
Improved HDAC Inhibition, Stronger Cytotoxic Effect and Higher Selectivity against Leukemias and Lymphomas of Novel, Tricyclic Vorinostat Analogues

 , , , , , and
, , , , , and
Abstract
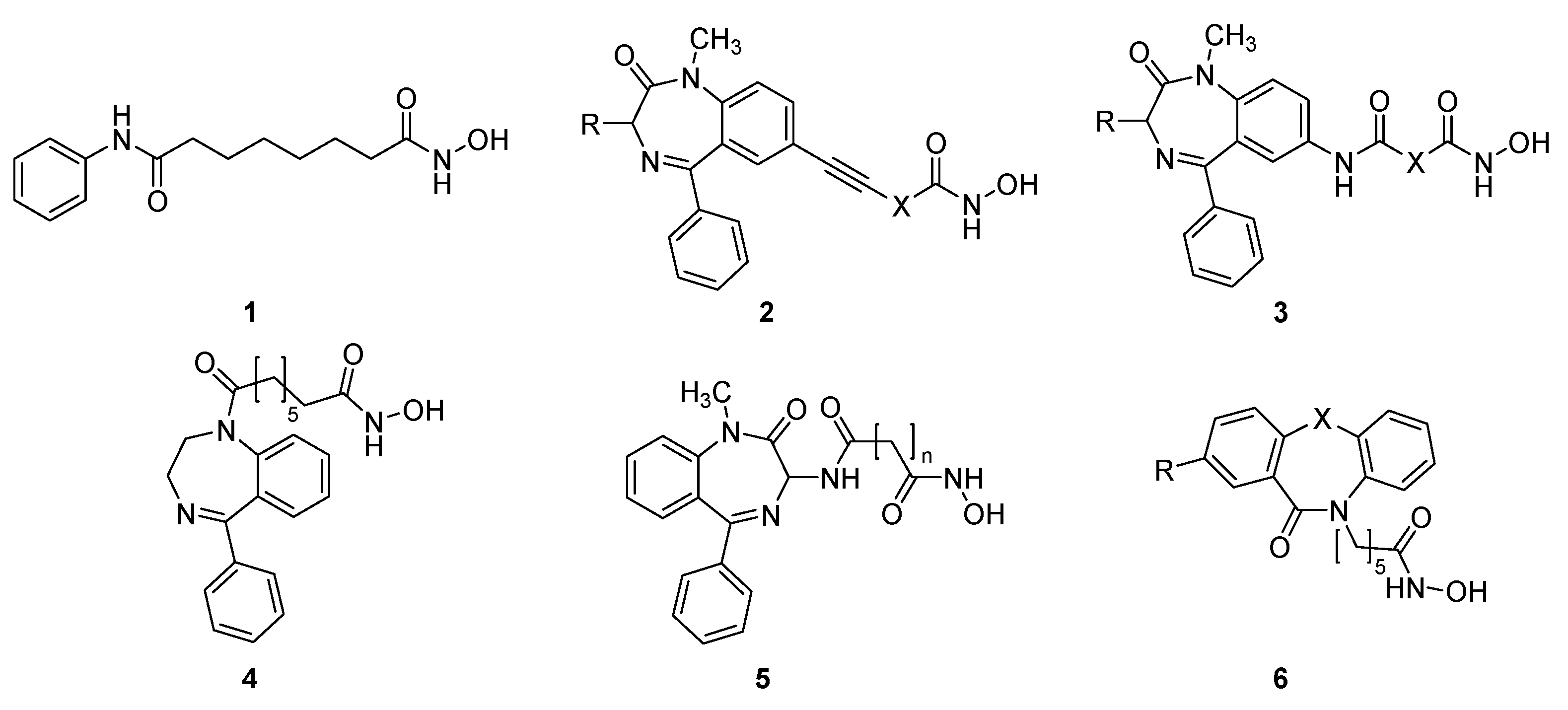
:1. Introduction
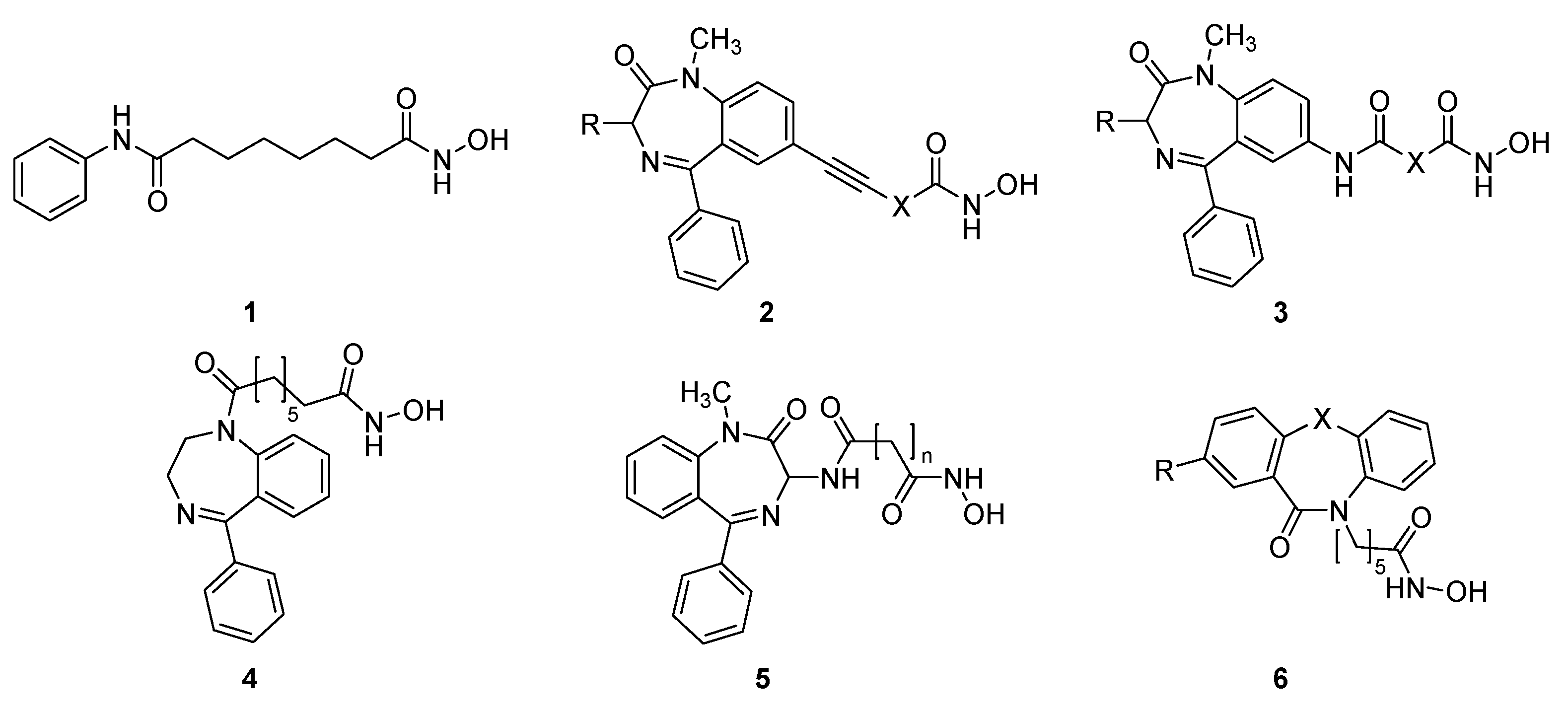
2. Results and Discussion
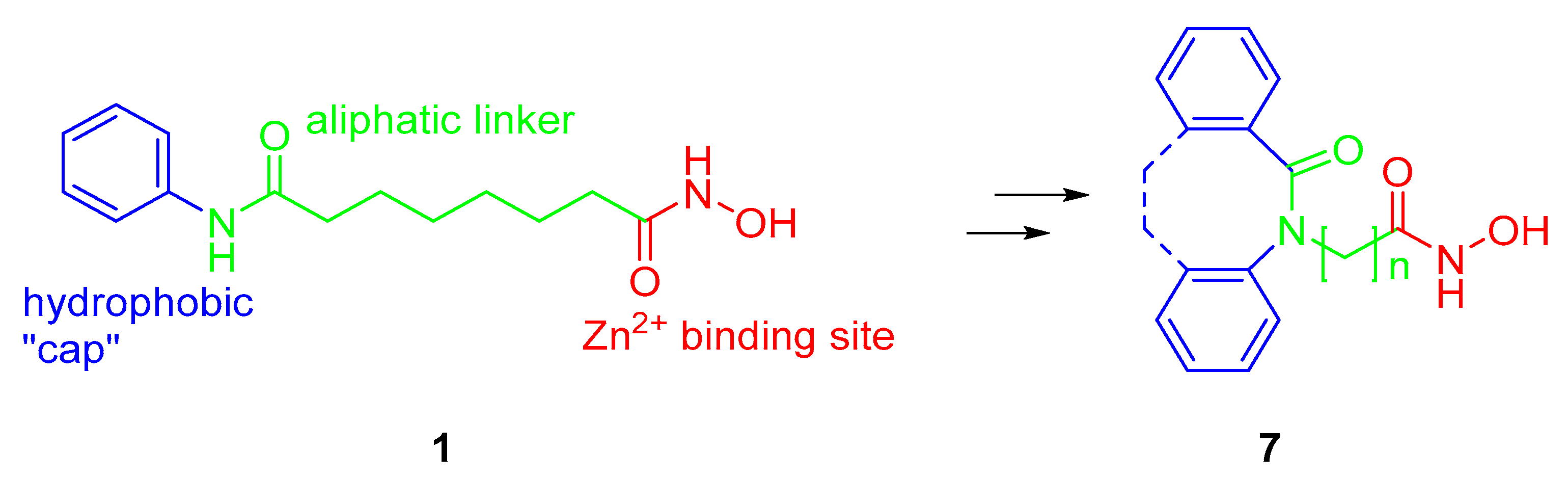
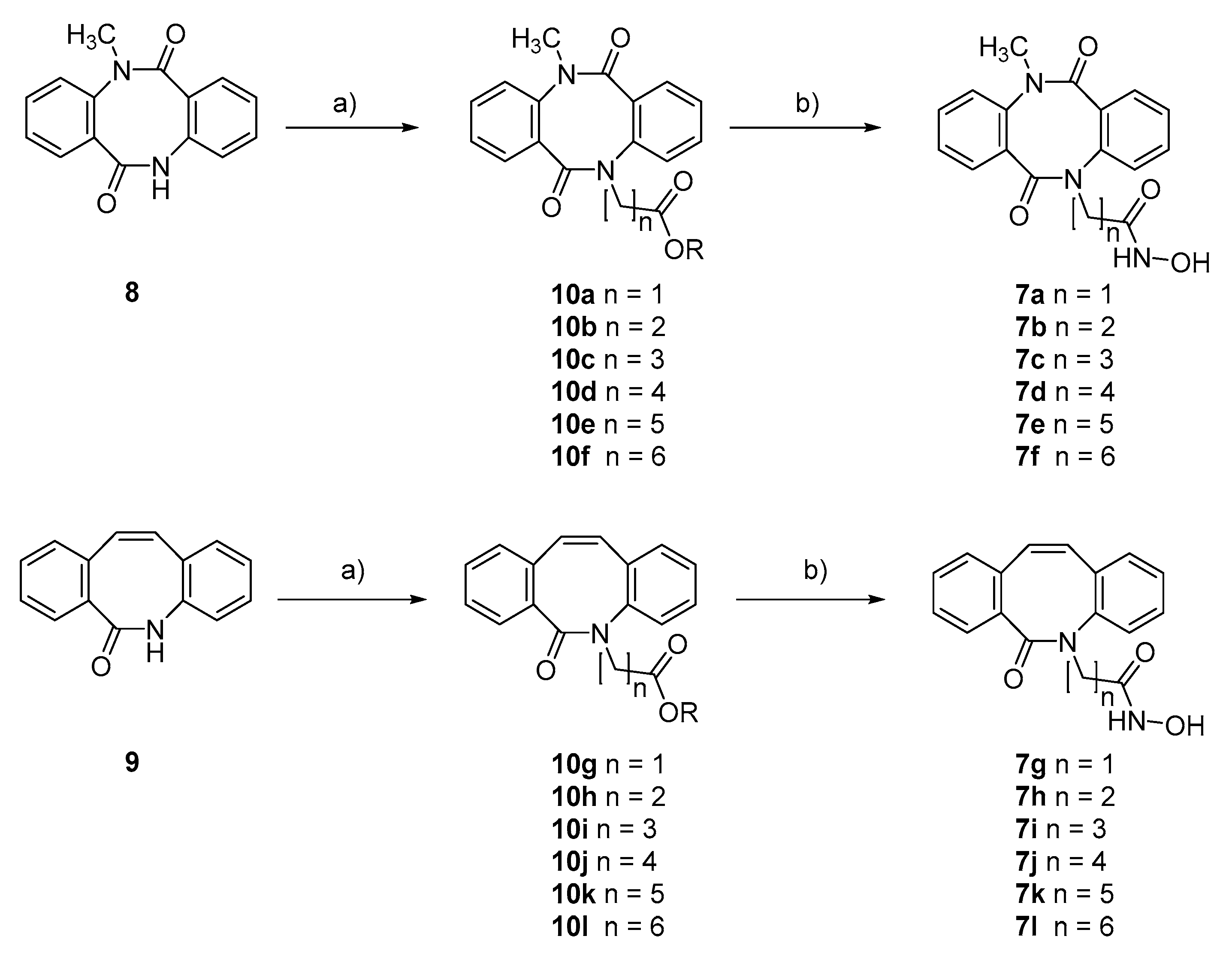
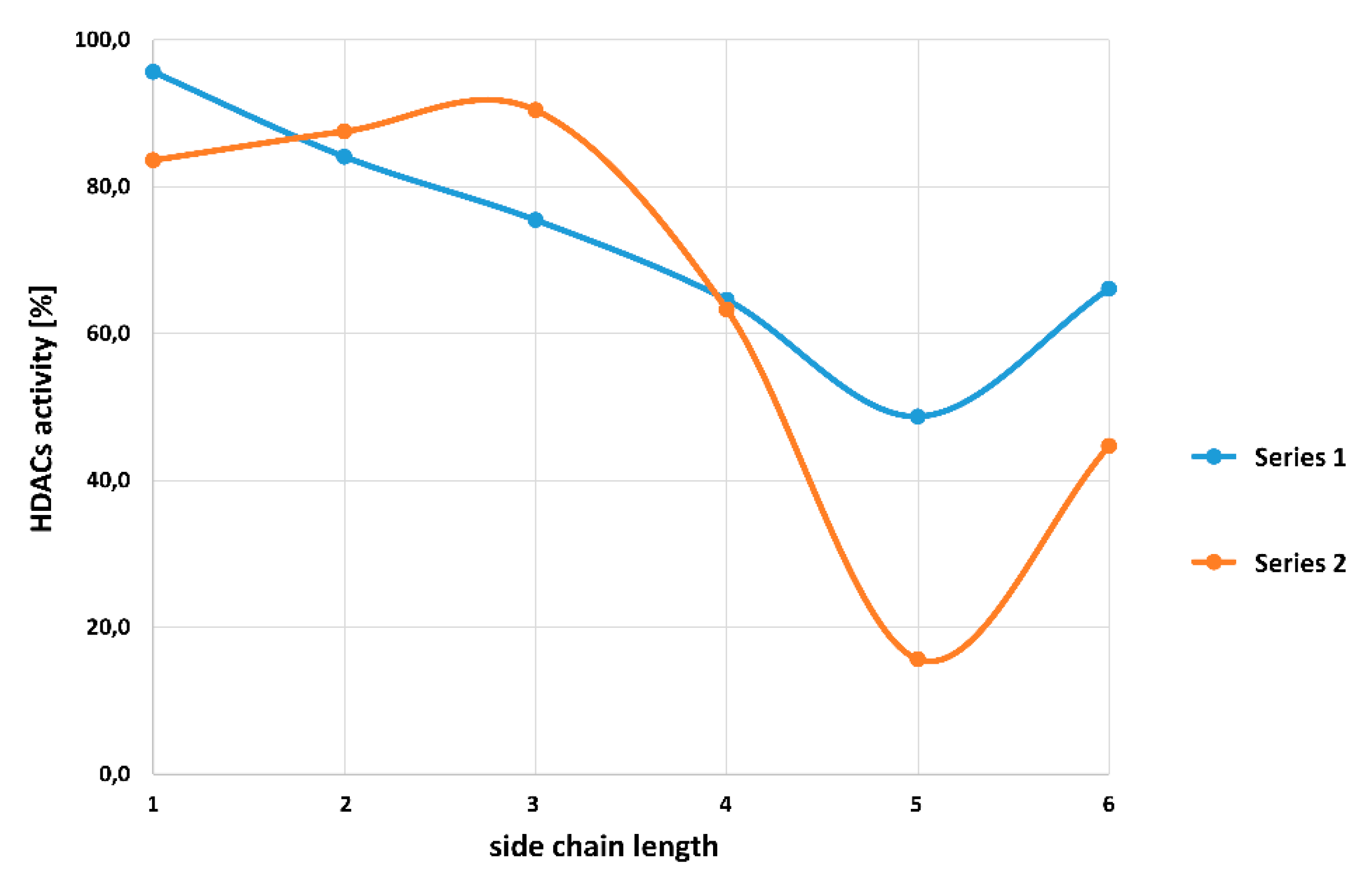
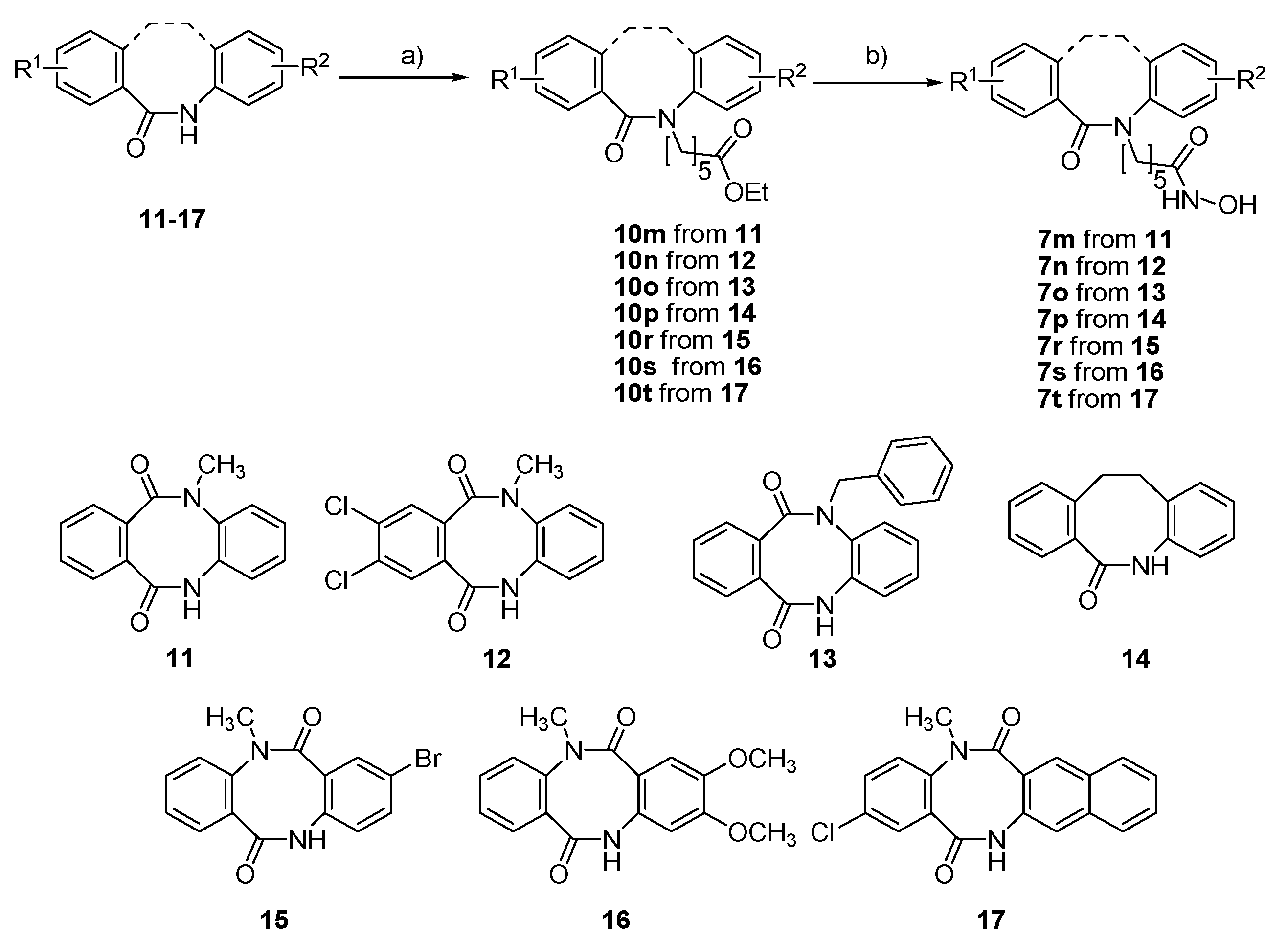
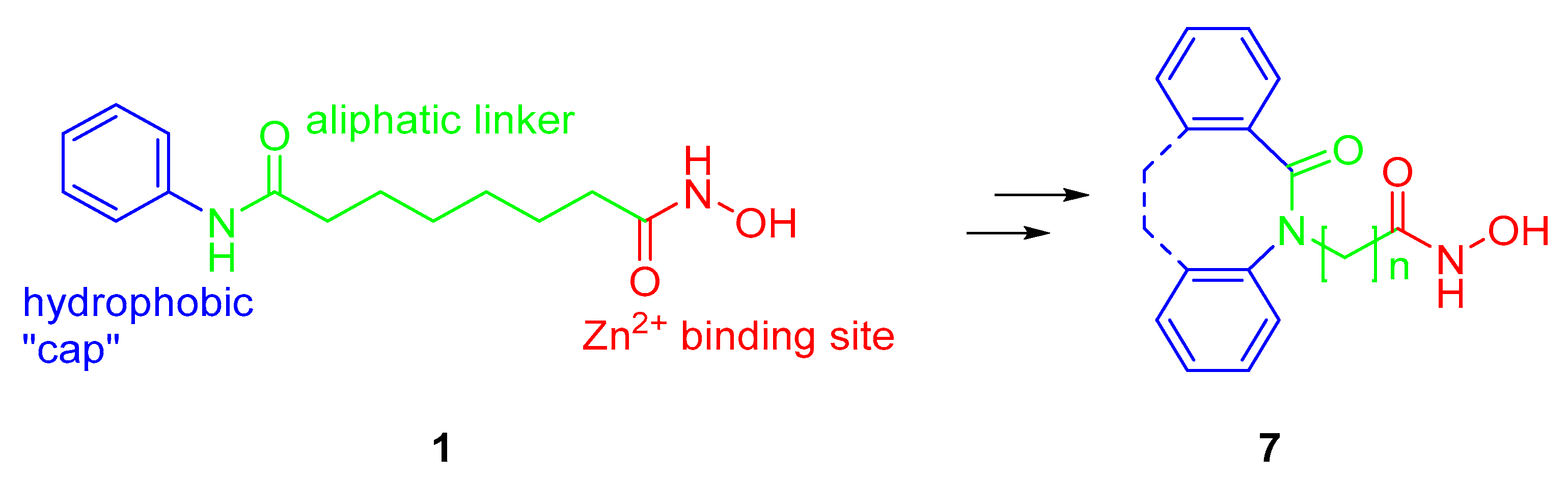
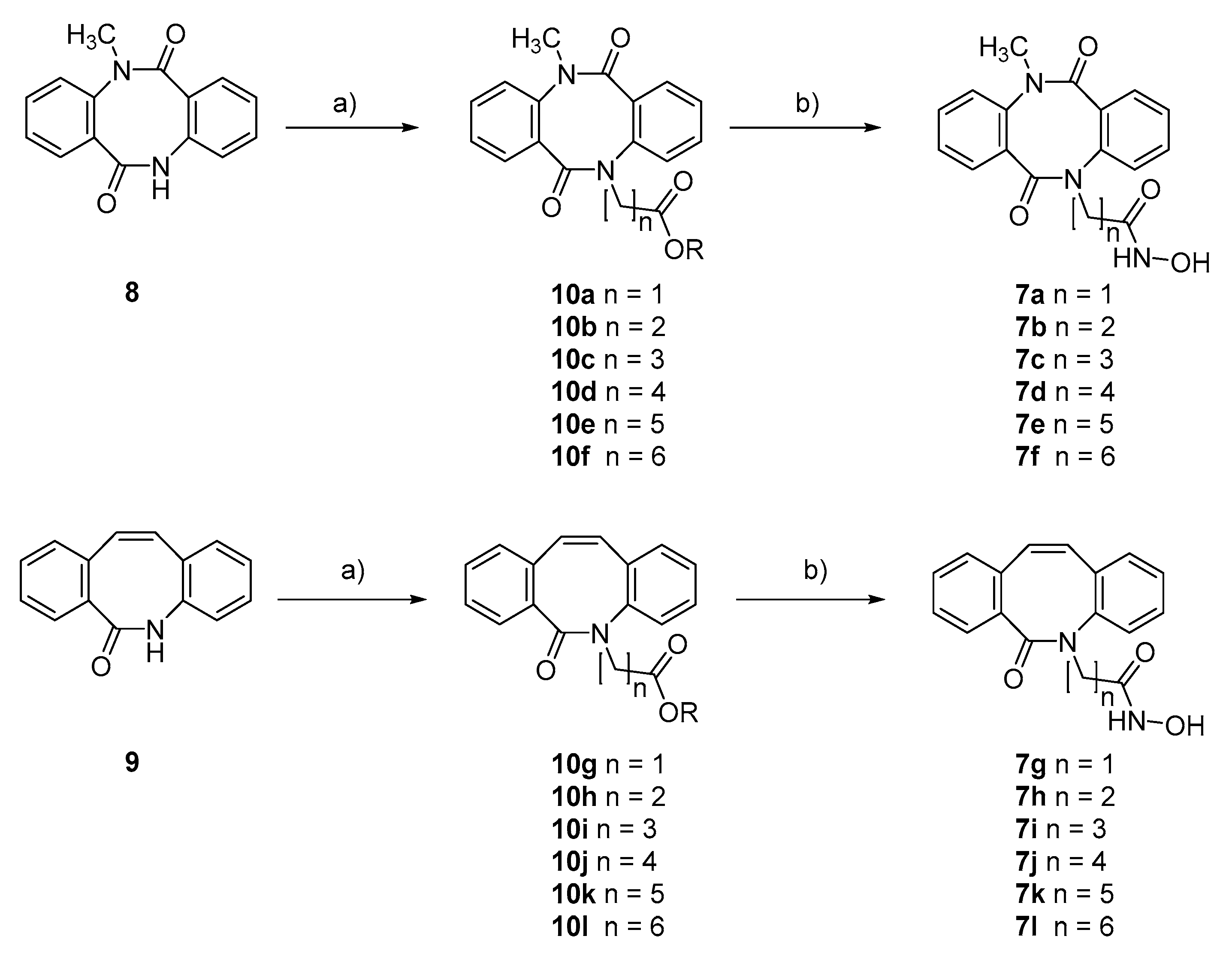
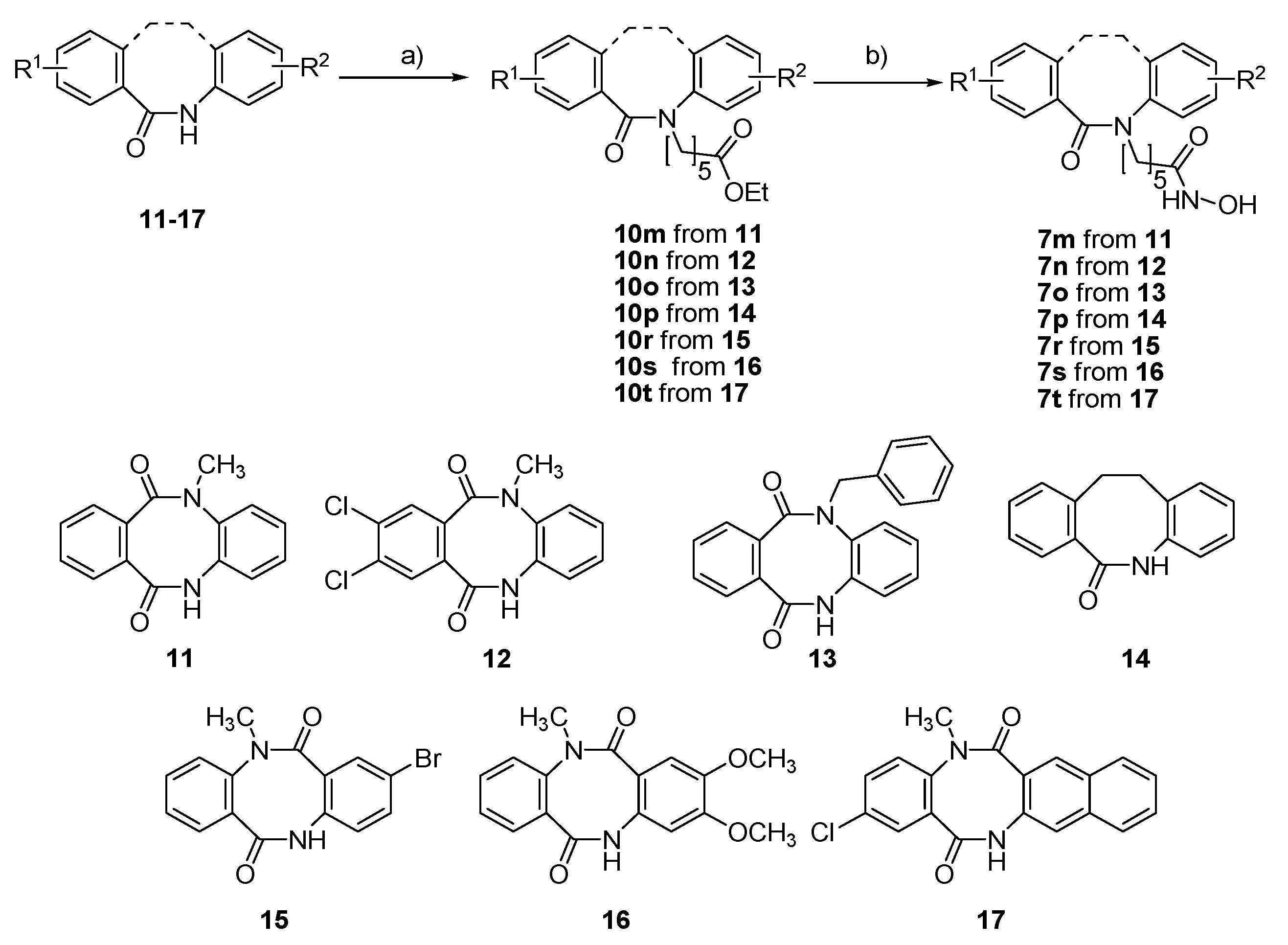
2.1. Synthesis and HDAC Inhibition
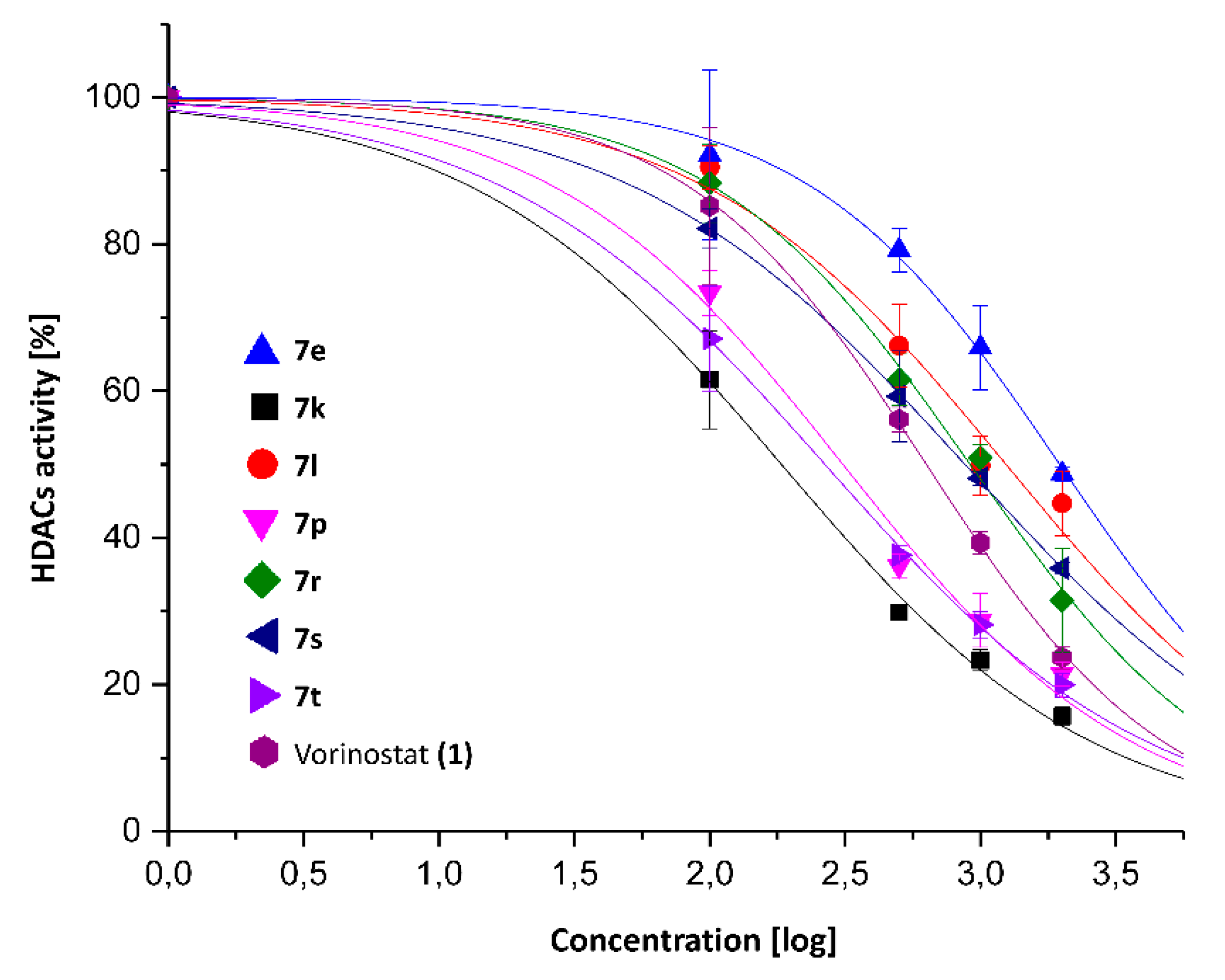
2.2. Cytotoxic Activity and Selectivity Index
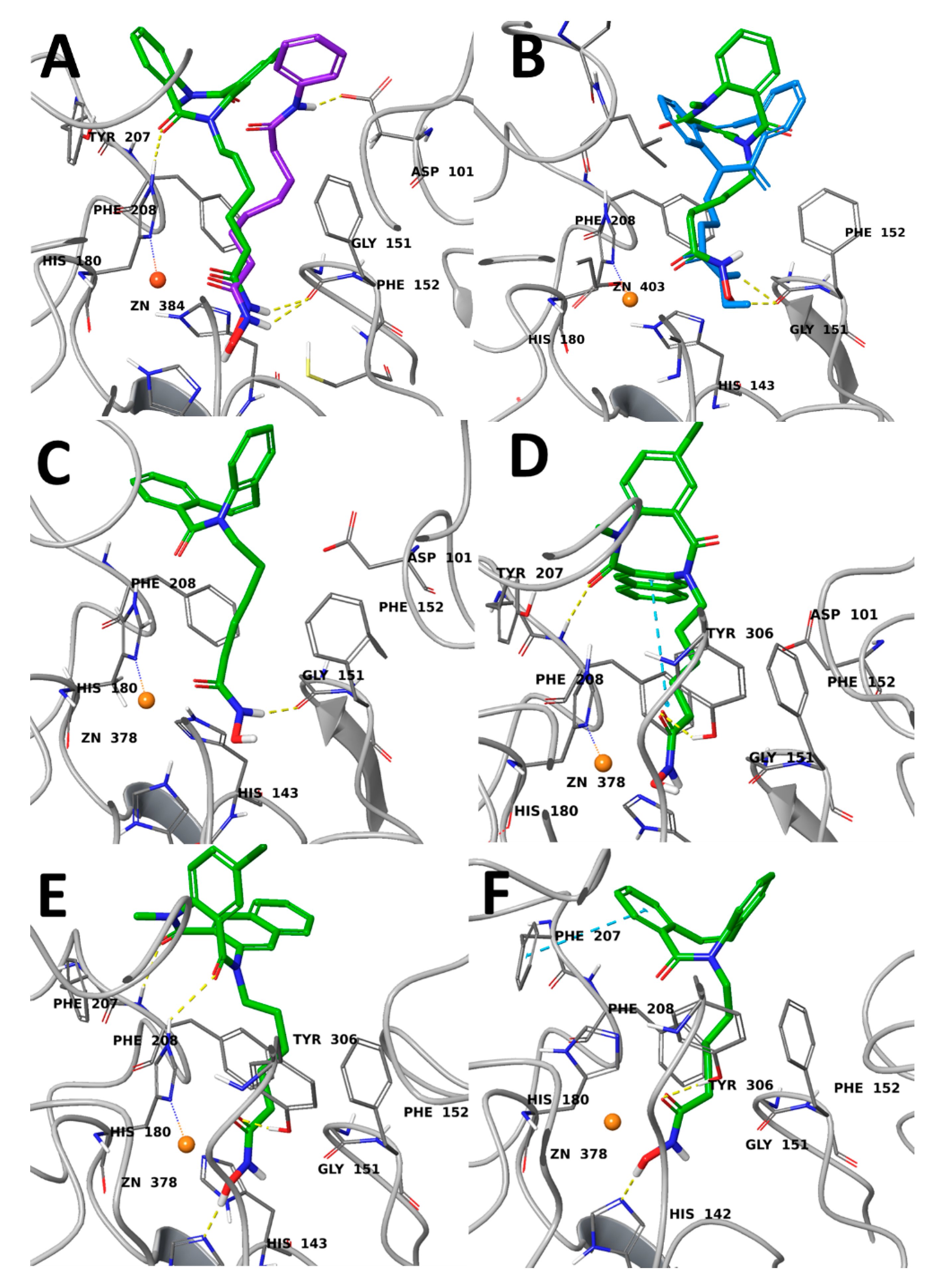
2.3. Molecular Modelling
3. Materials and Methods
3.1. Chemistry
3.2. Biology
3.2.1. Cell Culturing
3.2.2. Histone Deacetylase Activity Assay (HDACs Activity)
3.2.3. Histone Deacetylase 8 Activity Assay
3.2.4. Cytotoxicity Assay
3.3. In Silico Modelling
Compounds Preparation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, S.-Y.; Kim, J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Zwergel, C.; Stazi, G.; Valente, S.; Mai, A. Histone deacetylase inhibitors: Updated studies in various epigenetic-related diseases. J. Clin. Epigenet. 2016, 2, 7. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Hassel, K.N. Histone deacetylases and their inhibitors in cancer epigenetics. Diseases 2019, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Dong, H.; Xu, Q.; Zhang, Y. Histone deacetylase (HDAC) inhibitors in cancer: A patent review (2017-present). Expert Opin. Ther. Pat. 2020, 20, 263–374. [Google Scholar] [CrossRef]
- Chen, I.; Sethy, B.; Liou, J.-P. Recent update of HDAC inhibitors in lymphoma. Front. Cell Dev. Biol. 2020, 8, 576391. [Google Scholar] [CrossRef]
- Sinhg, A.K.; Bishayee, A.; Pandey, A.K. Targeting histone deacetylases with natural and synthetic agents: An emerging anticancer strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef] [Green Version]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent progress in histone deacetylase inhibitors as anticancer agents. Curr. Med. Chem. 2020, 27, 2449–2493. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Terasawa, K.; Kajikuri, J.; Kito, H.; Endo, K.; Jaikhan, P.; Suzuki, T.; Ohya, S. Histone deacetylases enhance Ca2+-activated K+ shannel KCa3.1 expression in murine inflammatory CD4+ T cells. Int. J. Mol. Sci. 2018, 19, 2942. [Google Scholar] [CrossRef] [Green Version]
- Delgado, F.G.; Cardenas, P.; Castellanos, J.E. Valproic acid downregulates cytokine expression in human macrophages infected with Dengue virus. Diseases 2018, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Jan, J.-S.; Chou, Y.-C.; Cheng, Y.-W.; Chen, C.-K.; Huang, W.-J.; Hsiao, G. The novel HDAC8 inhibitor WK2-16 attenuates lipopolysaccharide-activated matrix metalloproteinase-9 expression in human monocytic cells and improves hypercytokinemia in vivo. Int. J. Mol. Sci. 2017, 18, 1394. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.X.; Park, Y.-K.; Lee, J.-Y. Anti-inflammatory effects of Spirulina platensis extract via the modulation of histone deacetylases. Nutrients 2016, 8, 381. [Google Scholar] [CrossRef] [Green Version]
- Gatla, H.R.; Muniraj, N.; Thevkar, P.; Yavvari, S.; Sukhavasi, S.; Makena, M.R. Regulation of chemokines and cytokines by histone deacetylases and an update on histone decetylase inhibitors in human diseases. Int. J. Mol. Sci. 2019, 20, 1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, Y.; Mimura, T. The Histone modification code in the pathogenesis of autoimmune diseases. Mediators Inflamm. 2017, 2017, 2608605. [Google Scholar] [CrossRef]
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319862697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yooh, S.; Kang, G.; Eom, G.H. HDAC inhibitors: Therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.H.; Choi, H.S.; Kim, C.S.; Kim, I.J.; Cha, H.; Cho, J.M.; Ma, S.K.; Kim, S.W.; Bae, E.H. CG200745, a novel HDAC inhibitor, attenuates kidney fibrosis in a murine model of alport syndrome. Int. J. Mol. Sci. 2020, 21, 1473. [Google Scholar] [CrossRef] [Green Version]
- Pardo-Jimenez, V.; Navarrete-Encina, P.; Diaz-Araya, G. Synthesis and biological evaluation of novel thiazolyl-coumarin derivatives as potent histone deacetylase inhibitors with antifibrotic activity. Molecules 2019, 24, 739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nural-Guvener, H.; Zakharova, L.; Feehery, L.; Sljukic, S.; Gaballa, M. Anti-fibrotic effects of class I HDAC inhibitor, Mocetinostat is associated with IL-6/Stat3 signaling in ischemic heart failure. Int. J. Mol. Sci. 2015, 16, 11482–11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.S.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [Green Version]
- Mielcarek, M.; Landles, C.; Weiss, A.; Bradaia, A.; Seredina, T.; Inuabasi, L.; Osborne, G.; Wadel, K.; Touller, C.; Butler, R.; et al. HDAC4 reduction: A novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 2013, 11, e1001717. [Google Scholar] [CrossRef]
- D’Mello, S.R. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009, 22, 513–524. [Google Scholar] [CrossRef]
- Jeong, H.; Shin, S.; Lee, J.-S.; Lee, S.H.; BAik, J.-H.; Lim, S.; Kim, Y.K. Pan-HDAC inhibitors promote tau aggregation by increasing the level of acetylated tau. Int. J. Mol. Sci. 2019, 20, 4283. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, J.; Zabidi-Hussin, Z.A.M.H.; Sasongko, T.H. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet. Mol. Biol. 2013, 36, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Soragni, E.; Jacques, V.; Rusche, J.R.; Gottesfeld, J.M. Improved histone deacetylase inhibitors as therapeutics for the neurodegenerative disease Friedreich’s ataxia: A new synthetic route. Pharmaceuticals 2011, 4, 1578–1590. [Google Scholar] [CrossRef]
- Zwinderman, M.R.H.; de Weerd, S.; Dekker, F.J. Targeting HDAC complexes in asthma and COPD. Epigenomes 2019, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Oh, Y.J.; Park, B.; Lee, S.; Jeong, C.-H.; Lee, S.; Seo, J.H.; Seo, Y.H. Development of thiazolidinedione-based HDAC6 inhibitors to overcome methamphetamine addiction. Int. J. Mol. Sci. 2019, 20, 6213. [Google Scholar] [CrossRef] [PubMed]
- Habibian, J.; Ferguson, B.S. The crosstalk between acetylation and phosphorylation: Emerging new roles for HDAC inhibitors in the heart. Int. J. Mol. Sci. 2019, 20, 102. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.W.; Ferguson, B.S. Food bioactive HDAC inhibitors in the epigenetic regulation of heart failure. Nutrients 2018, 10, 1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Cattaneo, F.; Trimarco, B.; Esposito, G.; Perrino, C. Cardiovascular effects of histone deacetylase inhibitors epigenetic therapies: Systematic review of 62 studies and new hypotheses for future research. Int. J. Cardiol. 2016, 219, 396–403. [Google Scholar] [CrossRef]
- Cho, H.M.; Seok, Y.M.; Lee, H.A.; Song, M.; Kim, I. Repression of transcriptional activity of forkhead box O1 by histone deacetylase inhibitors ameliorates hyperglycemia in type 2 diabetic rats. Int. J. Mol. Sci. 2018, 19, 3539. [Google Scholar] [CrossRef] [Green Version]
- Bocchi, L.; Motta, B.M.; Savi, M.; Vilella, R.; Meraviglia, V.; Rizzi, F.; Galati, S.; Buschini, A.; Lazzaretti, M.; Pramstaller, P.P.; et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) restores cardiomyocyte contractility in a rat model of early diabetes. Int. J. Mol. Sci. 2019, 20, 1873. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Pacheco, H.; Picazo, O.; Lopez-Torres, A.; Morin, J.-P.; Castro-Cerritos, K.V.; Zepeda, R.C.; Roldán-Roldán, G. Biochemical and behavioral characterization of IN14, a new inhibitor of HDACs with antidepressant-like properties. Biomolecules 2020, 10, 299. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, R.L.; Daniels, E.G.; Molenaars, M.; Houtkooper, R.H.; Janssens, G.E. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol. Med. 2019, 11, e9854. [Google Scholar] [CrossRef]
- Herbein, G.; Wendling, D. Histone deacetylases in viral infections. Clin. Epigenet. 2010, 1, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Liu, N.; Zhan, P.; Liu, X. Repurposing of HDAC inhibitors toward anti-hepatitis C virus drug discovery: Teaching an old dog new tricks. Future Med. Chem. 2015, 7, 1367–1371. [Google Scholar] [CrossRef]
- Lu, C.-Y.; Chang, Y.-C.; Hua, C.-H.; Chuang, C.; Huang, S.-H.; Kung, S.-H.; Hour, M.-J.; Lin, C.-W. Tubacin, an HDAC6 selective inhibitor, reduces the replication of the japanese encephalitis virus via the decrease of viral RNA synthesis. Int. J. Mol. Sci. 2017, 18, 954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yedery, R.D.; Jerse, A.E. Augmentation of cationic antimicrobial peptide production with histone deacetylase inhibitors as a novel epigenetic therapy for bacterial infections. Antibiotics 2015, 4, 44–61. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Culf, A.S. Role of histone deacetylases in fungal phytopathogenesis: A review. Int. J. Mod. Bot. 2014, 4, 48–50. [Google Scholar] [CrossRef]
- Shanmugam, G.; Kim, T.; Jeon, J. In silico identification of potential inhibitor against a fungal histone deacetylase, RPD3 from Magnaporthe oryzae. Molecules 2019, 24, 2075. [Google Scholar] [CrossRef] [Green Version]
- De Souza, W.; Zuma, A.A. Histone deacetylases as targets for antitrypanosomal drugs. Future Sci. OA 2018, 4, FSO325. [Google Scholar] [CrossRef] [Green Version]
- Loeuillet, C.; Touquet, B.; Guichou, J.-F.; Labesse, G.; Sereno, D. A tiny change makes a big difference in the anti-parasitic activities of an HDAC inhibitor. Int. J. Mol. Sci. 2019, 20, 2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A novel class of Schistosoma mansoni histone deacetylase 8 (HDAC8) inhibitors identified by structure-based virtual screening and in vitro testing. Molecules 2018, 23, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yelton, C.J.; Ray, S.K. Histone deacetylase enzymes and selective histone deacetylase inhibitors for antitumor effects and enhancement of antitumor immunity in glioblastoma. Neuroimmunol. Neuroinflamm. 2018, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Shen, L.; Li, X.; Liu, J. Efficacy and toxicity of histone deacetylase inhibitors in relapsed/refractory multiple myeloma: Systematic review and meta-analysis of clinical trials. Exp. Ther. Med. 2019, 18, 1057–1068. [Google Scholar] [CrossRef] [Green Version]
- Tandon, N.; Ramakrishnan, V.; Kumar, S.K. Clinical use and applications of histone deacetylase inhibitors in multiple myeloma. Clin. Pharmacol. 2016, 8, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC inhibitors exert anti-myeloma effects through multiple modes of action. Cancers 2019, 11, 475. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone deacetylases (HDACs) guided novel therapies for T-cell lymphomas. Int. J. Med. Sci. 2019, 16, 424–442. [Google Scholar] [CrossRef] [Green Version]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Anthanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San José-Enriz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC inhibitors in acute myeloid leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [Green Version]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. HDAC6—An emerging target against chronic myeloid leukemia? Cancers 2020, 12, 318. [Google Scholar] [CrossRef] [Green Version]
- Cosenza, M.; Pozzi, S. The therapeutic strategy of HDAC6 inhibitors in lymphoproliferative disease. Int. J. Mol. Sci. 2018, 19, 2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesham, H.M.; Lasheen, D.S.; Abouzid, K.A.M. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.A.; Jordan, H.R.; Tollefbols, T.O. Effects of SAHA and EGCG on growth potentiation of triple-negative breast cancer cells. Cancers 2019, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Bandolik, J.J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-histone deacetylase (HDAC) inhibition is superior to pan-HDAC inhibition in modulating cisplatin potency in high grade serous ovarian cancer cell lines. Int. J. Mol. Sci. 2019, 20, 3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laschanzky, R.S.; Humphrey, L.E.; Ma, J.; Smith, L.M.; Enke, T.J.; Shukla, S.K.; Dasgupta, A.; Singh, P.K.; Howell, G.M.; Brattain, M.G.; et al. Selective inhibition of histone deacetylases 1/2/6 in combination with gemcitabine: A promising combination for pancreatic cancer therapy. Cancers 2019, 11, 1327. [Google Scholar] [CrossRef] [Green Version]
- Najem, S.A.; Khawaja, G.; Hodroj, M.H.; Babikian, P.; Rizk, S. Adjuvant epigenetic therapy of decitabine and suberoylanilide hydroxamic acid exerts anti-neoplastic effects in acute myeloid leukemia cells. Cells 2019, 8, 1480. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.K.; Yin, Y.; Kim, K.; Yang, G.-M.; Dayem, A.A.; Choi, H.Y.; Cho, S.-G. Valproic acid induces endocytosis-mediated doxorubicin internalization and shows synergistic cytotoxic effects in hepatocellular carcinoma cells. Int. J. Mol. Sci. 2017, 18, 1048. [Google Scholar] [CrossRef] [Green Version]
- Cerna, T.; Hrabeta, J.; Eckschlager, T.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborová, M. The histone deacetylase inhibitor valproic acid exerts a synergistic cytotoxicity with the DNA-damaging drug ellipticine in neuroblastoma cells. Int. J. Mol. Sci. 2017, 19, 164. [Google Scholar] [CrossRef] [Green Version]
- Cuperlovic-Culf, M.; Touaibia, M.; St-Coeur, P.-D.; Poitras, J.; Morin, P., Jr.; Culf, A.S. Metabolic effects of known and novel HDAC and SIRT inhibitors in glioblastomas independently or combined with temozolomide. Metabolites 2014, 4, 807–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, S. Novel proteasome inhibitors and histone deacetylase inhibitors: Progress in myeloma therapeutics. Pharmaceuticals 2017, 10, 40. [Google Scholar] [CrossRef]
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET proteins and histone deacetylase (HDACs): Crossing roads in cancer therapy. Cancers 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, U.; Venkatesan, T.; Radhakrishnan, V.; Samuel, S.; Rathinavelu, A. Differential mechanisms of cell death induced by HDAC inhibitor SAHA and MDM2 Inhibitor RG7388 in MCF-7 cells. Cells 2019, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Li, P.-T.; Tsai, Y.-J.; Lee, M.-J.; Chen, C.-T. Increased histone deacetylase activity involved in the suppressed invasion of cancer cells survived from ALA-mediated photodynamic treatment. Int. J. Mol. Sci. 2015, 16, 23994–24010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, H.-W.; Yeh, Y.-L.; Ho, S.-Y.; Wu, Y.-H.; Wang, B.-J.; Huang, W.-J.; Ho, Y.-S.; Wang, Y.-J.; Chen, L.-C.; Tu, S.-H. A new histone deacetylase inhibitor enhances radiation sensitivity through the induction of misfolded protein aggregation and autophagy in triple-negative breast cancer. Cancers 2019, 11, 1703. [Google Scholar] [CrossRef] [Green Version]
- Moertl, S.; Payer, S.; Kell, R.; Winkler, K.; Anastasov, N.; Atkinson, M.J. Comparison of radiosensitization by HDAC inhibitors CUDC-101 and SAHA in pancreatic cancer cells. Int. J. Mol. Sci. 2019, 20, 3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalal, B.S.; Pai, V.R.; Behera, S.K.; Somashekarappa, H.M. HDAC2 inhibitor valproic acid increases radiation sensitivity of drug-resistant melanoma cells. Med. Sci. 2019, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Fox, C.R.; Parks, G.D. Histone deacetylase inhibitors enhance cell killing and block interferon-beta synthesis elicited by infection with an oncolytic parainfluenza virus. Viruses 2019, 11, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, S.A.; Barnett, P.G. The Role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [Green Version]
- Merarchi, M.; Sethi, G.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Ahn, K.S. Role of natural products in modulating histone deacetylases in cancer. Molecules 2019, 24, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Liao, G.; Sun, P.; Yu, Z.; Chen, J. An overview of HDAC inhibitors and their synthetic routes. Curr. Top. Med. Chem. 2019, 19, 1005–1040. [Google Scholar] [CrossRef]
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat. Rev. Drug Discov. 2007, 6, 21–22. [Google Scholar] [CrossRef]
- McGuire, C.; Lee, J. Brief review of vorinostat. Clin. Med. Insights Ther. 2010, 2, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting class I histone deacetylases in a “complex” environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Degado, I.; Piekarz, R.L. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.R. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottamal, M.; Zhen, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [Green Version]
- Guandalini, L.; Cellai, C.; Laurenzana, A.; Scapecchi, S.; Paoletti, F.; Romanelli, M.N. Design, synthesis and preliminary biological evaluation of new hydroxamate histone deacetylase inhibitors as potential antileukemic agents. Bioorg. Med. Chem. Lett. 2008, 18, 5071–5074. [Google Scholar] [CrossRef]
- Cellai, C.; Balliu, M.; Laurenzana, A.; Guandalini, L.; Matucci, R.; Miniati, D.; Torre, E.; Nebbioso, A.; Carafa, V.; Altucci, L.; et al. The new low-toxic histone deacetylase inhibitor S-(2) induces apoptosis in various acute myeloid leukaemia cells. J. Cell. Mol. Med. 2012, 16, 1758–1765. [Google Scholar] [CrossRef]
- Guandalini, L.; Balliu, M.; Cellai, C.; Martino, M.V.; Nebbioso, A.; Mercurio, C.; Carafa, V.; Bartolucci, G.; Dei, S.; Manetti, D.; et al. Design, synthesis and preliminary evaluation of a series of histone deacetylase inhibitors carrying a benzodiazepine ring. Eur. J. Med. Chem. 2013, 66, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Binaschi, M.; Boldetti, A.; Gianni, M.; Maggi, C.A.; Gensini, M.; Bigioni, M.; Parlani, M.; Giolitti, A.; Fratelli, M.; Valli, C.; et al. Antiproliferative and differentiating activities of a novel series of histone deacetylase inhibitors. ACS Med. Chem. Lett. 2010, 1, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mieczkowski, A.; Koźmiński, W.; Jurczak, J. A traceless, solid-supported synthesis of b-turn mimetics based on the hexahydropyrazino[1,2-a]pyrazine-1,2-dione scaffold. Synthesis 2010, 221–232. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Jurczak, J. A Traceless solid-supported synthesis of novel pyrazinediazepinedione derivatives. Tetrahedron 2010, 66, 2514–2519. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Speina, E.; Trzybiński, D.; Winiewska-Szajewska, M.; Wińska, P.; Borsuk, E.M.; Podsiadła-Białoskórska, M.; Przygodzki, T.; Drabikowski, K.; Stańczyk, L.; et al. Diketopiperazine-based, flexible tadalafil analogues: Synthesis, crystal structures and biological activity profile. Molecules 2021, 26, 794. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Trzybiński, D.; Wilczek, M.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Woźniak, K. (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—synthesis and crystallographic studies. Molbank 2017, 2017, M964. [Google Scholar] [CrossRef] [Green Version]
- Mieczkowski, A.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Wilczek, M.; Czajkowska, J.; Trzybiński, D.; Woźniak, K.; Wietrzyk, J. Novel (S)-1,3,4,12a-tetrahydropyrazino[2,1-c][1,4]benzodiazepine-6,12(2H,11H)-dione derivatives: Selective inhibition of MV-4-11 biphenotypic B myelomonocytic leukemia cells’ growth is accompanied by reactive oxygen species overproduction and apoptosis. Bioorg. Med. Chem. Lett. 2018, 28, 618–625. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Frączyk, T.; Psurski, M.; Wińska, P.; Siedlecki, P.; Dziełak, M.; Trzybiński, D.; Wilczek, M.; Bagiński, M.; Bieszczad, B.; et al. Design and in vitro characterization of tricyclic benzodiazepine derivatives as potent and selective antileukemic agents. Chem. Biodivers. 2021, 19, e2000733. [Google Scholar] [CrossRef]
- Bieszczad, B.; Garbicz, D.; Trzybiński, D.; Mielecki, D.; Woźniak, K.; Grzesiuk, E.; Mieczkowski, A. Unsymmetrically substituted dibenzo[b,f][1,5]-diazocine-6,12(5H,11H)dione—A convenient scaffold for bioactive molecule design. Molecules 2020, 25, 906. [Google Scholar] [CrossRef] [Green Version]
- Bieszczad, B.; Garbicz, D.; Trzybiński, D.; Dudek, M.; Woźniak, K.; Grzesiuk, E.; Mieczkowski, A. Unsymetrically-substituted 5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione scaffold—A useful tool for bioactive molecules design. Molecules 2020, 25, 2855. [Google Scholar] [CrossRef] [PubMed]
- Bieszczad, B.; Pawlędzio, S.; Polak, P.; Antonowicz, J.; Mieczkowski, A.; Trzybiński, D. Influence of halogen size on the supramolecular and energy landscape of the THF solvates of the halogen derivatives of dianthranilide. CrystEngComm 2020, 22, 5389–5399. [Google Scholar] [CrossRef]
- Bieszczad, B.; Siwek, A.; Wilczek, M.; Trzybiński, D.; Woźniak, A.; Satała, G.; Bojarski, A.; Mieczkowski, A. Synthesis, crystal structure and biological activity of novel analogues of tricyclic drugs. Bioorg. Med. Chem. Lett. 2020, 30, 127493. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Vu, J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 2007, 16, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-2: Protein Preparation Wizard; Schrödinger, LLC: New York, NY, USA, 2020.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comp.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Nevozhay, D. Cheburator software for sutomatically calculating drug inhibitory concentrations from in vitro screening assays. PLoS ONE 2014, 9, e0106186. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
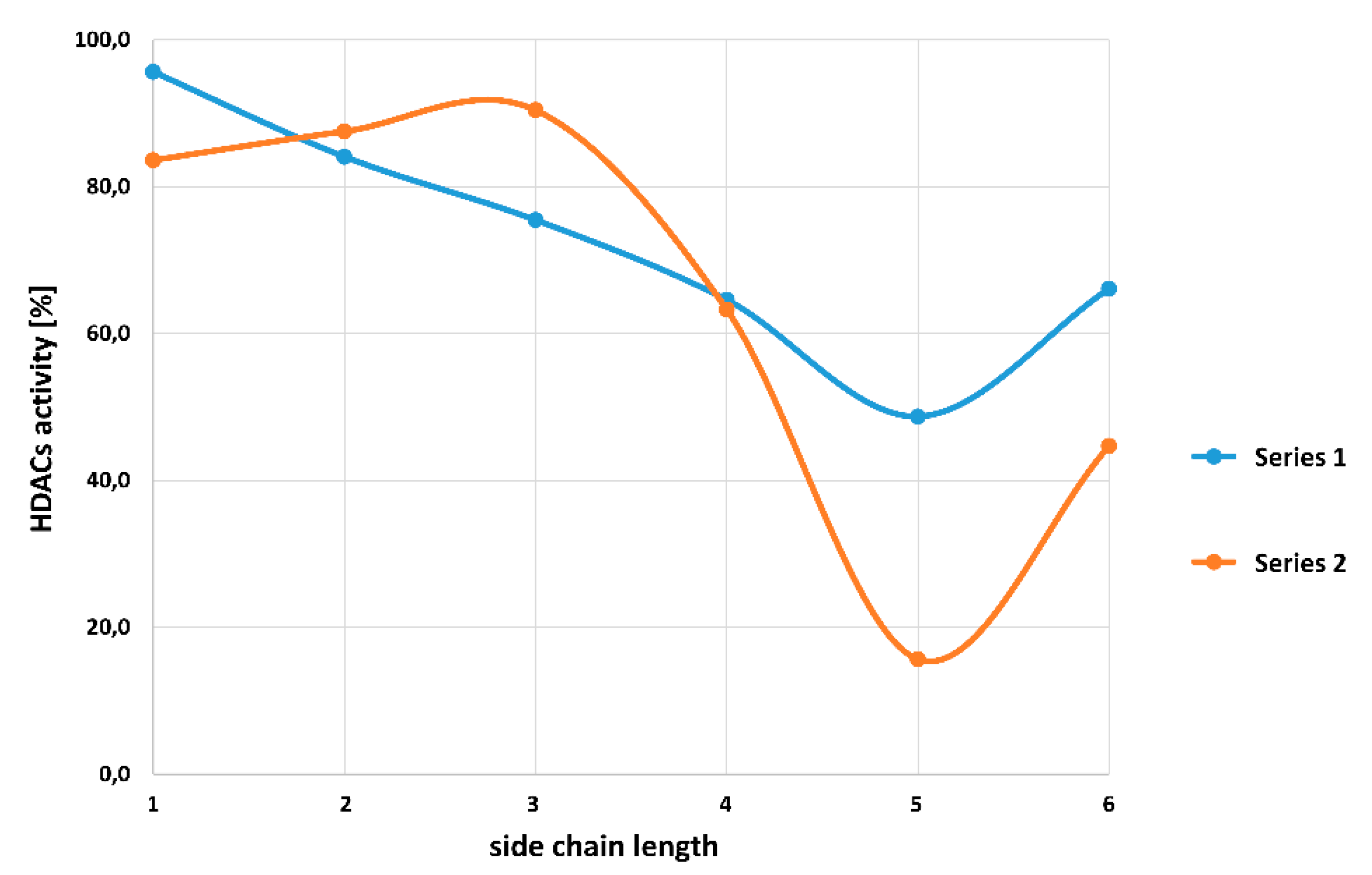
| Series 1 | |||||||
| Compound | 7a | 7b | 7c | 7d | 7e | 7f | |
| HDAC activity at 2 µM [%] | 95.6 ± 1.4 | 84.1 ± 2.3 | 75.4 ± 3.0 | 64.6 ± 3.6 | 48.7 ± 1.0 | 66.1 ± 4.1 | |
| Series 2 | |||||||
| Compound | 7g | 7h | 7i | 7j | 7k | 7l | |
| HDAC activity at 2 µM [%] | 83.6 ± 4.2 | 87.5 ± 1.2 | 90.4 ± 1.6 | 63.3 ± 0.5 | 15.7 ± 1.3 | 44.7 ± 4.4 | |
| Series 3 | |||||||
| Compound | 7m | 7n | 7o | 7p | 7r | 7s | 7t |
| HDAC activity at 2 µM [%] | 72.7 ± 5.2 | 68.3 ± 2.8 | 64.5 ± 2.1 | 21.5 ± 1.6 | 31.5 ± 7.1 | 35.9 ± 0.8 | 19.9 ± 1.6 |
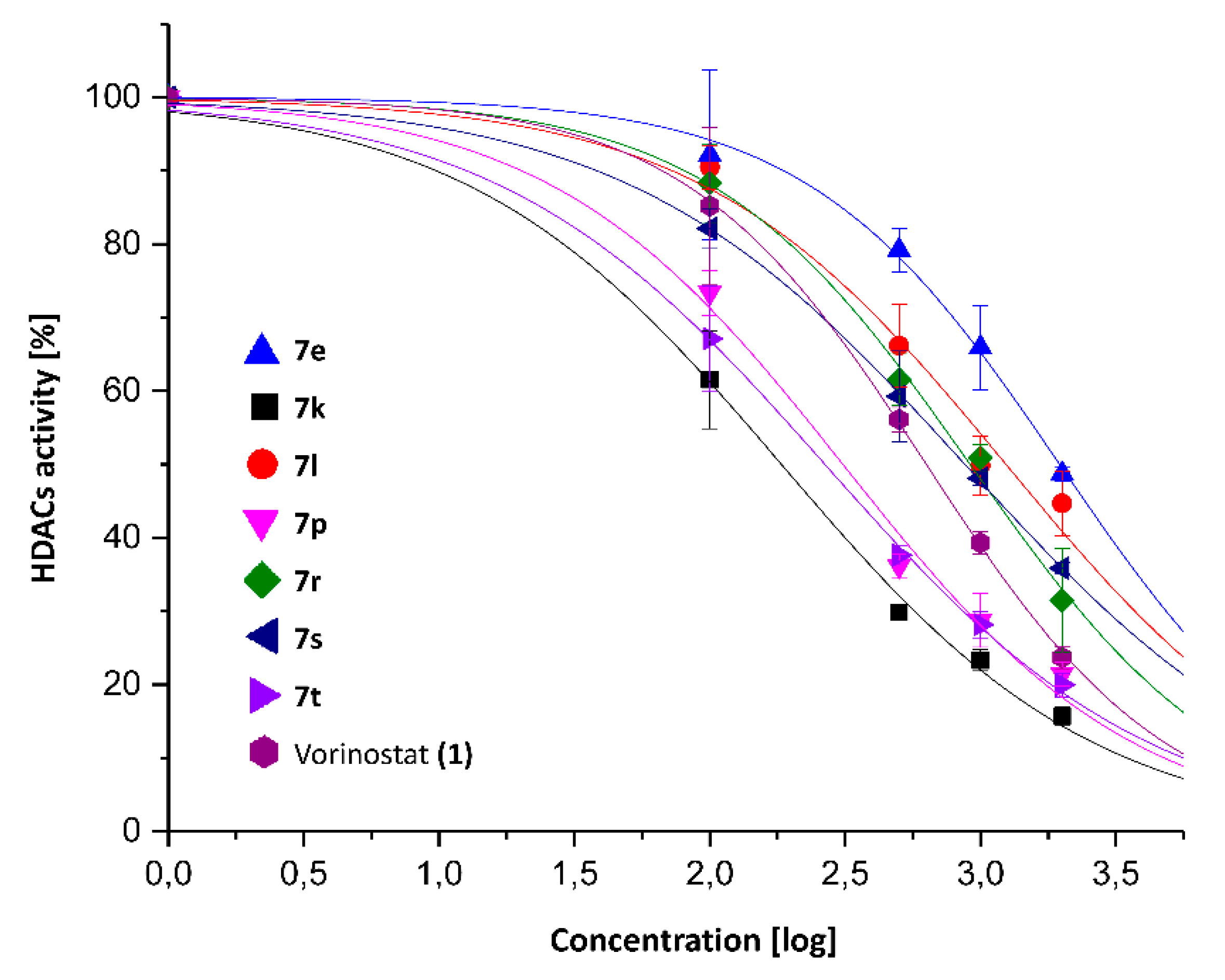
| Compound | HDACs | HDAC8 |
|---|---|---|
| 7e | 1.959 ± 0.105 | 5.67 ± 0.64 |
| 7k | 0.183 ± 0.015 | 3.37 ± 0.33 |
| 7l | 1.241 ± 0.155 | N/T |
| 7p | 0.309 ± 0.035 | 3.14 ± 0.42 |
| 7r | 0.914 ± 0.053 | N/T |
| 7s | 0.875 ± 0.017 | N/T |
| 7t | 0.266 ± 0.014 | 1.95 ± 0.17 |
| Vorinostat (1) | 0.630 ± 0.011 | 1.51 ± 0.13 |
| Compound | IC50 [µM] | |||||
|---|---|---|---|---|---|---|
| MV4-11 | Daudi | A549 | MCF-7 | BALB/3T3 | ||
| Series 1 | 7a | >50.00 | >50.00 | N/T | N/T | N/T |
| 7b | >50.00 | >50.00 | N/T | N/T | N/T | |
| 7c | >50.00 | >50.00 | N/T | N/T | N/T | |
| 7d | 7.58 ± 2.50 * | 9.6 ± 2.10 * | N/T | N/T | N/T | |
| 7e | 2.33 ± 0.64 * | 3.03 ± 0.55 * | N/T | N/T | N/T | |
| 7f | 4.45 ± 0.34 * | 4.83 ± 1.96 * | N/T | N/T | N/T | |
| Series 2 | 7g | 33.97 ± 0.75 * | 17.31 ± 6.2 * | N/T | N/T | N/T |
| 7h | >50.00 | 32.62 ± 4.78 * | N/T | N/T | N/T | |
| 7i | >50.00 | >50.00 | N/T | N/T | N/T | |
| 7j | 5.30 ± 1.36 * | 3.47 ± 0.66 * | N/T | N/T | N/T | |
| 7k | 0.220 ± 0.021 * | 0.460 ± 0.122 | 1.27 ± 0.42 | 0.618 ± 0.095 | 1.28 ± 0.15 | |
| 7l | 1.57 ± 0.12 * | 1.44 ± 0.61 * | N/T | N/T | N/T | |
| Series 3 | 7m | 4.45 ± 0.96 * | 5.09 ± 0.3 * | N/T | N/T | N/T |
| 7n | 2.85 ± 0.34 * | 2.56 ± 0.98 * | N/T | N/T | N/T | |
| 7o | 3.55 ± 0.75 * | 3.52 ± 0.80 * | N/T | N/T | N/T | |
| 7p | 0.200 ± 0.073 * | 0.318 ± 0.098 | 1.21 ± 0.24 | 0.661 ± 0.12 | 1.04 ± 0.28 | |
| 7r | 0.603 ± 0.132 | 0.785 ± 0.246 | 4.61 ± 0.38 * | 2.72 ± 0.57 * | 3.37 ± 0.87 * | |
| 7s | 0.692 ± 0.110 | 0.944 ± 0.167 * | 17.96 ± 5.77 * | 4.24 ± 1.03 * | 12.04 ± 5.9 * | |
| 7t | 0.093 ± 0.009 * | 0.137 ± 0.04 * | 1.05 ± 0.07 * | 0.368 ± 0.015 * | 0.69 ± 0.05 * | |
| Vorinostat (1) | 0.636 ± 0.092 | 0.493 ± 0.093 | 1.64 ± 0.32 | 0.685 ± 0.06 | 1.42 ± 0.23 | |
| Compound | MV4-11 | Daudi | A549 | MCF-7 |
|---|---|---|---|---|
| 7k | 5.82 | 2.78 | 1.01 | 2.07 |
| 7p | 5.20 | 3.27 | 0.86 | 1.57 |
| 7r | 5.59 | 4.29 | 0.73 | 1.24 |
| 7s | 17.4 | 12.75 | 0.67 | 2.84 |
| 7t | 7.42 | 5.04 | 0.66 | 1.88 |
| Vorinostat (1) | 2.23 | 2.88 | 0.87 | 2.07 |
| Histone Deacetylase | PDBid | Ligand | Resolution [Å] |
|---|---|---|---|
| HDAC1 | 5ICN | GAXRH (peptide) | 3.30 |
| HDAC2 | 4LXZ | Vorinostat | 1.85 |
| HDAC3 | 4A69 | I0P | 2.06 |
| HDAC4 | 2VQM | HA3 | 1.80 |
| HDAC6 | 5EDU | Trichostatin A | 2.79 |
| HDAC7 | 3C0Z | Vorinostat | 2.10 |
| HDAC8 | 1T69 | Vorinostat | 2.91 |
| Component | Assay Buffer [µL] | HDAC Inhibitor Solution [µL] | HeLa Lysate [µL] | DMSO [µL] | HDAC Substrate Solution [µL] |
|---|---|---|---|---|---|
| HDAC activity assay | 40 | - | 5 | 5 | 50 |
| HDAC activity inhibition assay | 40 | 5 | 5 | - | 50 |
| Blank | 45 | - | - | 5 | 50 |
| Component | HDAC8 Assay Buffer [µL] | HDAC Inhibitor Solution [µL] | HDAC8 | DMSO [µL] | HDAC8 Substrate Solution [µL] |
|---|---|---|---|---|---|
| HDAC activity assay | 43 | - | 2 | 5 | 50 |
| HDAC activity inhibition assay | 43 | 5 | 2 | - | 50 |
| Blank | 45 | - | - | 5 | 50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bieszczad, B.; Garbicz, D.; Świtalska, M.; Dudek, M.K.; Warszycki, D.; Wietrzyk, J.; Grzesiuk, E.; Mieczkowski, A. Improved HDAC Inhibition, Stronger Cytotoxic Effect and Higher Selectivity against Leukemias and Lymphomas of Novel, Tricyclic Vorinostat Analogues. Pharmaceuticals 2021, 14, 851. https://doi.org/10.3390/ph14090851
Bieszczad B, Garbicz D, Świtalska M, Dudek MK, Warszycki D, Wietrzyk J, Grzesiuk E, Mieczkowski A. Improved HDAC Inhibition, Stronger Cytotoxic Effect and Higher Selectivity against Leukemias and Lymphomas of Novel, Tricyclic Vorinostat Analogues. Pharmaceuticals. 2021; 14(9):851. https://doi.org/10.3390/ph14090851
Chicago/Turabian StyleBieszczad, Bartosz, Damian Garbicz, Marta Świtalska, Marta K. Dudek, Dawid Warszycki, Joanna Wietrzyk, Elżbieta Grzesiuk, and Adam Mieczkowski. 2021. "Improved HDAC Inhibition, Stronger Cytotoxic Effect and Higher Selectivity against Leukemias and Lymphomas of Novel, Tricyclic Vorinostat Analogues" Pharmaceuticals 14, no. 9: 851. https://doi.org/10.3390/ph14090851
APA StyleBieszczad, B., Garbicz, D., Świtalska, M., Dudek, M. K., Warszycki, D., Wietrzyk, J., Grzesiuk, E., & Mieczkowski, A. (2021). Improved HDAC Inhibition, Stronger Cytotoxic Effect and Higher Selectivity against Leukemias and Lymphomas of Novel, Tricyclic Vorinostat Analogues. Pharmaceuticals, 14(9), 851. https://doi.org/10.3390/ph14090851