
Genetic Modifiers and Phenotype of Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
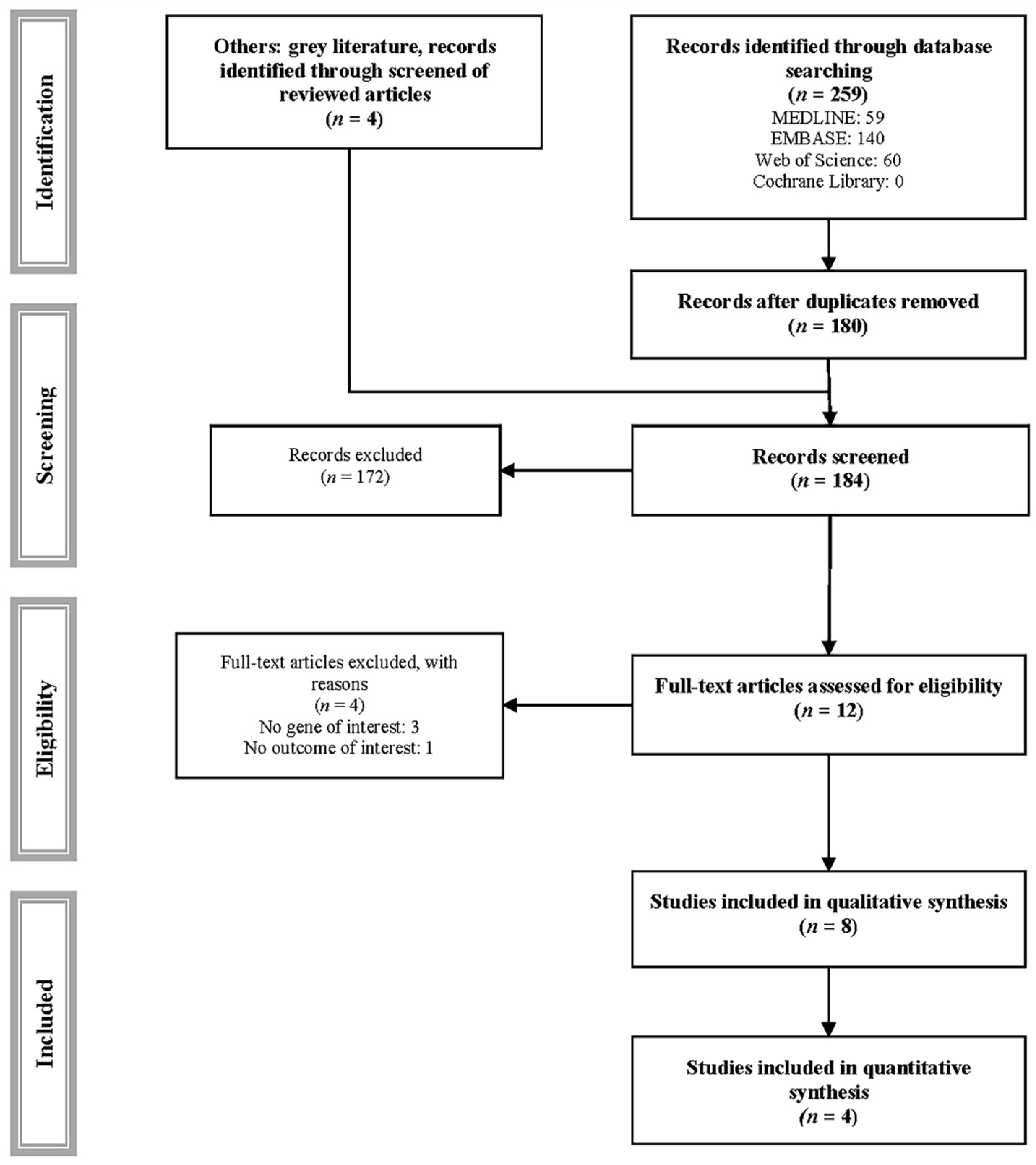
2.1. Search Strategy
2.2. Inclusion/Exclusion Criteria
2.3. Data Extraction
2.4. Risk of Bias Assessment
2.5. Grading the Quality of Evidence
2.6. Data Synthesis
2.7. Infographics
3. Results
3.1. Loss of Ambulation—Kaplan–Meier Analyses
3.2. Loss of Ambulation—Cox Regression Analyses
3.3. Cardiac Function
3.4. Assessment of the Risk of Bias
3.5. Evidence Assessment
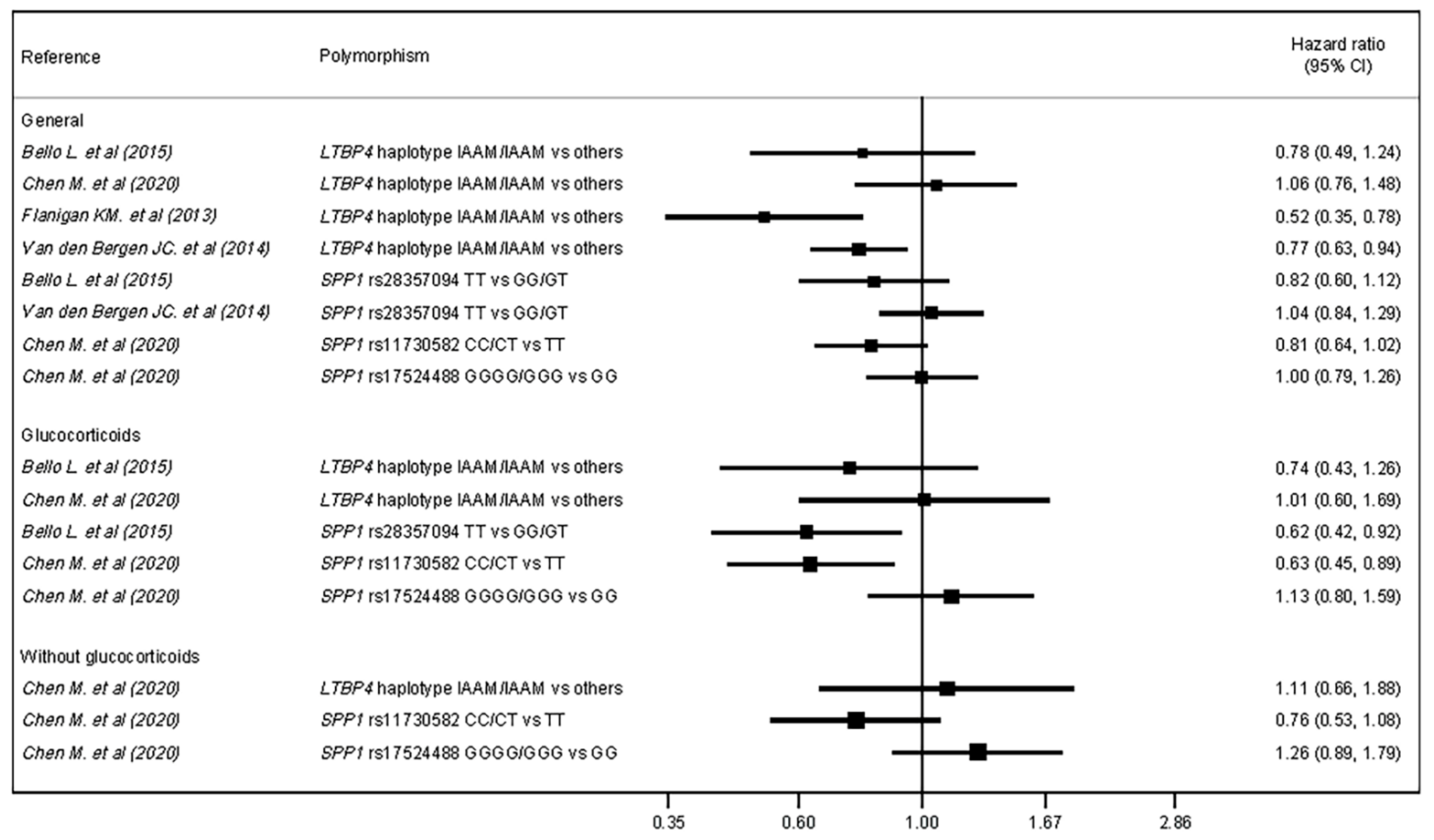
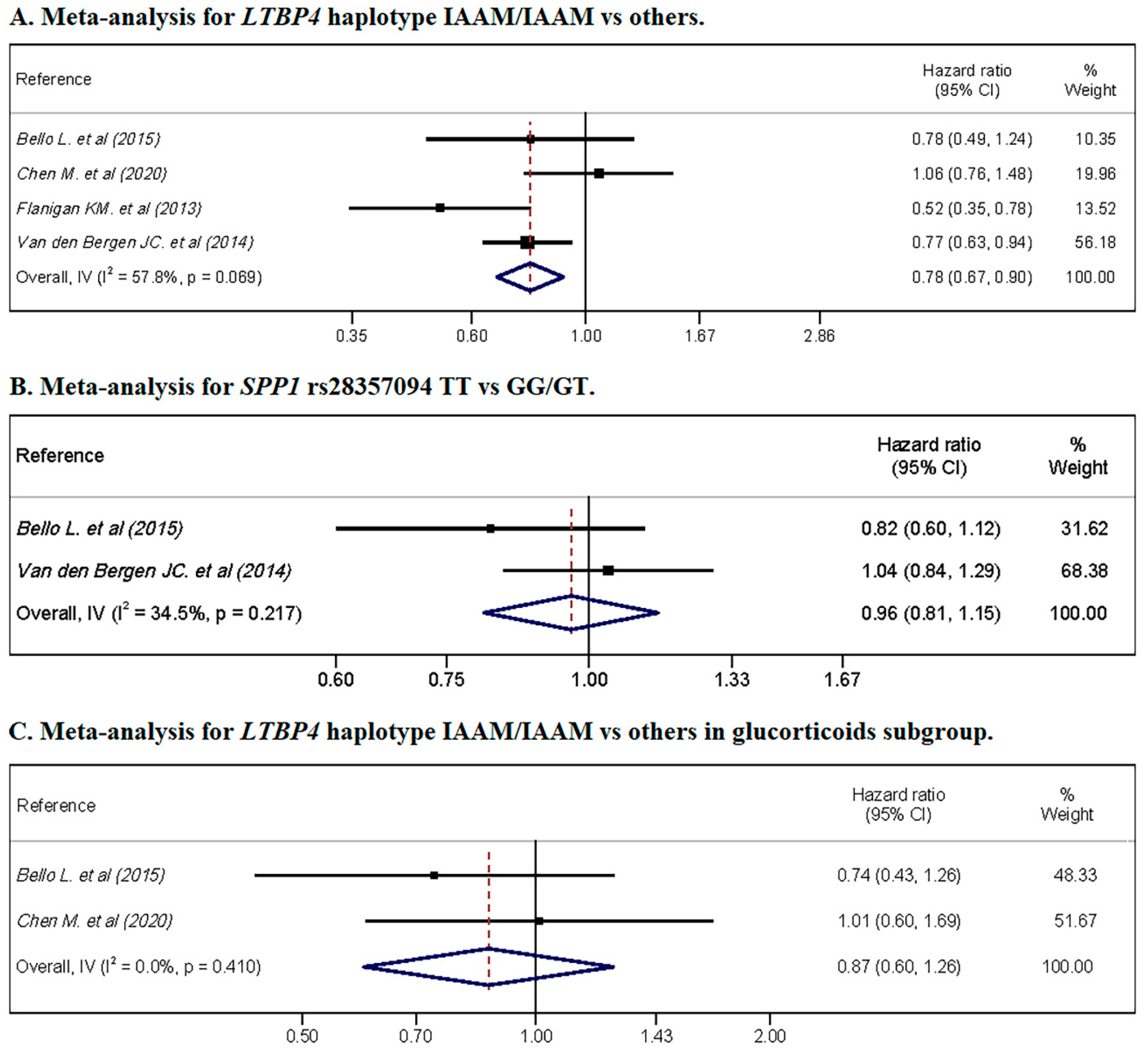
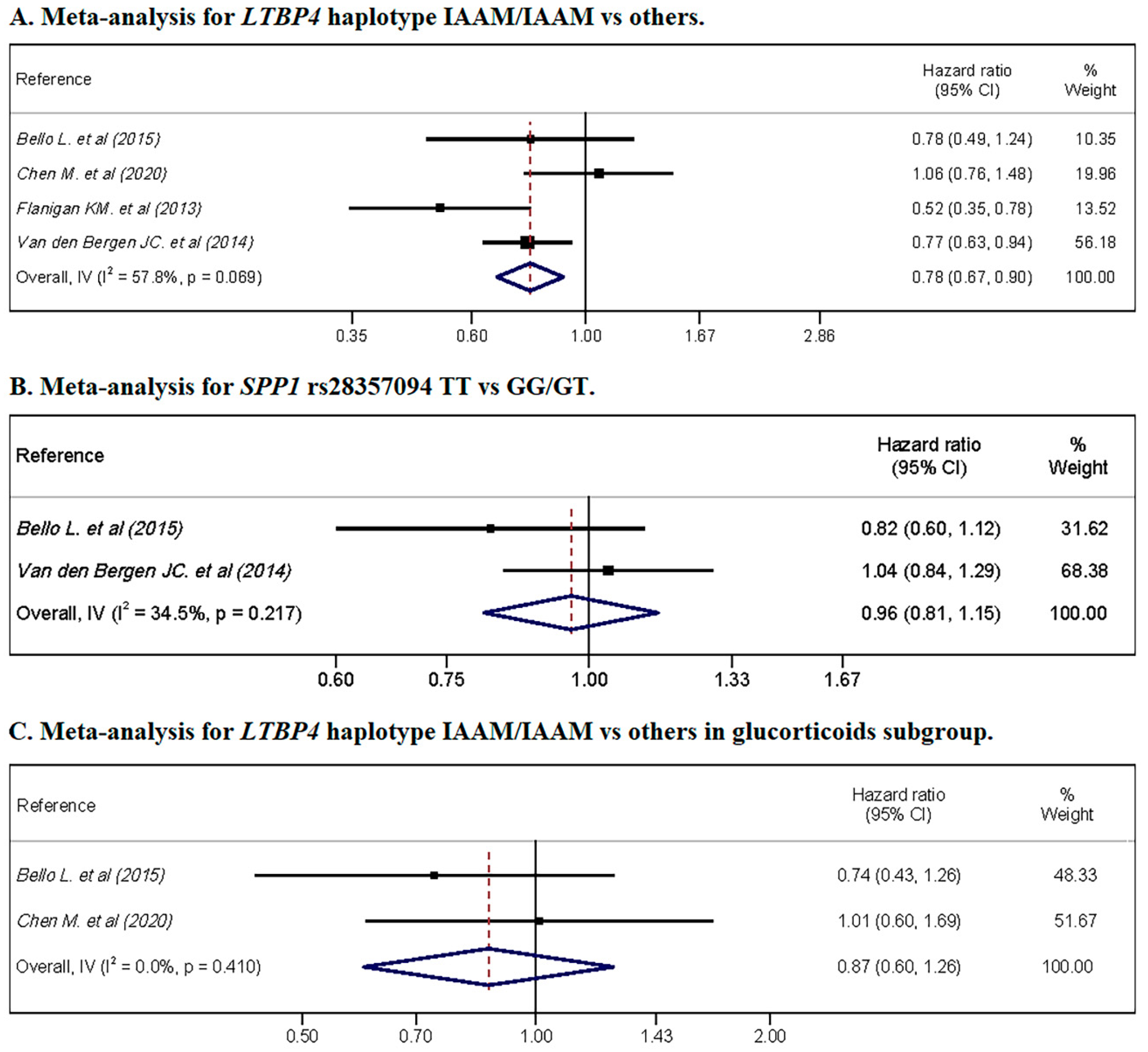
3.6. Meta-Analysis
4. Discussion
4.1. Main Findings
4.2. Interpretation
4.3. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Medline, EMBASE, Web of Science, Cochrane Library
Appendix A.2. Grey Literature
Appendix B

References
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A Systematic Review and Meta-Analysis on the Epidemiology of Duchenne and Becker Muscular Dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Holland, A.; Carberry, S.; Ohlendieck, K. Proteomics of the Dystrophin-Glycoprotein Complex and Dystrophinopathy. Curr. Protein Pept. Sci. 2014, 14, 680–697. [Google Scholar] [CrossRef]
- Warner, L.E.; DelloRusso, C.T.; Crawford, R.W.; Rybakova, I.N.; Patel, J.R.; Ervasti, J.M.; Chamberlain, J.S. Expression of Dp260 in Muscle Tethers the Actin Cytoskeleton to the Dystrophin-Glycoprotein Complex and Partially Prevents Dystrophy. Hum. Mol. Genet. 2002, 11, 1095–1105. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Goemans, N. How Glucocorticoids Change Life in Duchenne Muscular Dystrophy. Lancet 2018, 391, 406–407. [Google Scholar] [CrossRef]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A Current Approach to Heart Failure in Duchenne Muscular Dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Garg, S. Management of Scoliosis in Patients with Duchenne Muscular Dystrophy and Spinal Muscular Atrophy: A Literature Review. J. Pediatr. Rehabil. Med. 2016, 9, 23–29. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 2: Implementation of Multidisciplinary Care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Pascual-Morena, C.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Mesas, A.E.; Pozuelo-Carrascosa, D.; Martínez-Vizcaíno, V. Restorative Treatments of Dystrophin Expression in Duchenne Muscular Dystrophy: A Systematic Review. Ann. Clin. Transl. Neurol. 2020, 7, acn3.51149. [Google Scholar] [CrossRef]
- Souchelnytskyi, S.; Rönnstrand, L.; Heldin, C.H.; ten Dijke, P. Phosphorylation of Smad Signaling Proteins by Receptor Serine/Threonine Kinases. Methods Mol. Biol. 2001, 124, 107–120. [Google Scholar] [CrossRef]
- Gardner, S.; Alzhanov, D.; Knollman, P.; Kuninger, D.; Rotwein, P. TGF-β Inhibits Muscle Differentiation by Blocking Autocrine Signaling Pathways Initiated by IGF-II. Mol. Endocrinol. 2011, 25, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming Growth Factor (TGF)-β Signaling in Cardiac Remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming Growth Factor Beta (TGF-β) Mediates Cardiac Fibrosis and Induces Diabetic Cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef]
- Ismaeel, A.; Kim, J.S.; Kirk, J.S.; Smith, R.S.; Bohannon, W.T.; Koutakis, P. Role of Transforming Growth Factor-β in Skeletal Muscle Fibrosis: A Review. Int. J. Mol. Sci. 2019, 20, 2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quattrocelli, M.; Capote, J.; Ohiri, J.C.; Warner, J.L.; Vo, A.H.; Earley, J.U.; Hadhazy, M.; Demonbreun, A.R.; Spencer, M.J.; McNally, E.M. Genetic Modifiers of Muscular Dystrophy Act on Sarcolemmal Resealing and Recovery from Injury. PLoS Genet. 2017, 13, e1007070. [Google Scholar] [CrossRef]
- Vo, A.H.; McNally, E.M. Modifier Genes and Their Effect on Duchenne Muscular Dystrophy. Curr. Opin. Neurol. 2015, 28, 528–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrone, S.A.; Montecino-Rodriguez, E.; Kudryashova, E.; Kramerova, I.; Hoffman, E.P.; Liu, S.D.; Miceli, M.C.; Spencer, M.J. Osteopontin Promotes Fibrosis in Dystrophic Mouse Muscle by Modulating Immune Cell Subsets and Intramuscular TGF-β. J. Clin. Investig. 2009, 119, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Barakat-Haddad, C.; Shin, S.; Candundo, H.; Lieshout, P.; Van Martino, R. A Systematic Review of Risk Factors Associated with Muscular Dystrophies. Neurotoxicology 2017, 61, 55–62. [Google Scholar] [CrossRef]
- Barp, A.; Bello, L.; Politano, L.; Melacini, P.; Calore, C.; Polo, A.; Vianello, S.; Soraru, G.; Semplicini, C.; Pantic, B.; et al. Genetic Modifiers of Duchenne Muscular Dystrophy and Dilated Cardiomyopathy. PLoS ONE 2015, 10, e0141240. [Google Scholar] [CrossRef]
- Bello, L.; Kesari, A.; Gordish-Dressman, H.; Cnaan, A.; Morgenroth, L.P.; Punetha, J.; Duong, T.; Henricson, E.K.; Pegoraro, E.; McDonald, C.M.; et al. Genetic Modifiers of Ambulation in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study. Ann. Neurol. 2015, 77, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, L.; Li, Y.; Chen, Y.; Zhang, H.; Zhu, Y.; He, R.; Li, H.; Lin, J.; Zhang, Y.; et al. Genetic Modifiers of Duchenne Muscular Dystrophy in Chinese Patients. Front. Neurol. 2020, 11, 721. [Google Scholar] [CrossRef]
- Flanigan, K.M.; Ceco, E.; Lamar, K.-M.; Kaminoh, Y.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. LTBP4 Genotype Predicts Age of Ambulatory Loss in Duchenne Muscular Dystrophy. Ann. Neurol. 2013, 73, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, E.; Hoffman, E.P.; Piva, L.; Gavassini, B.F.; Cagnin, S.; Ermani, M.; Bello, L.; Soraru, G.; Pacchioni, B.; Bonifati, M.D.; et al. SPP1 Genotype is a Determinant of Disease Severity in Duchenne Muscular Dystrophy. Neurology 2011, 76, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bergen, J.C.; Hiller, M.; Bohringer, S.; Vijfhuizen, L.; Ginjaar, H.B.; Chaouch, A.; Bushby, K.; Straub, V.; Scoto, M.; Cirak, S.; et al. Validation of Genetic Modifiers for Duchenne Muscular Dystrophy: A Multicentre Study Assessing SPP1 and LTBP4 Variants. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Van Dorn, C.S.; Puchalski, M.D.; Weng, H.-Y.; Bleyl, S.B.; Butterfield, R.J.; Williams, R.V. DMD Mutation and LTBP4 Haplotype do not Predict Onset of Left Ventricular Dysfunction in Duchenne Muscular Dystrophy. Cardiol. Young 2018, 28, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.B.; Vieland, V.J.; Dunn, D.M.; Kaminoh, Y.; Flanigan, K.M. Long-Range Genomic Regulators of THBS1 and LTBP4 Modify Disease Severity in Duchenne Muscular Dystrophy. Ann. Neurol. 2018, 84, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Stroup, D.F.; Berlin, J.A.; Morton, S.C.; Olkin, I.; Williamson, G.D.; Rennie, D.; Moher, D.; Becker, B.J.; Sipe, T.A.; Thacker, S.B. Meta-Analysis of Observational Studies in Epidemiology: A Proposal for Reporting. J. Am. Med. Assoc. 2000, 283, 2008–2012. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P.; Green, S. Cochrane Handbook for Systematic Reviews of Interventions: Cochrane Book Series; John Wiley and Sons: Hoboken, NJ, USA, 2008; ISBN 9780470699515. [Google Scholar]
- Study Quality Assessment Tools|NHLBI, NIH. Available online: https://www.nhlbi.nih.gov/health-topics/study-quality-assessment-tools (accessed on 15 April 2021).
- Neumann, I.; Pantoja, T.; Peñaloza, B.; Cifuentes, L.; Rada, G. El sistema GRADE: Un Cambio en la Forma de Evaluar la Calidad de la Evidencia y la Fuerza de Recomendaciones. Rev. Med. Chil. 2014, 142, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Tufanaru, C.; Munn, Z.; Stephenson, M.; Aromataris, E. Fixed or Random Effects Meta-Analysis? Common Methodological Issues in Systematic Reviews of Effectiveness. Int. J. Evid. Based Healthc. 2015, 13, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Altman, D.G.; Bland, J.M. How to Obtain the Confidence Interval from a P Value. BMJ 2011, 343, d2090. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.P.T.; Thompson, S.G. Quantifying Heterogeneity in a Meta-Analysis. Stat. Med. 2002, 21, 1539–1558. [Google Scholar] [CrossRef]
- Bello, L.; Piva, L.; Barp, A.; Taglia, A.; Picillo, E.; Vasco, G.; Pane, M.; Previtali, S.C.; Torrente, Y.; Gazzerro, E.; et al. Importance of SPP1 Genotype as a Covariate in Clinical Trials in Duchenne Muscular Dystrophy. Neurology 2012, 79, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Flanigan, K.M.; Weiss, R.B.; Spitali, P.; Aartsma-Rus, A.; Muntoni, F.; Zaharieva, I.; Ferlini, A.; Mercuri, E.; Tuffery-Giraud, S.; et al. Association Study of Exon Variants in the NF-kappaB and TGFbeta Pathways Identifies CD40 as a Modifier of Duchenne Muscular Dystrophy. Am. J. Hum. Genet. 2016, 99, 1163–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, M.; Awano, H.; Yamamoto, T.; Bo, R.; Matsuo, M.; Iijima, K. The ACTN3 577XX Null Genotype Is Associated with Low Left Ventricular Dilation-Free Survival Rate in Patients with Duchenne Muscular Dystrophy. J. Card. Fail. 2020, 26, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, D.M.; Witchel, S.F.; Ermani, M.; Hoffman, E.P.; Angelini, C.; Pegoraro, E. The Glucocorticoid Receptor N363S Polymorphism and Steroid Response in Duchenne Dystrophy. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1177–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, L.; Pegoraro, E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J. Clin. Med. 2019, 8, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.F.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a Major Activator of TGF-β1 In Vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Lawler, P.R.; Lawler, J. Molecular Basis for the Regulation of Angiogenesis by Thrombospondin-1 and -2. Cold Spring Harb. Perspect. Med. 2012, 2, a006627. [Google Scholar] [CrossRef]
- Miyazaki, D.; Nakamura, A.; Fukushima, K.; Yoshida, K.; Takeda, S.; Ikeda, S.I. Matrix Metalloproteinase-2 Ablation in Dystrophin-Deficient mdx Muscles Reduces Angiogenesis Resulting in Impaired Growth of Regenerated Muscle Fibers. Hum. Mol. Genet. 2011, 20, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- Capote, J.; Kramerova, I.; Martinez, L.; Vetrone, S.; Barton, E.R.; Sweeney, H.L.; Miceli, M.C.; Spencer, M.J. Osteopontin Ablation Ameliorates Muscular Dystrophy by Shifting Macrophages to a Proregenerative Phenotype. J. Cell Biol. 2016, 213, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Giacopelli, F.; Marciano, R.; Pistorio, A.; Catarsi, P.; Canini, S.; Karsenty, G.; Ravazzolo, R. Polymorphisms in the Osteopontin Promoter Affect Its Transcriptional Activity. Physiol. Genom. 2005, 20, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Barfield, W.L.; Uaesoontrachoon, K.; Wu, C.S.; Lin, S.; Chen, Y.; Wang, P.C.; Kanaan, Y.; Bond, V.; Hoffman, E.P. Eccentric Muscle Challenge Shows Osteopontin Polymorphism Modulation of Muscle Damage. Hum. Mol. Genet. 2014, 23, 4043–4050. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.; Lorenz, P.; Ibrahim, S.M.; Kundt, G.; Gross, G.; Kunz, M. The Functional -443T/C Osteopontin Promoter Polymorphism Influences Osteopontin Gene Expression in Melanoma Cells via Binding of c-Myb Transcription Factor. Mol. Carcinog. 2009, 48, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Renault, M.A.; Robbesyn, F.; Réant, P.; Douin, V.; Daret, D.; Allières, C.; Belloc, I.; Couffinhal, T.; Arnal, J.F.; Klingel, K.; et al. Osteopontin Expression in Cardiomyocytes Induces Dilated Cardiomyopathy. Circ. Heart Fail. 2010, 3, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuña, M.J.; Pessina, P.; Olguin, H.; Cabrera, D.; Vio, C.P.; Bader, M.; Muñoz-canoves, P.; Santos, R.A.; Cabello-verrugio, C.; Brandan, E. Restoration of Muscle Strength in Dystrophic Muscle by Angiotensin-1-7 through Inhibition of TGF-β Signalling. Hum. Mol. Genet. 2014, 23, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Pines, M.; Halevy, O. Halofuginone and Muscular Dystrophy. Histol. Histopathol. 2011, 26, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Andreetta, F.; Bernasconi, P.; Baggi, F.; Ferro, P.; Oliva, L.; Arnoldi, E.; Cornelio, F.; Mantegazza, R.; Confalonieri, P. Immunomodulation of TGF-Beta1 in mdx Mouse Inhibits Connective Tissue Proliferation in Diaphragm but Increases Inflammatory Response: Implications for Antifibrotic Therapy. J. Neuroimmunol. 2006, 175, 77–86. [Google Scholar] [CrossRef]
- Lee, E.M.; Kim, D.Y.; Kim, A.Y.; Lee, E.J.; Kim, S.H.; Lee, M.M.; Sung, S.E.; Park, J.K.; Jeong, K.S. Chronic Effects of Losartan on the Muscles and the Serologic Profiles of mdx Mice. Life Sci. 2015, 143, 35–42. [Google Scholar] [CrossRef]
- Micheletto, M.L.J.; Hermes, T.A.; Bertassoli, B.M.; Petri, G.; Perez, M.M.; Fonseca, F.L.A.; Carvalho, A.A.S.; Feder, D. Ixazomib an Oral Proteasome Inhibitor, Exhibits Potential Effect in Dystrophin-Deficient mdx Mice. Int. J. Exp. Pathol. 2021, 102, 11–21. [Google Scholar] [CrossRef]
- Kramerova, I.; Marinov, M.; Owens, J.; Lee, S.J.; Becerra, D.; Spencer, M.J. Myostatin Inhibition Promotes Fast Fibre Hypertrophy but Causes Loss of AMP-Activated Protein Kinase Signalling and Poor Exercise Tolerance in a Model of Limb-Girdle Muscular Dystrophy R1/2A. J. Physiol. 2020, 598, 3927–3939. [Google Scholar] [CrossRef]
- St. Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A Mouse Anti-Myostatin Antibody Increases Muscle Mass and Improves Muscle Strength and Contractility in the mdx Mouse Model of Duchenne Muscular Dystrophy and its Humanized Equivalent, Domagrozumab (PF-06252616), Increases Muscle Volume in Cynomolgus Monkeys. Skelet. Muscle 2017, 7, 25. [Google Scholar] [CrossRef]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin Inhibitor ACE-031 Treatment of Ambulatory Boys with Duchenne Muscular Dystrophy: Results of a Randomized, Placebo-Controlled Clinical Trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Asari, T.; Saitoh, M.; Nirasawa, K.; Sasaki, E.; Roppongi, Y.; Nakamura, A.; Saga, Y.; Shimada, T.; Ikeyama, H.; et al. Chain-Shortened Myostatin Inhibitory Peptides Improve Grip Strength in Mice. ACS Med. Chem. Lett. 2019, 10, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Iskenderian, A.; Liu, N.; Deng, Q.; Huang, Y.; Shen, C.; Palmieri, K.; Crooker, R.; Lundberg, D.; Kastrapeli, N.; Pescatore, B.; et al. Myostatin and Activin Blockade by Engineered Follistatin Results in Hypertrophy and Improves Dystrophic Pathology in mdx Mouse more than Myostatin Blockade Alone. Skelet. Muscle 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Qiao, C.; Li, J.; Xiao, B.; Li, J.; Xiao, X. A GDF11/Myostatin Inhibitor, GDF11 Propeptide-Fc, Increases Skeletal Muscle Mass and Improves Muscle Strength in Dystrophic mdx Mice. Skelet. Muscle 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, R.; Yu, T.; Li, J.; Zhang, M.; Jiang, S.; Wang, L.; Zhang, G.; Li, R.; Zhu, B.; et al. Sudden Cardiac Death of Duchenne Muscular Dystrophy with NT-proBNP in Pericardial Fluid as a Useful Biomarker for Diagnosis of the Cause of Death: A Case Report. Forensic Sci. Res. 2020, 5, 165. [Google Scholar] [CrossRef]
- Nassoro, D.D.; Torres, L.; Marando, R.; Mboma, L.; Mushi, S.; Mwakyula, I.H. A Child with Duchenne Muscular Dystrophy: A Case Report of a Rare Diagnosis among Africans. Clin. Case Rep. 2020, 8, 2654–2660. [Google Scholar] [CrossRef]
- Wakefield, S.E.; Dimberg, E.L.; Moore, S.A.; Tseng, B.S. Dystrophinopathy Presenting with Arrhythmia in an Asymptomatic 34-Year-Old Man: A Case Report. J. Med. Case Rep. 2009, 3, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Cheang, I.F.; Li Li, X. Cardiac Injury from Asymptomatic Duchenne Muscular Dystrophy. J. Am. Coll. Cardiol. 2019, 73, 2190. [Google Scholar] [CrossRef]
- Navarro, G.C.; Poutvinski, V.; Alvarado, K.R.; Alvarado, F.; Padilla, C.J.I.; Rafael Calderón Guardia, H.A.; José, S.; Rica, C. Compromiso Cardiaco en Distrofias Musculares: A Propósito de un Caso. Rev. Costarric. Cardiol. 2020, 22, 35–40. Available online: http://www.scielo.sa.cr/scielo.php?script=sci_arttext&pid=S1409-41422020000100035&lng=en&nrm=iso> (accessed on 6 August 2021).
- Brogna, C.; Coratti, G.; Pane, M.; Ricotti, V.; Messina, S.; D’Amico, A.; Bruno, C.; Vita, G.; Berardinelli, A.; Mazzone, E.; et al. Long-Term Natural History Data in Duchenne Muscular Dystrophy Ambulant Patients with Mutations Amenable to Skip Exons 44, 45, 51 and 53. PLoS ONE 2019, 14, e218683. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.T.; Barthelemy, F.; Martin, A.S.; Douine, E.D.; Eskin, A.; Lucas, A.; Lavigne, J.; Peay, H.; Khanlou, N.; Sweeney, L.; et al. DMD Genotype Correlations from the Duchenne Registry: Endogenous Exon Skipping is a Factor in Prolonged Ambulation for Individuals with a Defined Mutation Subtype. Hum. Mutat. 2018, 39, 1193–1202. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Country | Gene | Genetic Variant | Sample | Design | Outcomes |
|---|---|---|---|---|---|---|
| Barp A. et al. (2015) [19] | Italy | SPP1 LTBP4 | rs28357094 rs10880 | 178 | Retrospective | CF LoA |
| Bello L. et al. (2015) [20] | CINRG | SPP1 LTBP4 | rs28357094 rs10880 | 340 | Prospective | LoA |
| Chen M. et al. (2020) [21] | China | SPP1 LTBP4 | rs28357094, rs11730582, and rs17524488 rs2303729, rs1131620, rs1051303, and rs10880 | 326 | Retrospective | LoA |
| Flanigan KM. et al. (2013) [22] | US | LTBP4 | Haplotype IAAM/IAAM vs. others | 254 | Retrospective | LoA |
| Pegoraro E. et al. (2011) [23] | Italy CINRG | SPP1 | rs28357094 | 262 | Retrospective | LoA |
| Van den Bergen JC. et al. (2015) [24] | France Italy The Netherlands UK | SPP1 LTBP4 | rs28357094 Haplotype IAAM/IAAM vs. others | 336 | Retrospective | LoA |
| Van Dorn CS et al. (2018) [25] | US | LTBP4 | rs10880 | 101 | Retrospective | CF |
| Weiss RB. et al. (2018) [26] | US | LTBP4 + THBS1 | rs710160 rs2725797 | 253 | Retrospective | LoA |
| Authors | Genetic Variant | Subgroup | Loss of Ambulation | |
|---|---|---|---|---|
| Effect Size | Statistical Significance | |||
| Barp A. et al. (2015) [19] | SPP1 rs28357094 | Total | LoATT = 10.0y.; LoAGG/GT = 10.5y. | p = ns |
| Glucocorticoids | LoATT = 11.3y.; LoAGG/GT = 10.9y. | p = ns | ||
| No glucocorticoids | LoATT = 9.9y.; LoAGG/GT = 10.3y. | p = ns | ||
| LTBP4 rs10880 | Total | LoATT = 9.9y.; LoACC/CT = 10.9y. | Log-rank p = 0.058 | |
| Glucocorticoids | LoATT = 10.9y.; LoACC/CT = 11.9y. | p = ns | ||
| No glucocorticoids | LoATT = 9.9y.; LoACC/CT = 9.9y. | p = ns | ||
| Bello L. et al. (2015) [20] | SPP1 rs28357094 | Total | LoATT = 13.0y; LoAGG/GT = 11.8y. | Log-rank p = 0.048 |
| Glucocorticoids | LoATT = 13.9y.; LoAGG/GT = 12.0y | Log-rank p = 0.032 | ||
| No glucocorticoids | LoATT = 10.0y.; LoAGG/GT = 10.0y | Log-rank p = 0.6 | ||
| LTBP4 rs10880 | Total | LoATT = 13.9y.; LoACC/CT = 12.0y. | Log-rank p = 0.20 | |
| Glucocorticoids | LoATT = 13.9y.; LoACC/CT = 13.3y. | Log-rank p = 0.27 | ||
| No glucocorticoids | LoATT = 9.1y.; LoACC/CT = 10.0y | NA | ||
| Chen M. et al. (2020) [21] | SPP1 rs11730582 | Total | LoACC/CT = 11.00y.: LoATT = 10.33y. | Log-rank p = 0.272 |
| Glucocorticoids † | LoACC/CT = 12.00y.; LoATT = 10.67y | Log-rank p = 0.006 | ||
| No glucocorticoids † | LoACC/CT = 9.92y.; LoATT = 9.33y. | Log-rank p = 0.104 | ||
| SPP1 rs17524488 | Total | LoAGGGG/GGG = 10.50y.; LoAGG = 10.67 | Log-rank p = 0.983 | |
| Glucocorticoids † | LoAGGGG/GGG = 11.42y.; LoAGG = 11.92y. | Log-rank p = 0.478 | ||
| No glucocorticoids † | LoAGGGG/GGG = 9.50y.; LoAGG = 10.00y. | Log-rank p = 0.173 | ||
| LTPB4 IAAM/IAAM vs. others | Total | LoAIAAM/IAAM = 10.50y.; LoAothers = 10.50y. | Log-rank p = 0.706 | |
| Glucocorticoids † | LoAIAAM/IAAM = 10.67y.; LoAothers = 11.58y. | Log-rank p = 0.960 | ||
| No glucocorticoids † | LoAIAAM/IAAM = 9.92y.; LoAothers = 9.83y. | Log-rank p = 0.676 | ||
| Flanigan KM. et al. (2013) [22] | LTBP4 IAAM/IAAM vs. others | Glucocorticoids | LoAIAAM/IAAM = 12.5y.; LoAothers = 10.7y. | SDIAAM/IAAM = 3.3y.; SDothers = 2.1y. |
| No glucocorticoids | LoAIAAM/IAAM = 11.2y.; LoAothers = 9.8y. | SDIAAM/IAAM = 2.7y. SDothers = 2.0y. | ||
| Pegoraro E. et al. (2011) [23] | SPP1 rs28357094 | Total | Group GG/GT earlier loss of ambulation that TT At 14 years, 20% TT were ambulant, nobody of GG/GT | pkaplan meyer = 0.035 |
| Weiss RB. et al. (2018) [26] | LTBP4 rs710160 and THBS1 rs2725797 | Total | Interactions between LTBP4 and THBS1 (∆means) LTBP4 rs710160 TT + THBS1 rs2725797 TT: ∆LoA = 0y. LTBP4 rs710160 CC + THBS1 rs2725797 CC: ∆LoA = 1.2y. LTBP4 rs710160 CC + THBS1 rs2725797 TT: ∆LoA = 6.8y. | NA |
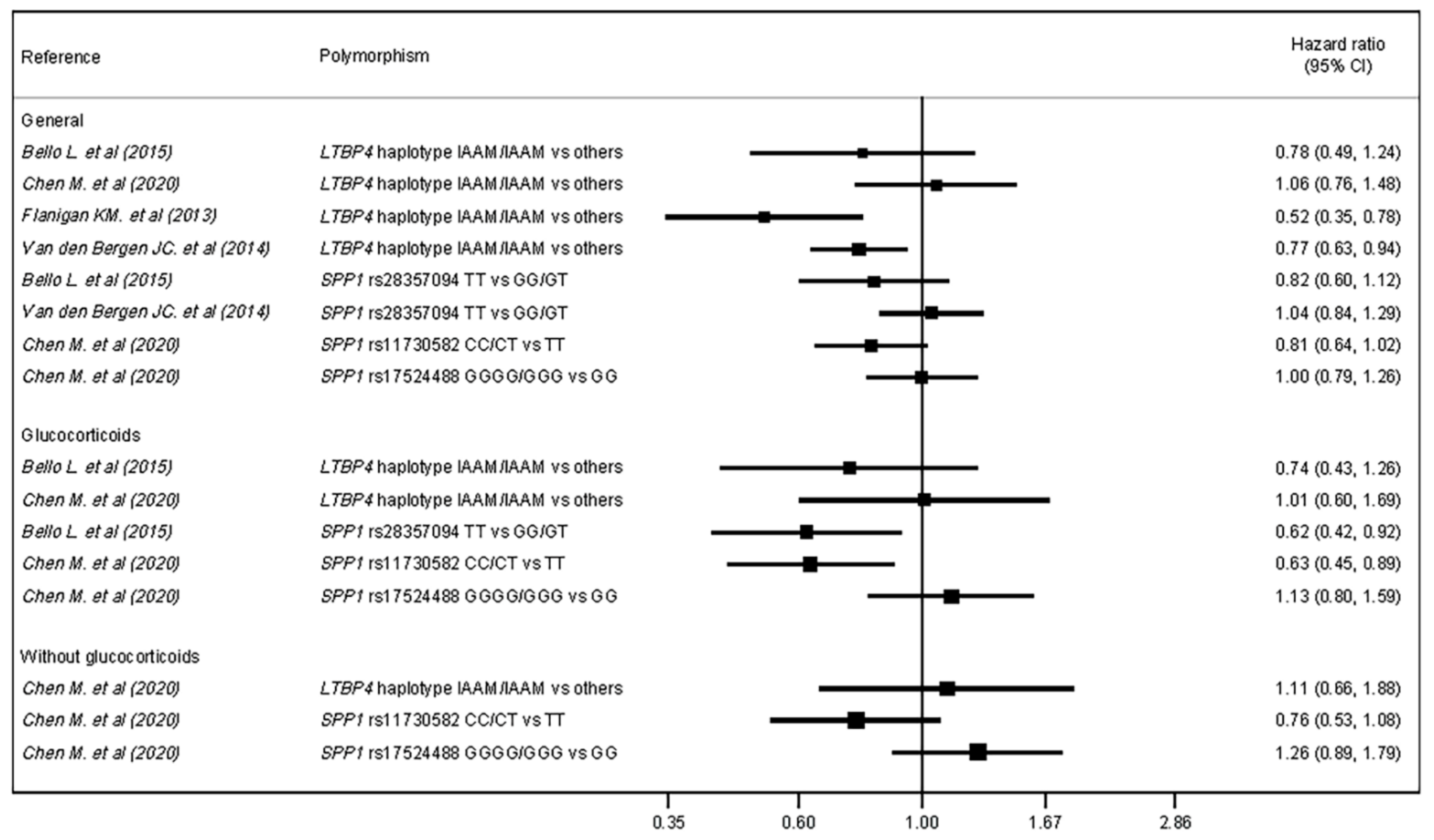
| Authors | Genetic Variant | Subgroup | Hazard Ratio for LoA | |
|---|---|---|---|---|
| Effect Size | Statistical Significance | |||
| Bello L. et al. (2015) [20] | SPP1 rs28357094 | Total | HR = 0.82 | 95% CI = 0.59–1.12 |
| Glucocorticoids | HR = 0.62 | 95% CI = 0.42–0.92 | ||
| LTBP4 rs10880 | Total | HR = 0.78 | 95% CI = 0.49–1.24 | |
| Glucocorticoids | HR = 0.74 | 95% CI = 0.44–1.26 | ||
| Chen M. et al. (2020) [21] | SPP1 rs11730582 | Total | HR = 0.81 | 95% CI = 0.64–1.02 |
| Glucocorticoids † | HR = 0.63 | 95% CI = 0.45–0.89 | ||
| No glucocorticoids † | HR = 0.76 | 95% CI = 0.53–1.08 | ||
| SPP1 rs17524488 | Total | HR = 1.00 | 95% CI = 0.80–1.26 | |
| Glucocorticoids † | HR = 1.13 | 95% CI = 0.80–1.59 | ||
| No glucocorticoids † | HR = 1.26 | 95% CI = 0.89–1.79 | ||
| LTPB4 IAAM/IAAM vs. others | Total | HR = 1.06 | 95% CI = 0.76–1.48 | |
| Glucocorticoids † | HR = 1.01 | 95% CI = 0.61–1.69 | ||
| No glucocorticoids † | HR = 1.11 | 95% CI = 0.66–1.88 | ||
| Flanigan KM. et al. (2013) [22] | LTBP4 IAAM/IAAM vs. others | Total | HR = 0.52 | 95% CI = 0.34–0.78 |
| Van den Bergen JC. et al. (2015) [24] | SPP1 rs28357094 | Total | HR = 1.04 | p = 0.73 |
| LTBP4 IAAM/IAAM vs. others | Total | HR = 0.77 | p = 0.01 | |
| Reference | Genetic Variant | Subgroup | Cardiac Function |
|---|---|---|---|
| Barp A. et al. (2015) [19] | SPP1 rs28357094 | Total | DCMTT = 19.1y. DCMGG/GT = 24.1y. (p = ns) |
| Glucocorticoids | DCMTT = 17.0y. DCMGG/GT = 24.0y. (p = ns) | ||
| LTBP4 rs10880 | Total | DCMTT = 29.5y. DCMCC/CT = 19.0y. (Log-rank p = 0.13) | |
| Glucocorticoids | DCMTT = >50% without DCM in the end DCMCC/CT = 17.9y. (Log-rank p < 0.05) | ||
| Van Dorn CS et al. (2018) [25] | LTBP4 rs10880 | Total | Myocardial dysfunction: CC (N = 20): 14.5 ± 3.2y. CT (N = 12): 13.1 ± 3.2y. TT (N = 2): 11.0 ± 2.8y. p = 0.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascual-Morena, C.; Cavero-Redondo, I.; Saz-Lara, A.; Sequí-Domínguez, I.; Lucerón-Lucas-Torres, M.; Martínez-Vizcaíno, V. Genetic Modifiers and Phenotype of Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis. Pharmaceuticals 2021, 14, 798. https://doi.org/10.3390/ph14080798
Pascual-Morena C, Cavero-Redondo I, Saz-Lara A, Sequí-Domínguez I, Lucerón-Lucas-Torres M, Martínez-Vizcaíno V. Genetic Modifiers and Phenotype of Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis. Pharmaceuticals. 2021; 14(8):798. https://doi.org/10.3390/ph14080798
Chicago/Turabian StylePascual-Morena, Carlos, Iván Cavero-Redondo, Alicia Saz-Lara, Irene Sequí-Domínguez, Maribel Lucerón-Lucas-Torres, and Vicente Martínez-Vizcaíno. 2021. "Genetic Modifiers and Phenotype of Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis" Pharmaceuticals 14, no. 8: 798. https://doi.org/10.3390/ph14080798
APA StylePascual-Morena, C., Cavero-Redondo, I., Saz-Lara, A., Sequí-Domínguez, I., Lucerón-Lucas-Torres, M., & Martínez-Vizcaíno, V. (2021). Genetic Modifiers and Phenotype of Duchenne Muscular Dystrophy: A Systematic Review and Meta-Analysis. Pharmaceuticals, 14(8), 798. https://doi.org/10.3390/ph14080798