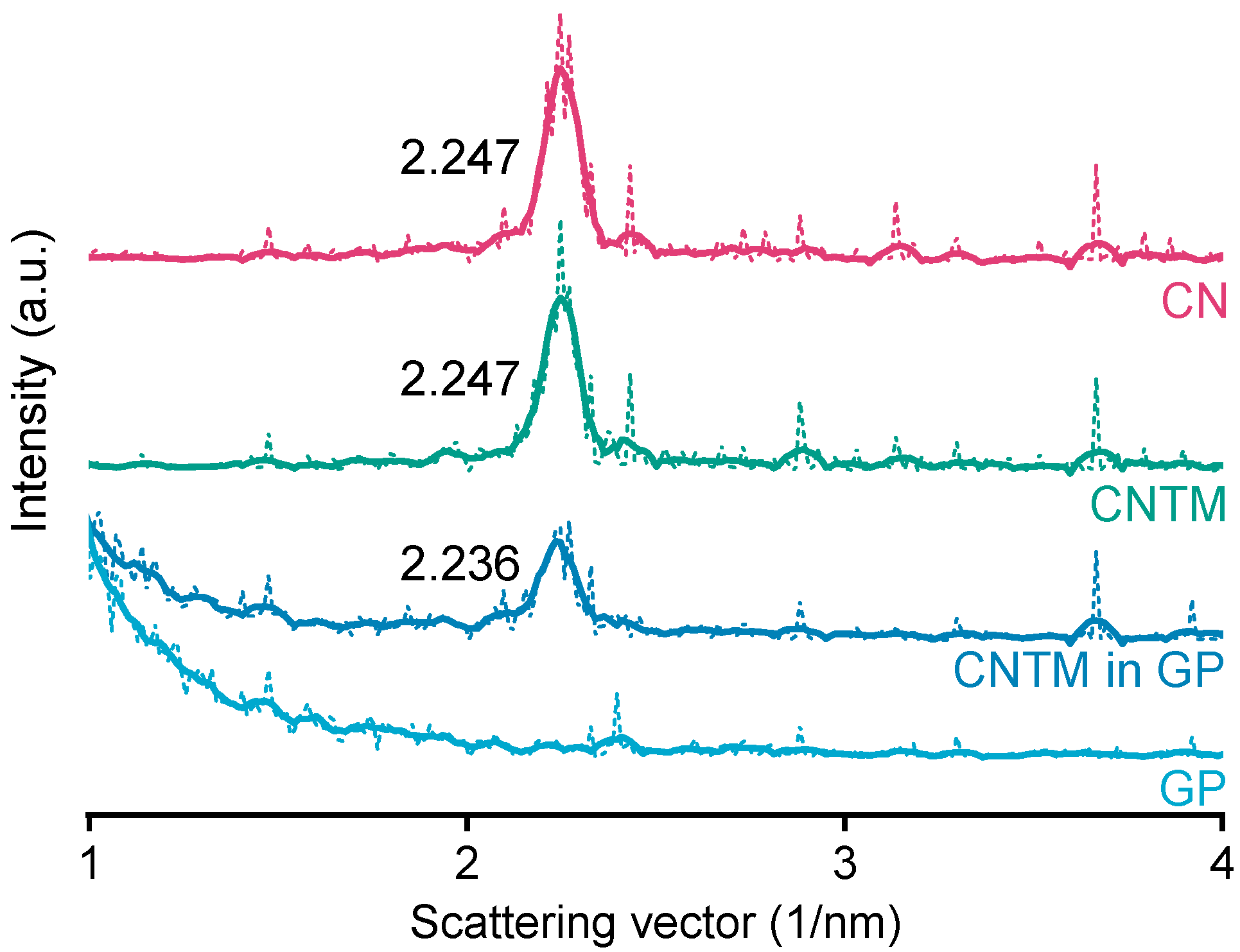
Small-angle X-ray scattering (SAXS) was performed to investigate the liquid crystalline structure of the cholesteryl-nonanoate-containing dispersions. The measurements were conducted with a SAXSess mc2 system (Anton Paar GmbH, Graz, Austria) using Cu Kα radiation (λ = 0.154) and a CCD detector (measurement range: q = 0 − 6 nm−1). The nanodispersions, as well as hydrogel beads (lipid-containing and placebo), were measured at room temperature in a 1 mm capillary, which was positioned in the beam path at a distance of 309 mm to the CCD detector.
Background and dark current subtraction, as well as desmearing, were performed using the SAXSquant software (Anton Paar GmbH, Graz, Austria). The raw data (dotted lines in
Figure 3) were appropriately smoothed (solid lines;
Figure 3), and the scattering vector q (nm
−1) was determined using OriginLab 2018. The position of the reflections was used to calculate the layer spacing (d) according to Bragg’s law: d = 2π/q.
3.9. Investigation of Drug Transfer
3.9.1. CNTM-Containing Hydrogel Beads as Acceptor
The procedure for drug transfer investigations using lipid-containing hydrogel beads as an acceptor was described earlier [
29,
33]. Briefly, drug-loaded nanoemulsions were mixed with the microsphere dispersion in 3 mL glass vials in a donor (d) to acceptor (a) lipid mass ratio of 1 + 9 (based on the results of lipid determination by DSC). This lipid ratio was chosen to ensure comparability of the present transfer results with those of a previous study [
29]. The transfer started when the donor emulsion (~70 µL) was added to the required amount of acceptor particle dispersion (resulting in a total volume of approximately 1.5 mL in each transfer vial). For each time point of sampling, a separate transfer vial was used. During transfer, the samples were placed on a horizontal shaker (Vibrax VXR Basic, IKA-Werke GmbH & Co. KG, Staufen, Germany) and agitated with 300 rpm at ~23 °C. Samples were withdrawn using a 2 mL plastic syringe via filtration at predetermined time points. For this purpose, a polyethersulfone (PES) membrane with 1.2 μm pore size (Pieper Filter GmbH, Bad Zwischenahn, Germany) was mounted into a custom-built screw cap that was attached to the transfer vial shortly before sampling.
For fenofibrate and retinyl acetate, the drug load of the donor nanoemulsions, as well as the remaining amount of drug in the nanoemulsions during transfer experiments, was quantified via UV spectroscopy (Specord 40, Analytik Jena AG, Jena, Germany). Samples were dissolved in tetrahydrofuran/water 9/1 (v/v) and measured at wavelengths of 287 nm (fenofibrate) or 360 nm (retinyl acetate) three times. Where required, the measured absorptions were corrected for the blank absorptions of the dissolved unloaded nanoemulsion that had been treated in the same way as the respective drug-containing nanoemulsion. Calibration curves for each drug were obtained by preparing at least six different dilutions containing varying amounts of the respective drug. The amount of transferred drug was calculated by subtracting the amount in the sampled aqueous donor system from the originally applied one.
Orlistat could not be quantified via UV spectroscopy in the presence of trimyristin because of overlapping absorption signals. Thus, orlistat transfer from the trimyristin nanoemulsion into the lipophilic acceptor was investigated by DSC. The change in crystallization temperature (ΔT
cryst., determined upon cooling) is in a linear relation to the decrease in drug content [
22]. The respective donor emulsion, diluted with water in the same volume as that of the acceptor system (which corresponded to 0% drug transfer), and an unloaded trimyristin nanoemulsion with comparable characteristics (corresponding to 100% drug transfer; measured as control) were used to calculate the transferred amount of orlistat by applying the rule of three. Control experiments were performed by incubating unloaded trimyristin emulsion with the lipid-containing microsphere dispersion under the same conditions. Fluctuations in the crystallization temperature of the donor emulsion that were not caused by drug transfer could thus be identified and included into the calculations of the transferred amount of drug [
22].
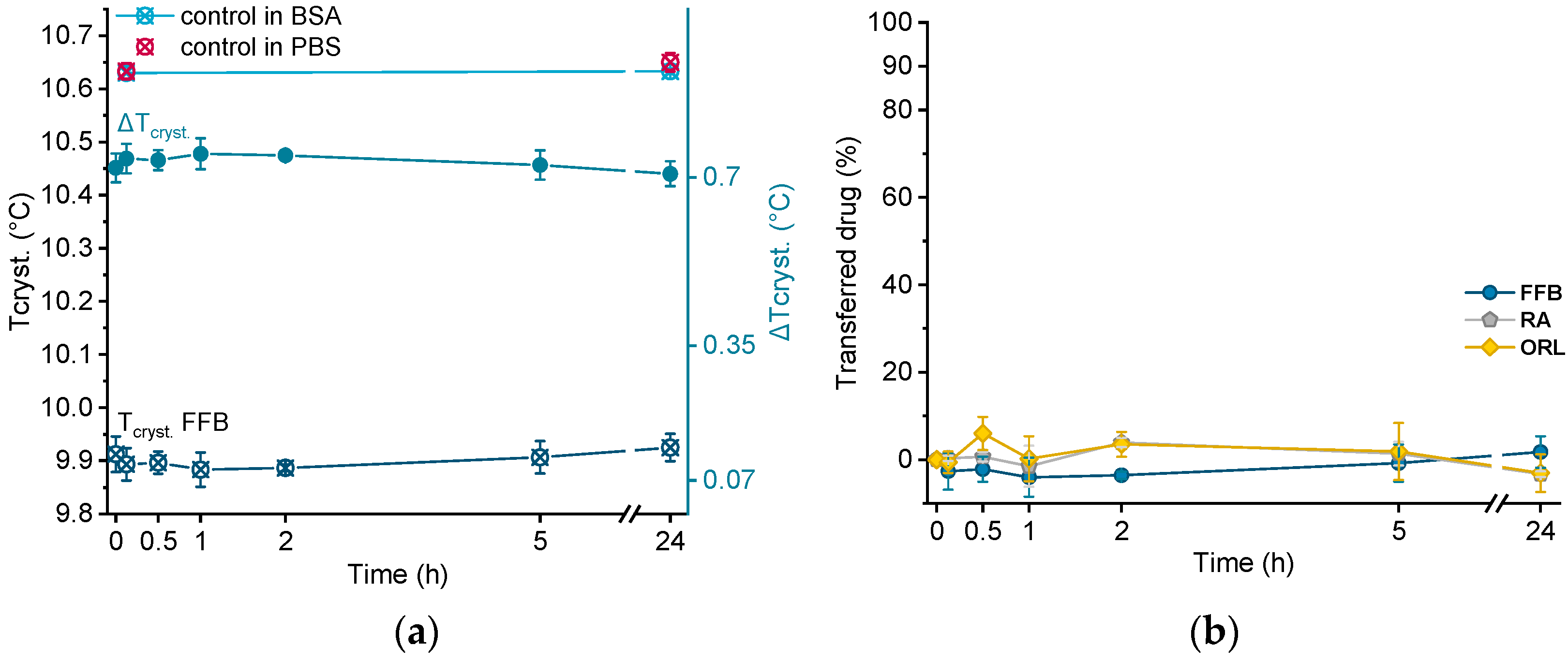
3.9.2. Albumin Solution as Acceptor
The overall amino acid sequence identity of BSA in comparison to human serum albumin (HSA) is ~76%, leading to a similar tertiary structure of these molecules, with similar binding sites [
40,
41]. In order to evaluate the contribution of albumin as a potential acceptor for lipophilic drugs in blood, BSA was used for screening purposes in this study.
BSA solution was freshly prepared before use by dissolving 43 mg/mL BSA in PBS buffer under continuous stirring at 200 rpm. This concentration was chosen for two reasons. First, the concentration of albumin in human plasma is 35–50 mg/mL [
23] and, second, the concentration of lipid acceptor in the studies performed here with CNTM-containing dispersion that was incorporated in microspheres was in the same range (cf.
Section 2.1.3). Thus, the chosen BSA concentration offered the possibility to compare the contribution of (different) available acceptor structures used in this study on the one hand, and also provided a realistic approach to the albumin concentration in vivo.
A total of 70 µL of the drug-loaded donor emulsions were mixed in a ratio of 1 + 9 (ratio adjusted based on the lipid quantification in the donor emulsion, as determined using DSC) with the acceptor solution in a 3 mL vial and incubated for 24 h in the same way as described above (total volume ~1.5 mL/vial). At various time points, samples were withdrawn using an Eppendorf pipette and directly measured by DSC. In contrast to the hydrogel-bead-based setup, no filtration step was necessary using this method. The change in crystallization temperature (ΔT
cryst.) determined upon cooling was used to calculate the transferred amount of each drug under investigation, as described in
Section 3.9.1 for orlistat. For the control experiments, an unloaded trimyristin emulsion with comparable characteristics was incubated in the acceptor solution and in PBS buffer, and measured by DSC as well.
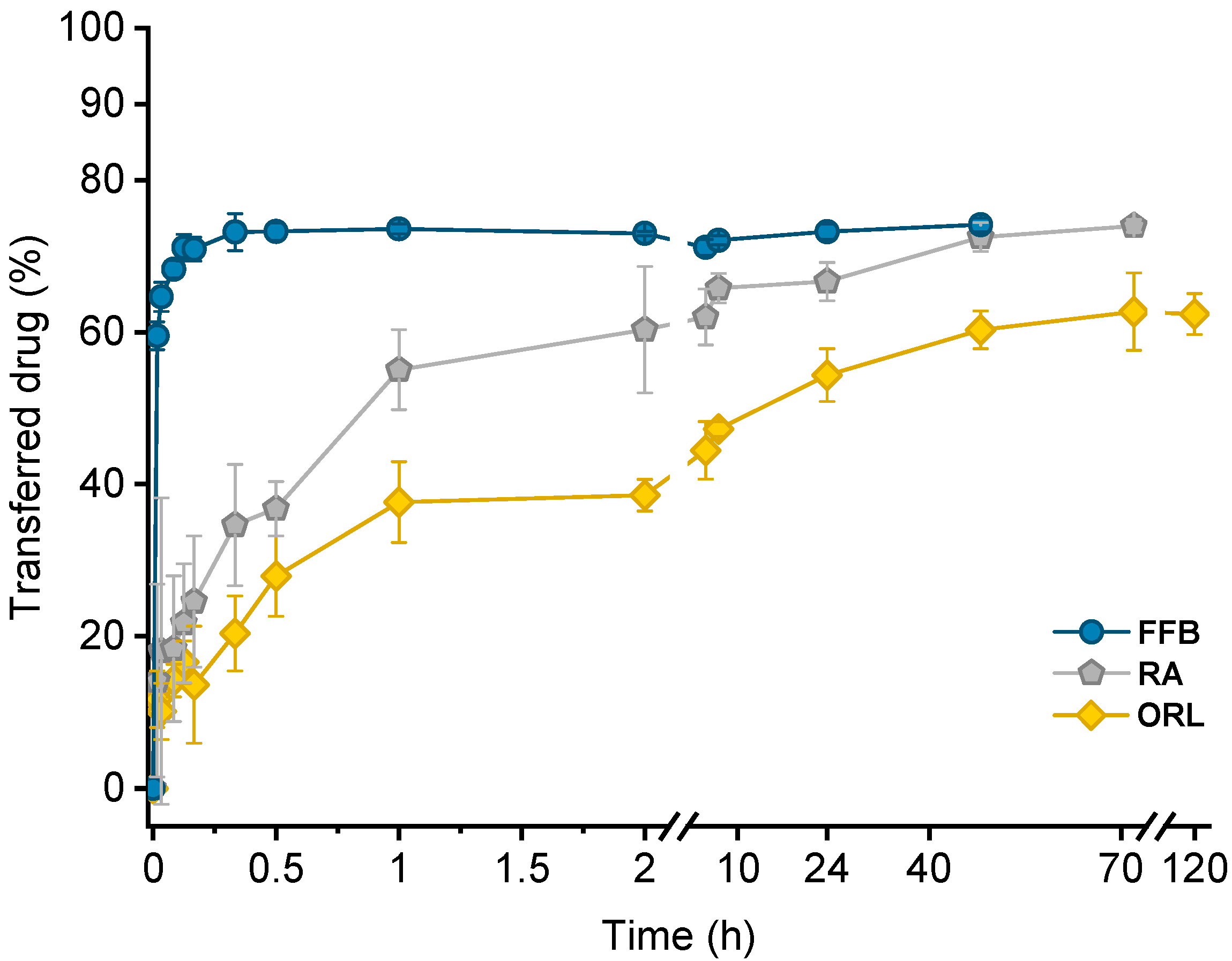
All transfer results are presented by plotting the fraction of transferred drug (%) against the time (hours, h). All transfer studies were performed in triplicate.
4. Conclusions
The liquid (trimyristin) and liquid crystalline (cholesteryl nonanoate) state, as well as the integrity of the nanoparticles, could be preserved during hydrogel bead production, and the resulting system was successfully applied as lipophilic acceptor in transfer studies. The course of transfer observed using the lipid-containing hydrogel particles as the acceptor was in relation to the lipophilicity of the drugs: the higher the logP value, the slower the transfer. In all cases, the partition equilibrium of the drugs under investigation was found to be in favor of the trimyristin emulsion droplets. Given that there is no officially approved method to investigate the release of lipophilic drugs from nanosized carriers, the hydrogel-bead-based setup can be helpful in order to compare the contribution of different lipophilic acceptors to the release performance of colloidal drug delivery systems. The nature of the lipophilic acceptor in release studies is essential, as it strongly affects the release behavior. No detectable fraction of the drugs was transferred to BSA, demonstrating clearly that albumin seemed to be of minor importance as lipophilic acceptor for the drugs under investigation in the present study. Albumin as solubilizing agent to be used in transfer experiments should thus be evaluated thoughtfully, especially for drugs with high lipophilicities. The lipophilic substances used in the present study as an acceptor were selected as model compounds in order to mimic different lipophilic acceptors present in the blood. Thus, a closer approach to the physiological environment was provided than with many other release media currently applied. However, many other aspects, e.g., the physical state of the acceptor particles and, with special regard to the intravenous route of administration, a realistic dilution of the donor system, should be taken into consideration for future investigations, as they may also affect the drug release performance.




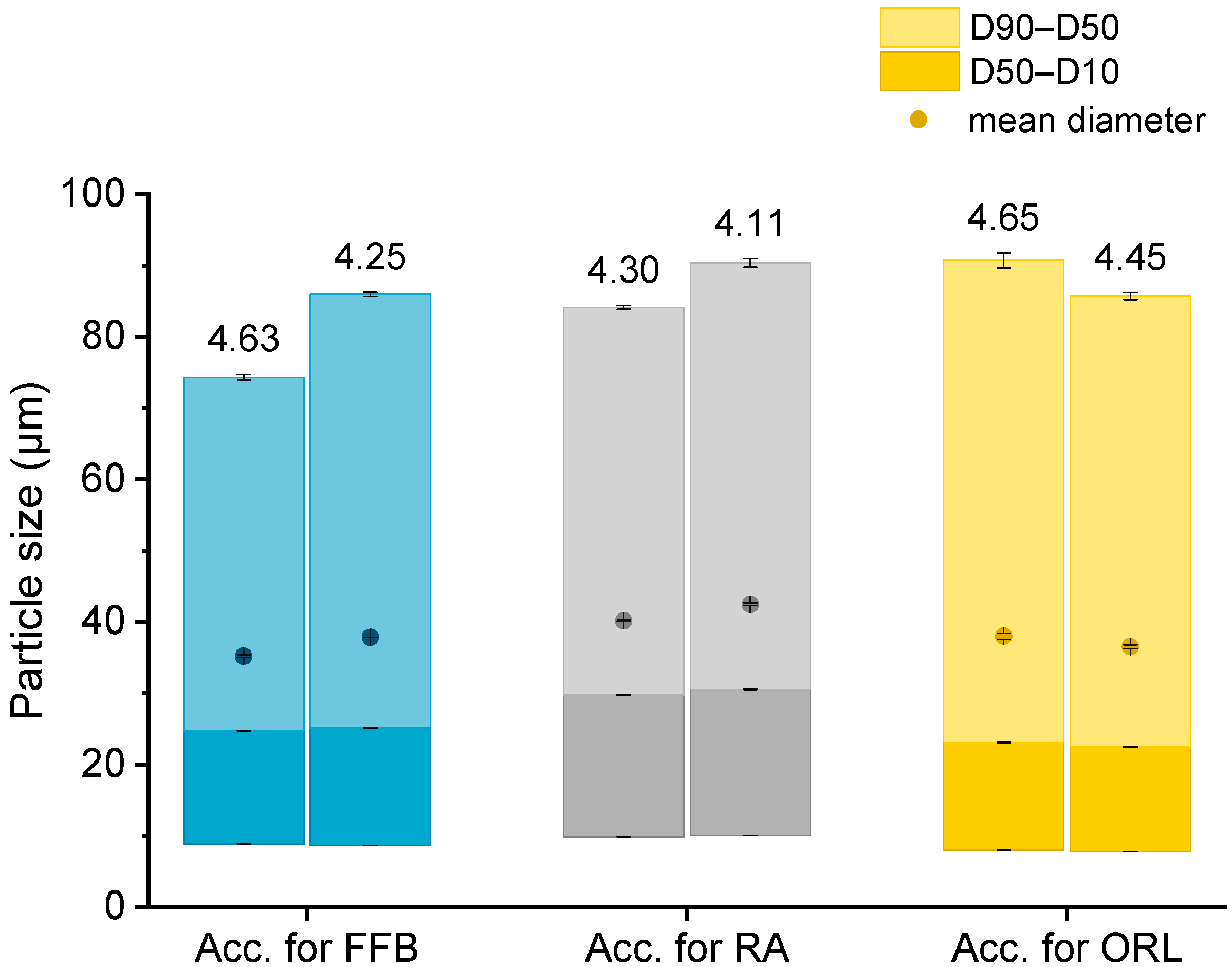



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}