Abstract
Influenza viruses are one of the major causative agents for human respiratory infections. Currently, vaccines and antivirals approved for preventing and treating viral infections are available. However, limited protection efficacy and frequent emergence of drug-resistant viruses stand for a need for the development of antivirals with different chemical skeletons from existing drugs. Screening of a chemical library identified an isoquinolone compound (1) as a hit with 50% effective concentrations (EC50s) between 0.2 and 0.6 µM against the influenza A and B viruses. However, it exhibited severe cytotoxic effects with a 50% cytotoxic concentration (CC50) of 39.0 µM in canine kidney epithelial cells. To address this cytotoxic issue, we synthesized an additional 22 chemical derivatives. Through structure-activity, as well as structure-cytotoxicity relationship studies, we discovered compound 21 that has higher EC50 values ranging from 9.9 to 18.5 µM, but greatly alleviated cytotoxicity with a CC50 value over 300 µM. Mode-of-action and cell type-dependent antiviral experiments indicated that it targets viral polymerase activity and functions also in human cells. Here, we present a new class of viral polymerase inhibitors with a core skeleton of isoquinolone, of which antiviral activity could be better improved through following design and synthesis of its derivatives for drug development.
1. Introduction
Influenza A and B viruses, belonging to the family Orthomyxoviridae, are the main causative agents of human infectious respiratory diseases during seasonal epidemics and unpredictable pandemics [1]. The viral genome comprises eight negative-sense RNA segments that are coated with nucleoprotein (NP). The 5′- and 3′-termini of genomic RNA are bound to polymerase subunits composed of polymerase basic protein 2 (PB2), PB1, and polymerase acidic protein (PA), which are required for immediate initiation of RNA replication and transcription in the nucleus of an infected cell [2]. Currently, quadrivalent vaccines are available to prevent infection by H1N1 and H3N2 subtypes of influenza A virus, as well as by both the Victoria and Yamagata lineages of influenza B virus. However, protective efficacy of these vaccines is no greater than 50% per year on average [3]. Moreover, several months are required to produce a vaccine against newly emerging and re-emerging viruses that are less efficiently or not neutralized by the existing vaccines. Accordingly, as an alternative way to respond to the influenza outbreaks that lack vaccine preparedness, antiviral treatments play a pivotal role in preventing the spread of viral infections.
Three different classes of antivirals have been developed to treat influenza viruses; all comprise small molecules that target viral matrix protein 2 (M2), neuraminidase (NA), or viral polymerase complex [1,4]. Adamantanes, such as amantadine and rimantadine, are potent inhibitors that block the M2 protein of influenza A, but not influenza B, viruses. They are no longer recommended for clinical use due to the prevalence of drug-resistant viruses harboring single or multiple substitutions (S31N, L26I, and/or V27A) in M2. NA inhibitors, including oseltamivir, zanamivir, peramivir, and laninamivir, are licensed in many countries. Of these, oseltamivir phosphate, a pro-drug of oseltamivir carboxylate, is prescribed most globally. Recently circulating influenza A and B viruses remain sensitive to oseltamivir. However, the possible emergence of oseltamivir-resistant or less sensitive viruses harboring the H275Y mutation or additional substitutions in NA, as occurred in 2008, is a threat to public health through increased pathogenicity and transmissibility [5]. The last class of antivirals, favipiravir and baloxavir (formally known as baloxavir marboxil), affect the viral polymerase subunits PB1 (RNA-dependent RNA polymerase) and PA (cap-dependent endonuclease) activities, respectively [6,7]. Favipiravir has gained only limited approval for use in pandemic situations in Japan due to concerns about its potential embryotoxicity [8]. Studies of the clinical safety and efficacy of baloxavir show more promising results. Nevertheless, they have identified amino acid substitutions (mainly I38X in PA), which may reduce drug susceptibility [9]. These studies suggest that the influenza viral polymerase complex is an attractive target for drug development. However, we need to ensure the diversity of available polymerase inhibitors if we are to overcome the issues associated with drug resistance.
We found an isoquinolone compound (1) from a cell-based, high-throughput screening approach that shows inhibitor activity against both the influenza A and B viruses. The compound efficiently suppresses the viral RNA replication step, but has cytotoxic effects. To improve this limitation, we synthesized 22 chemical derivatives and identified a less toxic antiviral agent, compound 21. Here, we show that compound 21 could act as the backbone for development of an antiviral drug inhibiting the influenza viral polymerase activity without exerting cytotoxic effects.
2. Results
2.1. Identification of Compound 1 with Anti-Influenza Viral Activity
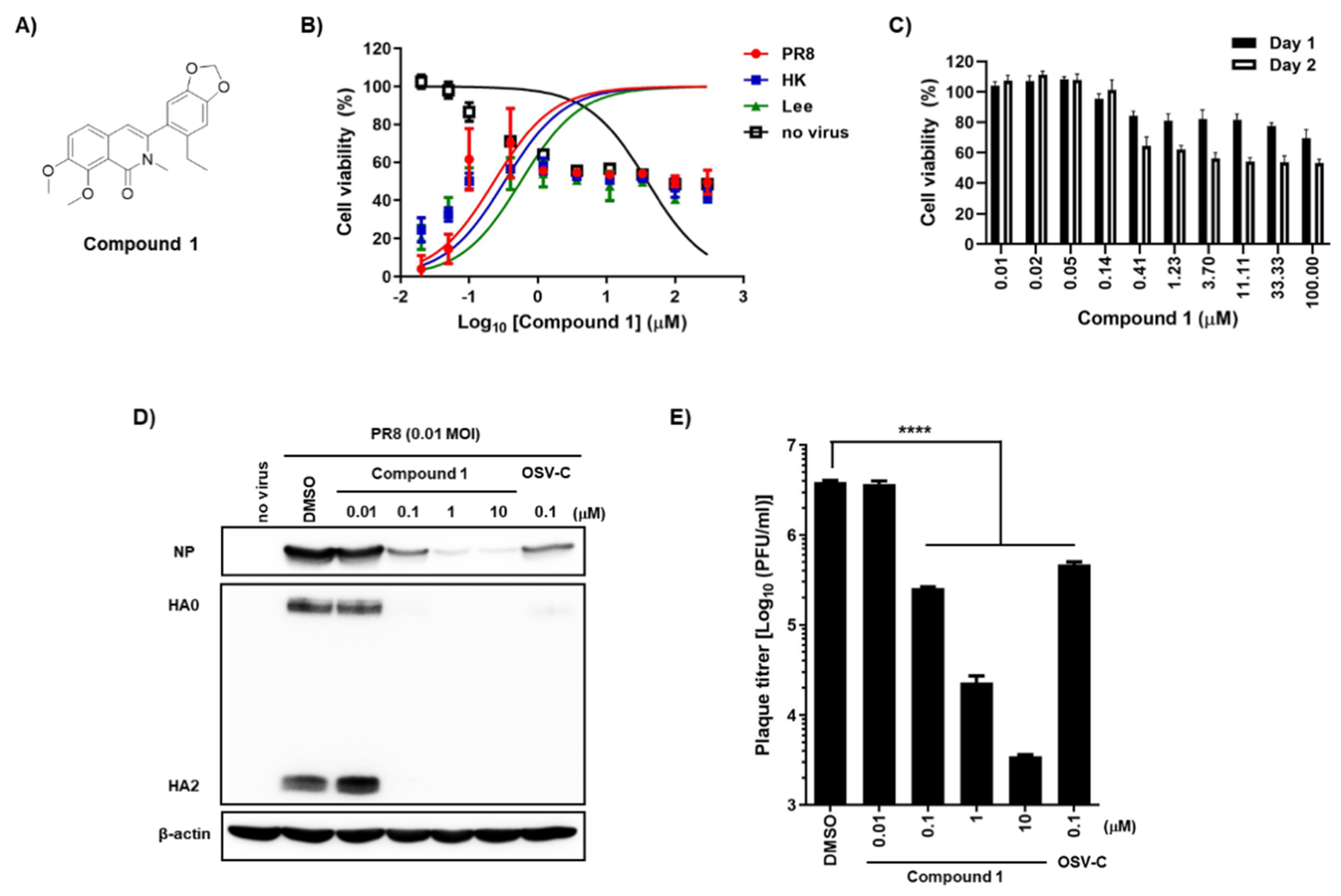
To secure compounds with anti-influenza activity, we screened a chemical library from the Korea Chemical Bank (Daejeon, Republic of Korea), which comprised 7000 different small molecules deposited by medicinal chemists. The fluorescent diacetate-based antiviral assay [10] identified 3-(6-ethylbenzo[d][1,3]dioxol-5-yl)-7,8-dimethoxy-2-methylisoquinolin-1(2H) (named compound 1) as a hit compound, with 50% effective concentration (EC50) values ranging from 0.2 to 0.6 μM against both influenza A [A/Puerto Rico/8/34 (PR8) and A/Hong Kong/8/68 (HK)] and B/Lee/40 (Lee) viruses, and a 50% cytotoxic concentration (CC50) value of 39.0 μM in MDCK cells (Figure 1A,B; Table 1). Despite an excellent selective index (S.I.) of ≥65, incubation for 2 days led to >40% cell death at concentrations > 0.41 μM (Figure 1B). To exclude the possibility that the antiviral efficacy was mixed with cytotoxicity, it was needed to evaluate antiviral activity of compound 1 under subtoxic concentrations. To find this condition, we compared its cytotoxicity between days 1 and 2 using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Figure 1C). Consistent with the fluorescein diacetate assay (Figure 1B), the MTT assay revealed that a 2-day treatment of compound 1 at concentrations above 0.41 µM led to cell death more than 40%, while 1-day treatment below 11.1 µM resulted in a cell viability higher than 80%. On the basis of these subtoxic concentrations, both Western blot analysis and plaque titration assays were performed, showing that compound 1 suppressed influenza viral infection by reducing the expression of viral proteins, including NP and HA, in cells, as well as by reducing the number of viral progeny released into the culture supernatant (Figure 1D,E).
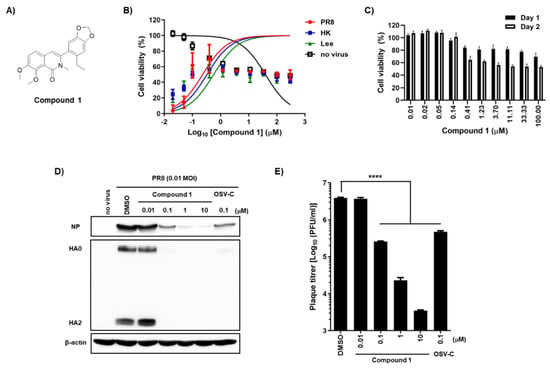
Figure 1.
Antiviral activity of compound 1 against influenza A and B viruses and its toxicity in MDCK cells. (A) Chemical structure of compound 1. (B) Antiviral activity and cytotoxicity of compound 1. MDCK cells were mock-infected (no virus) or infected individually with A/Puerto Rico/8/34 (H1N1; PR8), A/Hong Kong/8/68 (H3N2; HK), or B/Lee/40 (Lee) in the presence of increasing concentrations of compound 1. On day 2, cell viability was measured using fluorescein diacetate; the fluorescence intensity of mock-infected DMSO-treated cells and virus-infected DMSO-treated cells was set as 100% and 0%, respectively. (C) Comparison of the cytotoxicity of compound 1 on different days. The percentage viability of MDCK cells was measured in an MTT assay after treatment with increasing concentrations of compound 1 on days 1 (black bar) and 2 (white bar). (D) Inhibition of viral protein expression. MDCK cells were infected with PR8 for 1 h at a multiplicity of infection (MOI) of 0.01 and then treated with increasing concentrations of compound 1 or 0.1 µM OSV-C. On the next day, cell lysates were harvested for Western blot analysis using anti-NP and anti-HA2 antibodies. β-Actin was used as a loading control. Each protein is denoted on the left-handed side of the blots. (E) Plaque assay. Culture supernatants from samples prepared in (D) were serially diluted 10-fold and used to infect fresh MDCK cells. On day 3 post-infection, the number of plaque-forming units (PFU) was counted by crystal violet staining. In panels (B), (C), and (E), values are expressed as the means ± standard deviation from three different experiments. ****, p < 0.0001.

Table 1.
Antiviral activity of phenylisoquinolone derivatives against influenza viruses and toxicity in MDCK cells.
2.2. Compound 1 Inhibits Viral Polymerase Activity
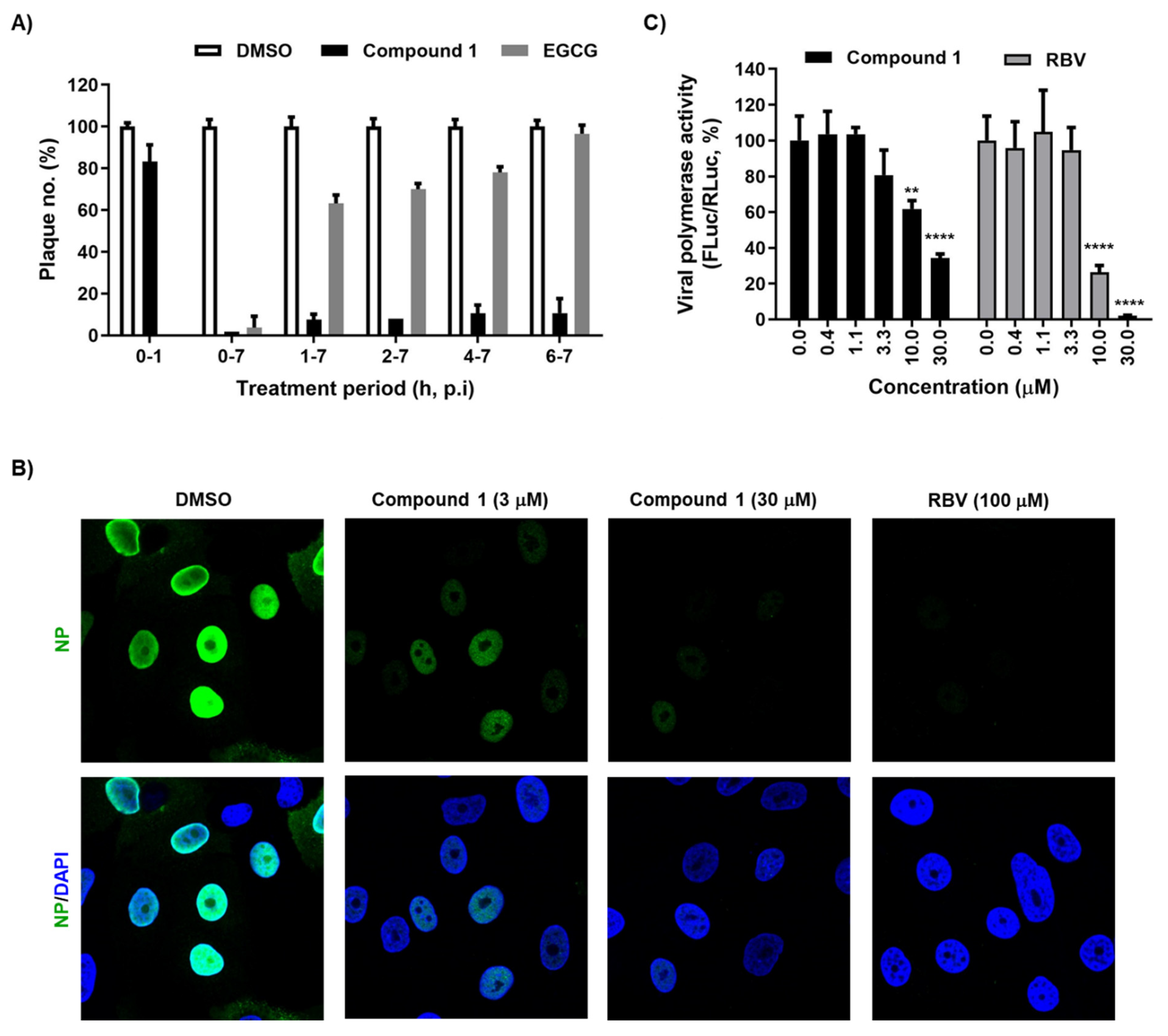
Next, we performed a time-of-addition experiment to determine the step in the influenza virus life cycle targeted by compound 1. Plaque titration assays revealed that exposure to compound 1 (10 μM) during viral adsorption (0–1 h) at 4 °C failed to induce antiviral activity, whereas EGCG, an entry inhibitor, blocked PR8 infection completely at the same condition (Figure 2A). However, treatment with compound 1 starting at any time point until 7 h post-infection consistently inhibited plaque formation by over 80%. This suggests that compound 1 could target virus penetration into cells or genome replication, rather than its attachment to cellular sialic acid receptors. To further evaluate this hypothesis, we compared the intracellular distribution of NP at 5 h post-infection in the presence or absence of compound 1. Confocal microscopy showed few or no NP-positive cells when compound 1 was treated at concentrations of 3 or 30 μM (Figure 2B). To collect a more direct evidence, we adopted a cell-based reporter assay for influenza viral RdRp activity in which a negative strand of the firefly luciferase (FLuc) RNA transcript flanked with viral 5′ and 3′ UTRs is used as a template for synthesis of positive sense RNA, as well as mRNA, in the presence of viral polymerase complex. To address a concern that antiviral function observed could be induced limitedly in canine cells in a species-specific manner (Figure 1), a human cell line, HeLa, was applied to this polymerase assay. The results of the dual luciferase assay clarified that compound 1 suppressed viral RNA replication/transcription in a similar way to RBV (Figure 2C). Taken together, these mode-of-antiviral action experiments suggested that compound 1 works at the viral genome replication step.
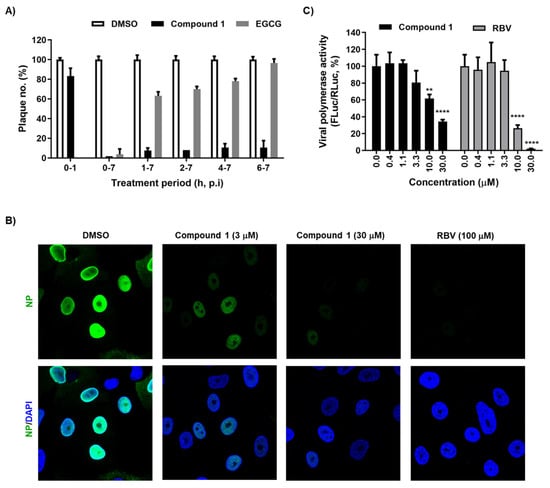
Figure 2.
Inhibition of viral genome replication by compound 1. (A) Time-of-addition experiment. MDCK cells in 48-well plates were infected with PR8 (MOI, 0.01) at 4 °C for 1 h. Unabsorbed virus was removed by washing with PBS. Compound 1 (10 µM) was added at different time points. EGCG (1 µM), which blocks virus entry, was used as a control. At 7 h post-infection, the supernatant was removed and overlay medium containing 0.6% CMC was overlaid for 3 days at 33 °C. Plaque numbers in DMSO-treated samples under each condition were set as 100%. (B) Confocal microscopy showing nuclear localization of NP. MDCK cells were infected with PR8 at an MOI of 3 in the presence of DMSO, compound 1 (3 or 30 µM), or ribavirin (RBV; 100 µM) at 33 °C for 5 h. Viral NP was visualized using an anti-NP antibody and Alexa 488-conjugated goat anti-mouse IgG (green), while cellular nuclei were counterstained with DAPI (blue). Original magnification, ×60. (C) Inhibition of viral polymerase activity. HeLa cells were transfected with plasmids expressing viral polymerase complex, including pVP-PB2, -PB1, -PA, or -NP (200 ng in well of a 24-well plate), together with reporter plasmids pHH21-FLuc (200 ng per well) and phRL-CMV (10 ng per well). At 6 h after transfection, cells were treated with increasing concentrations of compound 1 or RBV. On day 1, viral polymerase activity (%) was determined by measuring FLuc expression from pHH21-FLuc [normalized to Renilla luciferase (RLuc) expression from phRL-CMV]. In (A) and (C), values are expressed as the mean ± standard deviation from three different experiments. **, p < 0.01; ****, p < 0.0001.
2.3. Synthesis of Isoquinolone Derivatives
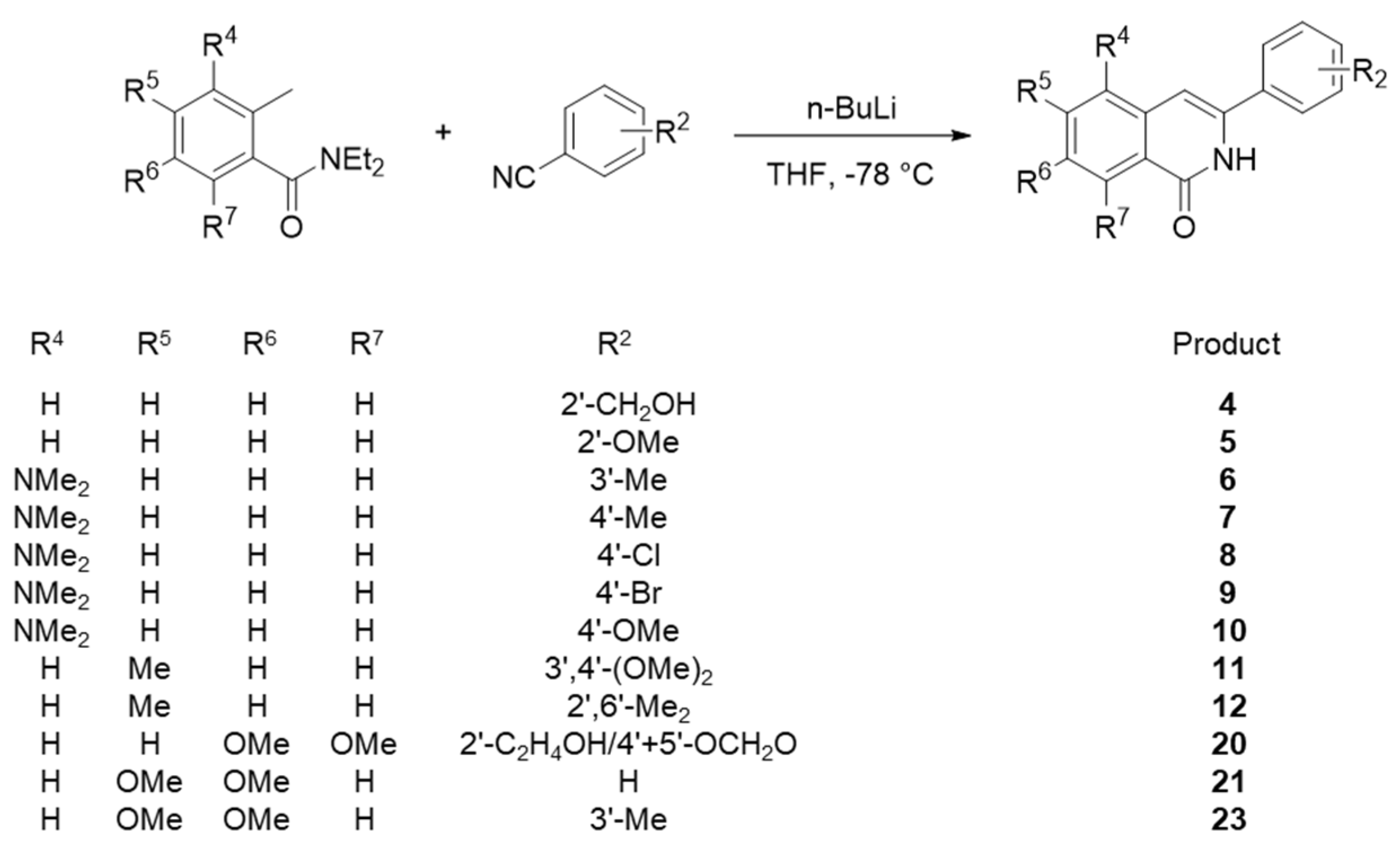
Among 23 isoquinolone compounds, 1 and 17–19 have been synthesized according to our established protocol (Supplementary Scheme S1) [11,12]. Compounds 2, 3, 13–15 and 22 without the methylenedioxy group at R2 were separately synthesized (Supplementary Scheme S2) [13,14,15,16]. A newly designed synthetic method was used for phensylisoquinolone derivatives 7–10 and 21, and these compounds were prepared in a one-step cyclization reaction using n-BuLi to link the N,N-diethyl-2-methylbenzamide and benzonitrile moieties (Scheme 1). These products were characterized by 1H NMR (Supplementary Figures S1 to S5). The remaining compounds, 4, 5, 6, 11, 12, 20, and 23, were also synthesized according to Scheme 1 and our previous reports [15,16,17,18,19].
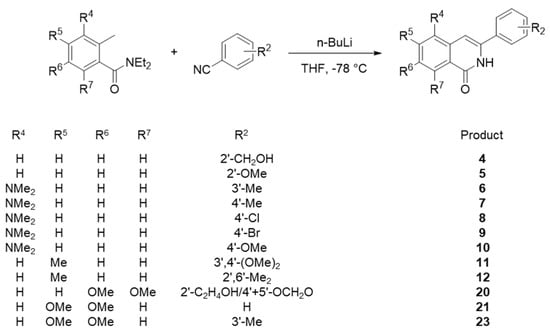
Scheme 1.
Synthesis of phenylisoquinolones 4–12, 20, 21, and 23.
2.4. Structure-Activity Relationship: Isoquinolone Derivatives
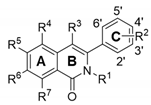
As mentioned above, although compound 1 is a highly potent antiviral agent, it showed severe cytotoxic effects after treatment for 2 days (Figure 1B,C). Thus, we wondered which moiety within compound 1 was responsible for the cytotoxicity, and whether it could be improved by chemical modifications. To investigate the structure-activity relationship (SAR), as well as the structure-cytotoxicity relationship analyses, we chemically modified three rings (A, B, and C) of phenylisoquinolone, either independently or simultaneously (Table 1).
Taking the simplest compound with a basic skeleton, 3-phenylisoquinolone (2) insertion of substituents into Ring C revealed that a hydroxyl (3) or hydroxymethyl group (4) at the ortho position inhibited replication of the PR8, HK, and Lee influenza viral strains, with less cytotoxicity than compound 1. The methoxy group (5) suppressed the Lee strain slightly, but was still cytotoxic. When the dimethylamino group was introduced at R4 in phenyl Ring C, the m-methyl derivative (6) was effective against Lee, but not against PR8 and HK. Interestingly, antiviral activity disappeared in derivatives with a para-substitution in Ring C (e.g., p-methyl (7), p-chloro (8), p-bromo (9), or p-methoxy (10) substitutions). Although none of these dimethylamino-substituted compounds showed cytotoxicity at the maximum concentration of 300 µM, additional dimethylamino derivatives were not considered for further modification and testing due to expectation of their loss of antiviral activities.
To investigate the influence of substitutions at R5 on cytotoxicity and antiviral activity, a methyl group was first introduced at that position, while R2 was modified. When dimethoxy moieties were substituted at the 3′ and 4′ positions of R2, the resulting compound 11 was non-toxic and showed considerable antiviral activity against the PR8 strain (S.I., >9.3), but its activity against the HK and Lee strains was weak (S.I., >3.2). Meanwhile, as shown for compound 12, dimethyl modification at the 2′ and 6′ positions of the phenyl ring led to complete loss of antiviral activity. Bromination at R3 in compounds 11 and 12 generated 13 and 14, respectively, which did not show significant antiviral activity. As another approach, derivatives methylated at R1 but not at R5 were synthesized, resulting in compounds 15, 16, and 17. Compound 15 with a formyl group at the 2′ position showed neither antiviral activity nor severe toxicity. Even though its cytotoxicity was not fully reflected in the CC50 calculation, approximately 30–40% of cells were dead at concentrations > 100 µM (data not shown). Compound 16, with a hydroxyl substitution at 2′ but a formyl group at R3, was not effective at subtoxic concentrations. Compound 17, with a methoxy group at R6, was partially active against influenza A viral strains (EC50 values between 5.8 and 36.8 µM) but not against the influenza B virus. Notably, its cytotoxicity (CC50, 109.0 µM) was lower than that of compound 1 (CC50, 39.0 µM). Taken together, the results from active but less toxic compounds 2, 3, 4, 11, and 17 suggest that the intrinsic antiviral activity of the hit compound 1 can be maintained with much lower cytotoxicity.
2.5. Structure-Activity Relationship: Dimethoxy Isoquinolone Derivatives
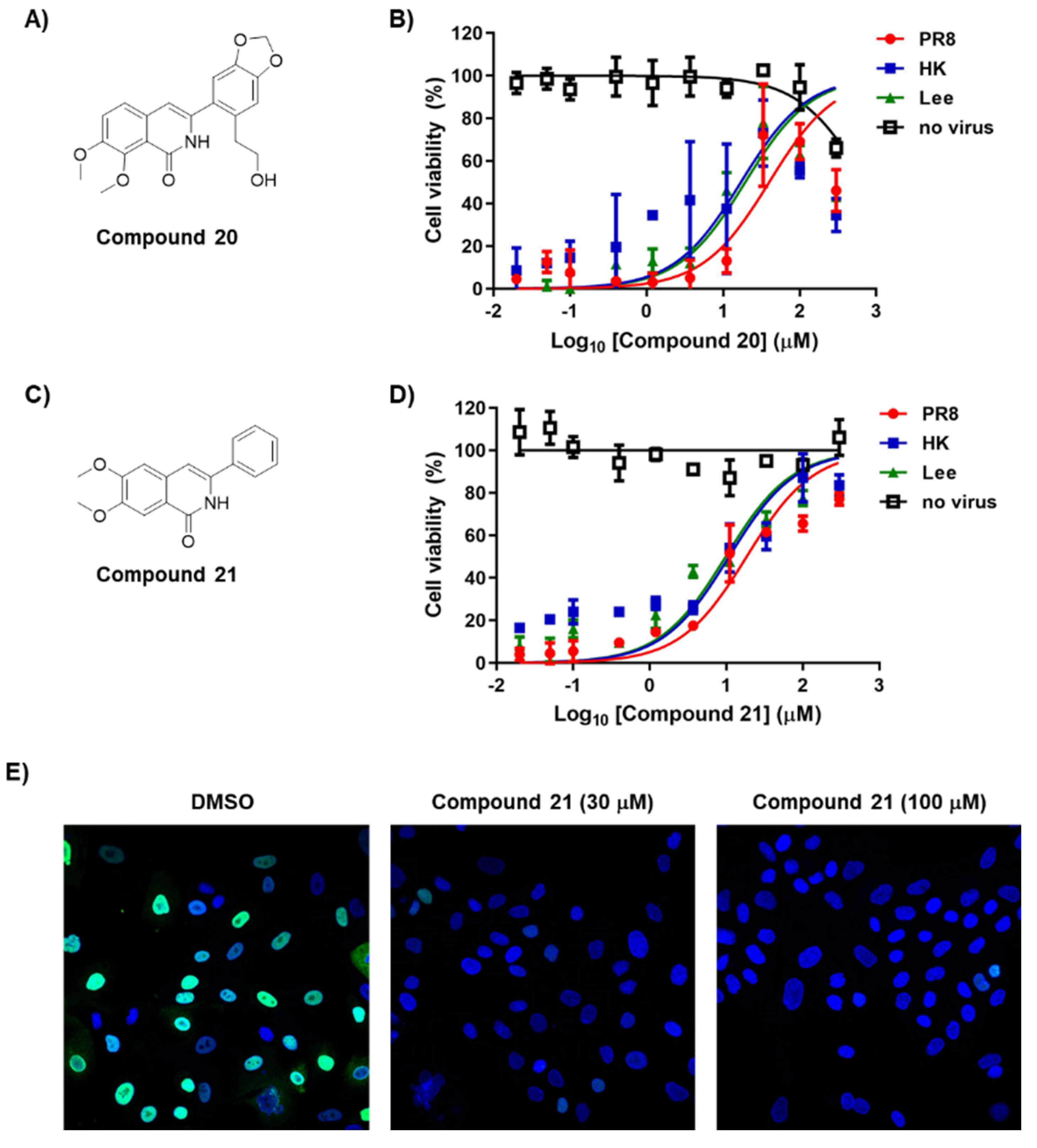
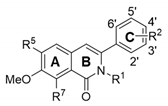
Based on previous conjecture, we asked if it is possible to discover an active polymerase inhibitor (S.I., >15) without cytotoxicity at 300 µM, as observed in RBV. It was interesting that compound 17 exhibited considerable antiviral activity against the HK strain (S.I., 18.8) and was less cytotoxic than compound 1 (Table 1). From a comparison of their chemical structures between compounds 1 and 17, we assumed that deleting the methoxy group at R7 would be associated directly with increased cell viability while maintaining antiviral activity. For an in depth SAR analysis, we focused on synthesizing additional dimethoxy derivatives of phenylisoquinolone, starting from compound 1 (Table 2). The ethyl group at the 2′ position of R2 in compound 1 was replaced by ethylene (18) and hydroxyethyl (19) groups. As expected, compound 18 had properties very similar to those of compound 1, i.e., it was active but highly toxic, with a CC50 value of 40.3 µM. Surprisingly, compound 19 was not only active against all three strains, even with relatively high EC50 values ranging from 22.2 to 59.9 µM, but also highly improved the cytotoxicity profile (CC50, >300 µM). A comparative SAR analysis of 19 with 1 or with 18 indicated that the magnitude of R7-OMe-derived cytotoxicity depends on 2′ modification of R2. Notably, the demethylated derivative at R1 of non-toxic 19, which generated compound 20, increased the antiviral activity against the influenza A and B viruses by 2–3-fold. However, latent toxicity was still detected at the highest concentration, resulting in approximately 30% cell death at 300 µM (Figure 3A,B). It confirmed that being linked with a chemical entity at the 2′ position of R2, the methoxy group at R7 could be attributed to cytotoxicity.
Table 2.
Antiviral activity of dimethoxyphenylisoquinolone derivatives against influenza viruses and toxicity in MDCK cells.
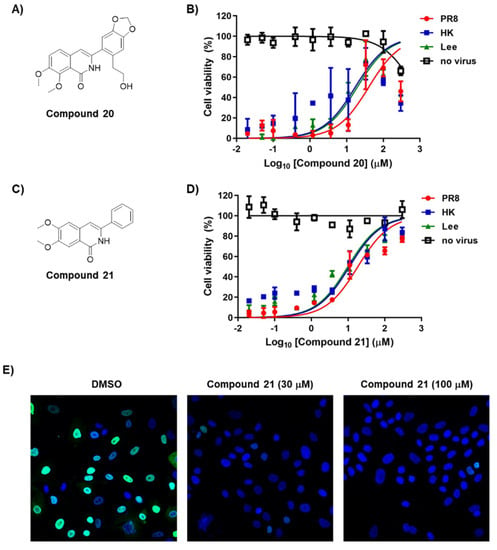
Figure 3.
Antiviral activity and cytotoxicity of compounds 20 and 21. (A,C) Chemical structure of compounds 20 and 21. (B,D) Antiviral activity and cytotoxicity of compounds 20 and 21. MDCK cells were mock-infected (no virus) or infected individually with influenza A viruses (PR8 and HK) or influenza B virus (Lee) in the presence of increasing concentrations of each compound. On day 2, percentage cell viability was measured using fluorescein diacetate; the fluorescence intensity of mock-infected DMSO-treated cells and virus-infected, DMSO-treated cells was set as 100% and 0%, respectively. (E) Confocal microscopy showing reduction of viral NP amounts by compound 21. MDCK cells were infected with PR8 at an MOI of 3 in the presence of 0.2% DMSO or compound 21 (30 or 100 µM) at 33 °C for 5 h. Viral NP was visualized using an anti-NP antibody and Alexa 488-conjugated goat anti-mouse IgG (green), while cellular nuclei were counterstained with DAPI (blue). Original magnification, ×40.
We tried to synthesize more primitive phenylisoquinolone derivatives in which all side chains were removed except for the dimethoxyl groups at R5 and R6. Compound 21 exhibited ideal antiviral activity profiles, including a dose-dependent response, as well as tolerable cytotoxicity (Table 2; Figure 3C,D). It was active against all three viral strains tested, with EC50 values ranging from 9.9 to 18.5 µM, and with higher potency than a polymerase inhibitor, ribavirin (RBV). Most importantly, it was not cytotoxic, even at the highest concentration (300 µM). We evaluated its specific antiviral activity by visualizing the reduction of viral NP expression in PR8-infected cells using fluorescence microscopy (Figure 3E). By contrast, hydroxylation (22) or methylation (23) of the phenyl ring resulted in complete loss of activity (Table 2). This finding was intriguing because the 2′-OH of compound 3 hardly affected antiviral activity when compared with that of compound 2. Collectively, it stresses again that the 2′ position in Ring C is involved in both antiviral activity and cytotoxicity, depending on the methoxylation stoichiometry of Ring A. In summary, the chemical modification of cytotoxic antiviral compound 1 identified seven safer (CC50, >300 µM) compounds (2–4, 11, and 19–21) exhibiting a broad spectrum antiviral activity. Among these, 6,7-dimethoxy-3-phenylisoquinolone (compound 21) was most potent in the cell culture system, satisfying S.I. values over 15 against all three viral strains tested.
2.6. Anti-Influenza Viral Activity of Compound 21 in Human Cells by Suppressing Viral Polymerase Activity
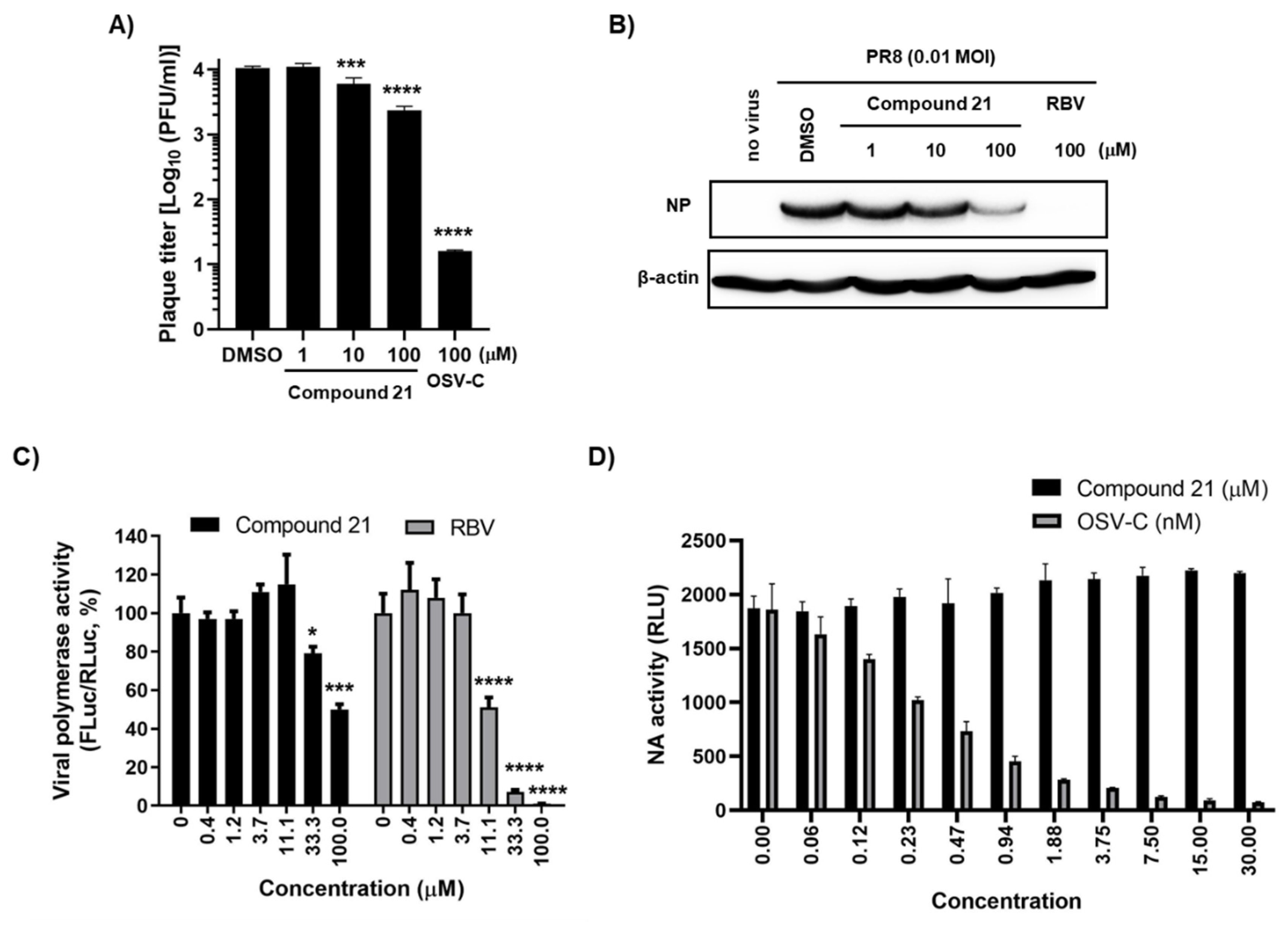
A plaque assay confirmed that compound 21 inhibited progeny virus production into MDCK culture supernatants in a dose-dependent manner (Figure 4A). We wondered whether its antiviral efficacy can be reproducible in human cells. Human lung epithelial cells, A549, were infected with PR8 and treated with the compound for one day. Western blot analysis with the cell lysates revealed an inhibition of NP expression (Figure 4B). Furthermore, like the primary hit compound 1 (Figure 2C), it efficiently suppressed viral polymerase activity in HeLa cells (Figure 4C). However, our previous time-of-addition experiment with compound 1, still showing antiviral activity between 6 and 7 h post-infection, motivatied us to investigate whether a later step of the virus life cycle can be influenced. As NA is a representative viral protein playing a pivotal role in the virus release, its activity was tested with a purified PR8 virus. The result showed that it had no inhibitory effect at a maximum concentration of 30 µM, while OSV-C efficiently blocked NA activity with a 50% inhibitory concentration (IC50) of 0.30 ± 0.04 nM (Figure 4D). These data clearly suggested that compound 21 affects viral polymerase activity, and its antiviral activity is reliable in human cells, ensuring its potential for the development of antivirals to treat human influenza infections through additional chemical modifications.
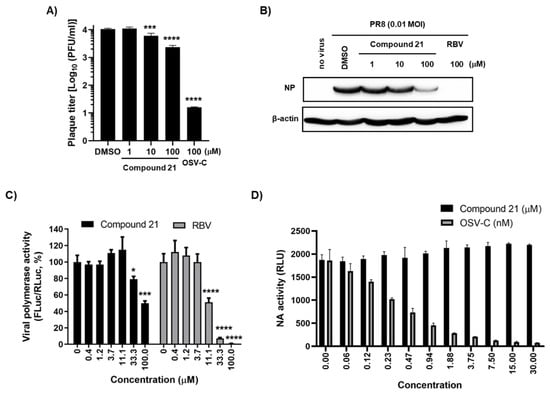
Figure 4.
Antiviral activity of compound 21 in human cells. (A) Plaque assay. PR8-infected MDCK cells (MOI, 0.01) were treated with compound 21 or OSV-C, as a control, for one day. Culture supernatants were serially diluted 10-fold and used to infect fresh MDCK cells. On day 3, the number of plaques was counted by crystal violet staining. (B) Antiviral activity in A549 cells. Cells were infected with PR8 (MOI, 0.01) and treated with compound 21 or RBV, as a control. After SDS-PAGE and electrotransfer, viral NP protein was detected with anti-NP antibody and HRP-conjugated goat anti-rabbit IgG. β-Actin was used as a loading control. (C) Viral polymerase activity assay in HeLa cells. Cells were transfected with plasmids expressing viral polymerases, including pVP-PB2, -PB1, -PA, or -NP (200 ng in well of a 24-well plate), together with reporter plasmids pHH21-FLuc (200 ng per well) and phRL-CMV (10 ng per well). At 6 h after transfection, cells were treated with increasing concentrations of compound 1 or RBV. On day 1, viral polymerase activity (%) was determined by measuring FLuc normalized to RLuc. (D) NA activity assay. PR8 virus (2.8 × 103 PFU) was pre-incubated with increasing concentrations of compound 21 or OSV-C at 37 °C for 20 min in 96-well plates. At 30 min after addition of a chemiluminescent NA substrate, luminescence was measured and expressed as relative luminescence units (RLU). In (A,C,D), values are expressed as the mean ± standard deviation from three different experiments. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
3. Discussion
Screening a chemical library identified that compound 1 had an inhibitory effect against both influenza A and B viruses with EC50 values ranging from 0.2 to 0.6 μM. It exhibited highly selective antiviral efficacy; nevertheless, it showed severe cytotoxicity (inducing > 40% cell death at concentrations > 0.41 μM on day 2). This was improved by reducing incubation time for 1 day, which resulted in 80% average viability at a concentration of 11.11 µM. However, the problem of intrinsic cytotoxicity had to be solved to make it more druggable. Therefore, we synthesized additional isoquinolone derivatives. Among the 22 modified compounds, we found that compound 21 did not affect cell viability when incubated with cells for 2 days at the maximum tested concentration of 300 µM. Although this modification reduced antiviral efficacy by 16.5–92.5-fold compared with compound 1, compound 21 sufficiently suppressed influenza viral infection at subtoxic concentrations, showing a desirable antiviral profile with high S.I. values (>15) against all three viral strains tested (Figure 3D and Table 2). A time-of-addition study and enzymatic assay revealed that these isoquinolone compounds target the viral RNA replication step during the virus life cycle, not HA-mediated viral adsorption onto the cell surface or NA-mediated progeny viral release. It was pondered when compound 21 is a direct acting polymerase inhibitor, which viral protein among PB2, PB1 and PA could be a putative target molecule. To make rational conjecture, an in silico docking study was performed to compare the structural similarities and docking patterns by using the three polymerase inhibitors including VX787 (PB2 inhibitor), T705 (favipiravir, PB1 inhibitor) and Baloxavir (PA inhibitor) as controls. Interestingly, the modeling data displayed that compound 21 could interact with PB2 in a similar way to VX787 (Supplementary Figures S6–S8) [20,21,22]. In detail, compound 21 formed π-π stacked interactions with Phe104 and Phe23 of PB2 as observed in the binding pattern of VX787 to PB2. It also generated a hydrogen bond to Glu61 and H2O4140, which could increase its binding affinity to the protein. On the basis of the in silico docking analysis, it is expected that compound 21 could inhibit the polymerase activity by binding to PB2. However, at present, it is not yet experimentally determined whether it directly binds to one of the vRNP components, i.e., PB2, PB1, PA, or NP, or to cellular factors that are participated in viral genome amplification. Particularly, in the latter case, to address any concerns regarding the species-specific antiviral activity of the compounds which were discovered from MDCK cell-based screening, we evaluated its antiviral efficacy and polymerase inhibitory effect in human epithelial cells, A549 and HeLa, respectively (Figure 4B,C). The results ensured that even when it recognizes a host factor, it could not be a matter for antiviral drug development in further extensive chemical modifications.
The structure-activity and structure-cytotoxicity relationship studies of isoquinolone derivatives had four outcomes: (i) the simplest core skeleton, phenylisoquinolone (2) and its 2′-substituted derivatives (3 and 4, but not 5) are non-toxic but marginally active; (ii) complete loss of antiviral activity is induced by dimethylamino substitutions at R4 (as shown for 6 to 10); (iii) methylation at R1 increases cytotoxicity (as shown for 1 and 16–18), although this can be moderated by substitution with either formyl (15) or hydroxyethanol (19) groups; and (iv) most of the compounds with mono-methoxy at R6 (17) or dimethoxy at both R6 and R7 (18 to 20) are effective but intrinsically toxic, while R5- and R6-substituted dimethoxy compounds are not toxic, but their antiviral activity is absolutely dependent on chemical substituents on R2 (21 to 23). Collectively, the data presented herein show that strategic chemical optimization of hit compound 1 generated compound 21, which may be the basis for development of a novel therapeutic agent affecting the viral RNA replication step of different (sub)types of influenza viruses.
4. Materials and Methods
4.1. Chemical Synthesis
Isoquinolone derivatives were synthesized as described previously [11,12,13,14,15,16,17,18,19]. The structure of newly synthesized compounds 7–10 and 21 was confirmed by 300 MHz (Varian Unity Plus 300 MHz; Varian Inc., Palo Alto, CA, USA) and 400 MHz (Bruker Ascend 400 MHz; Bruker Daltonik, Bremen, Germany) nuclear magnetic resonance (NMR) spectrometry. Purity was estimated to be ≥99%.
4.2. Cells, Viruses, and Antiviral Compounds
Madin-Darby canine kidney (MDCK) cells, human cervical cancer HeLa cells and human lung epithelial A549 cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured at 37 °C/5% CO2 in Minimum Essential Medium (MEM; Invitrogen, Carlsbad, CA, USA), Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen) or Roswell Park Memorial Institute 1640 Medium (RPMI; Hyclone, Logan, UT, USA), respectively, supplemented with 10% FBS (Invitrogen). Influenza viruses A/Puerto Rico/8/34 (PR8), A/Hong Kong/8/68 (HK), and B/Lee/40 (Lee) were obtained from the ATCC. The two type A influenza strains were amplified in 10-day-old embryonated chicken eggs, while the type B strain was inoculated into MDCK cells in the presence of 2 μg/mL TPCK-treated trypsin at 37 °C for 3 days. The NA inhibitor, oseltamivir carboxylate (OSV-C; ≥98%), was purchased from United States Biological (Swampscott, MA, USA). The cell entry blocker (-)-epigallocatechin-3-gallate (EGCG, ≥95%) and the polymerase inhibitor ribavirin (RBV) were from Sigma-Aldrich (St. Louis, MO, USA). Stocks were prepared in water (for EGCG) or DMSO (for OSV-C and RBV); the final concentration of each was 50 mM.
4.3. Cytopathic Effect Assay
The fluorescein diacetate-based cell viability assay was performed as described previously [23]. Briefly, MDCK cells seeded on 96-well plates (3 × 104 cells per well) were grown to confluency and then mock-infected or infected with the influenza A or B virus at a multiplicity of infection (MOI) of 0.0001. After viral absorption for 1 h, cells were washed with PBS and treated for 3 days at 33 °C with 3-fold serial dilutions of each compound in the presence of 2 μg/mL TPCK-treated trypsin (Sigma-Aldrich). Finally, they were incubated for 15 min at 37 °C with 30 μg/mL fluorescein diacetate. Cell viability was quantified using a Spectra Max M3 plate reader (Molecular Devices, Sunnyvale, CA) at an excitation wavelength of 485 nm and an emission wavelength of 538 nm. Both CC50 and EC50 values were determined by comparison with cell viability values from mock-infected and virus-infected cells, respectively, using GraphPad Prism 8.4.2 (GraphPad Software, La Jolla, CA, USA).
4.4. Immunoassay
For Western blot analysis, MDCK or A549 cells in 6-well plates (9 × 105 cells per well) were infected with the PR8 virus at an MOI of 0.01 for 1 h and then treated with increasing concentrations of compound 1 or OSV-C in the presence of TPCK-trypsin (2 µg/mL for MDCK cells at 33 °C or 0.1 µg/mL for A549 cells at 35 °C). On the next day, cell lysates were subjected to 10% SDS-PAGE (30 µg per well), followed by electrotransfer to a PVDF membrane. Viral proteins NP and HA, and the cellular β-actin protein (a loading control) were detected using specific antibodies as described previously [24].
For fluorescence microscopy, PR8 virus-infected MDCK cells (MOI, 3) seeded onto 4-well slides (8.5 × 104 cells per well) were treated with DMSO (delivery vehicle), compound 1, or RBV at 33 °C for 5 h. After fixing and permeabilization, viral NP was visualized using an anti-NP antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by an Alexa 488-conjugated goat anti-mouse IgG (Invitrogen) [10]. Cells were counterstained with a Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA, USA). Images were captured under a Zeiss LSM 700 confocal microscope (Carl Zeiss, Thornwood, NY, USA).
4.5. Plaque Titration
Cell culture supernatants harvested on day 1 post-viral infection in the presence or absence of compound 1 or OSV-C were centrifuged for 5 min at 3000 g and then serially diluted (10−1 to 10−6) in MEM. Fresh MDCK cells were seeded into 48-well plates (1 × 105 cells per well) and incubated with the diluted samples for 1 h at 33 °C. Next, the cells were washed with PBS and incubated at 33 °C in the serum-free overlay medium [MEM containing 0.6% carboxymethyl cellulose (CMC; Sigma-Aldrich)] supplemented with 2 µg/mL TPCK-trypsin. On day 3, viral plaques were counted by staining with crystal violet.
In a time-of-addition experiment, the PR8 virus (MOI, 0.01) was loaded onto MDCK cells cultured in 48-well plates (1.5 × 105 cells per well) for 1 h at 4 °C. After washing away unabsorbed virus, the cells were incubated at 33 °C for a further 7 h. In parallel with virus infection, 10 µM compound 1 or 1 µM EGCG (as a control for entry blocking) was added to infected cells during adsorption or at different times (0, 1, 2, 4, and 6 h) post-infection. At 7 h post-infection, all compounds were removed by washing twice with PBS prior to plaque titration as described above.
4.6. Viral Polymerase Assay
The influenza A viral polymerase activity assay was performed as described previously, with some modifications. Briefly, HeLa cells were seeded in 24-well plates (1 × 105 cells per well). On the next day, the cells were transfected with plasmids expressing viral polymerases and NP (pVP-PB2, -PB2, -PA, or NP; 200 ng of each), together with reporter plasmids pHH21-FLuc (200 ng per well) and phRL-CMV (10 ng per well), which transcribe the negative-sense RNA of the FLuc gene flanked by UTRs from the influenza viral NS segment from RNA polymerase I and the RLuc mRNA from RNA polymerase II, respectively [10]. At 6 h post-transfection, the cells were treated with compound 1 or 21 or RBV. After 1 day, the cell lysates were harvested and tested in a dual luciferase assay (Promega, Madison, WI, USA).
4.7. NA Activity Assay
An NA activity assay was performed by using the NA-XTD Influenza Neuraminidase assay kit (Applied Biosystems, Foster City, CA, USA) [25,26]. The influenza A virus PR8 (2.8 × 103 PFU) was mixed with the same volume of compounds at 37 °C for 20 min. After an addition of NA-XTD substrate, the mixture was incubated at room temperature for 30 min. The luminescent signal was maximized by the addition of an NA-XTD accelerator according to the manufacturer’s instructions and read using a microplate luminometer (Centro LB960 system; Berthold Technologies, Bad Wildbad, Germany). Half maximum inhibitory concentrations (IC50s) were determined using GraphPad Prism 8.4.2.
4.8. Statistical Analysis
Statistical analysis was performed using a two-tailed Student’s t-test. p values < 0.05 were considered significant.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ph14070650/s1, Scheme S1: Synthesis of phenylisoquinolones 1 and 17–19; Scheme S2: Synthesis of phenylisoquinolones 2, 3, 13–15 and 22; Scheme S3: Synthesis of phenylisoquinolone 16; Figure S1: 1H NMR (400 MHz, DMSO-d6) spectrum of 7; Figure S2: 1H NMR (400 MHz, DMSO-d6) spectrum of 8; Figure S3: 1H NMR (400 MHz, DMSO-d6) spectrum of 9; Figure S4: 1H NMR (400 MHz, DMSO-d6) spectrum of 10; Figure S5: 1H NMR (400 MHz, DMSO-d6) spectrum of 21; Figure S6. Docking pattern comparison of VX787 (A and B) and compound 21 (C and D) to PB2; Figure S7: Docking pattern comparison of T705 (A and B) and compound 21 (C and D) to PB1; Figure S8. Docking pattern comparison of Baloxavir (A and B) and compound 21 (C and D) to PA.
Author Contributions
Conceptualization, M.K. and W.-J.C.; methodology, Y.J. and J.H.; formal analysis, X.L. and H.S.; investigation, Y.J., J.H., X.L. and H.S.; writing—original draft preparation, M.K. and W.-J.C.; writing—review and editing, H.S. and J.H.; visualization, Y.J.; supervision, M.K. and W.-J.C.; project administration, M.K. and W.-J.C.; funding acquisition, M.K. and W.-J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIT) [NRF-2018M3A9H4089601 to M.K., NRF- 2018M3A9H4089602 to W.-J.C.] and by an intramural fund from KRICT (Grant Number SI2132-20 to M.K.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Acknowledgments
We would like to thank the Korea Chemical Bank for providing a chemical library and the hit compound 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. 4), D49–D53. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Walker, A.P.; Carrique, L.; Keown, J.R.; Serna Martin, I.; Karia, D.; Sharps, J.; Hengrung, N.; Pardon, E.; Steyaert, J.; et al. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 2019, 573, 287–290. [Google Scholar] [CrossRef] [PubMed]
- US Flu VE Data for 2019–2020. Available online: https://www.cdc.gov/flu/vaccines-work/2019-2020.html#Influenza-Positive (accessed on 29 April 2021).
- Duwe, S. Influenza viruses—Antiviral therapy and resistance. GMS Infect. Dis. 2017, 5. [Google Scholar] [CrossRef]
- Dharan, N.J.; Gubareva, L.V.; Meyer, J.J.; Okomo-Adhiambo, M.; McClinton, R.C.; Marshall, S.A.; St George, K.; Epperson, S.; Brammer, L.; Klimov, A.I.; et al. Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States. JAMA 2009, 301, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.C.; Pascua, P.N.Q.; Fabrizio, T.P.; Marathe, B.M.; Seiler, P.; Barman, S.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. Influenza A and B viruses with reduced baloxavir susceptibility display attenuated in vitro fitness but retain ferret transmissibility. Proc. Natl. Acad. Sci. USA 2020, 117, 8593–8601. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.Y.; Lee, H.W.; Shin, J.S.; Kim, P.; Jung, Y.S.; Jeong, H.S.; Hyun, J.K.; Lee, C.K. Inhibition of influenza virus internalization by (-)-epigallocatechin-3-gallate. Antivir. Res. 2013, 100, 460–472. [Google Scholar] [CrossRef]
- Cho, W.J.; Yoo, S.J.; Park, M.J.; Chung, B.H.; Lee, C.O. Synthesis and antitumor activity of 3-arylisoquinoline derivatives. Arch. Pharm. Res. 1997, 20, 264–268. [Google Scholar] [CrossRef]
- Le, T.N.; Gang, S.G.; Cho, W.J. A versatile total synthesis of benzo[c]phenanthridine and protoberberine alkaloids using lithiated toluamide-benzonitrile cycloaddition. J. Org. Chem. 2004, 69, 2768–2772. [Google Scholar] [CrossRef]
- Van, H.T.; Le, Q.M.; Lee, K.Y.; Lee, E.S.; Kwon, Y.; Kim, T.S.; Le, T.N.; Lee, S.H.; Cho, W.J. Convenient synthesis of indeno [1,2-c]isoquinolines as constrained forms of 3-arylisoquinolines and docking study of a topoisomerase I inhibitor into DNA-topoisomerase I complex. Bioorg. Med. Chem. Lett. 2007, 17, 5763–5767. [Google Scholar] [CrossRef]
- Van, H.T.; Cho, W.J. Structural modification of 3-arylisoquinolines to isoindolo[2,1-b]isoquinolinones for the development of novel topoisomerase 1 inhibitors with molecular docking study. Bioorg. Med. Chem. Lett. 2009, 19, 2551–2554. [Google Scholar] [CrossRef]
- Jin, Y.; Khadka, D.B.; Yang, S.H.; Zhao, C.; Cho, W.-J. Synthesis of novel 5-oxaprotoberberines as bioisosteres of protoberberines. Tetrahedron Lett. 2014, 55, 1366–1369. [Google Scholar] [CrossRef]
- Khadka, D.B.; Woo, H.; Yang, S.H.; Zhao, C.; Jin, Y.; Le, T.N.; Kwon, Y.; Cho, W.J. Modification of 3-arylisoquinolines into 3,4-diarylisoquinolines and assessment of their cytotoxicity and topoisomerase inhibition. Eur. J. Med. Chem. 2015, 92, 583–607. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.J.; Kim, E.K.; Park, M.J.; Choi, S.U.; Lee, C.O.; Cheon, S.H.; Choi, B.G.; Chung, B.H. Synthesis and comparative molecular field analysis (CoMFA) of antitumor 3-arylisoquinoline derivatives. Bioorg. Med. Chem. 1998, 6, 2449–2458. [Google Scholar] [CrossRef]
- Yang, S.H.; Van, H.T.; Le, T.N.; Khadka, D.B.; Cho, S.H.; Lee, K.T.; Chung, H.J.; Lee, S.K.; Ahn, C.H.; Lee, Y.B.; et al. Synthesis, in vitro and in vivo evaluation of 3-arylisoquinolinamines as potent antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 5277–5281. [Google Scholar] [CrossRef] [PubMed]
- Khadka, D.B.; Yang, S.H.; Cho, S.H.; Zhao, C.; Cho, W.-J. Synthesis of 12-oxobenzo[c]phenanthridinones and 4-substituted 3-arylisoquinolones via Vilsmeier–Haack reaction. Tetrahedron 2012, 68, 250–261. [Google Scholar] [CrossRef]
- Goldhill, D.H.; Te Velthuis, A.J.W.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.P.; Ledeboer, M.W.; Davies, I.; Byrn, R.A.; Jones, S.M.; Perola, E.; Tsai, A.; Jacobs, M.; Nti-Addae, K.; Bandarage, U.K.; et al. Discovery of a novel, first-in-class, orally bioavailable azaindole inhibitor (VX-787) of influenza PB2. J. Med. Chem. 2014, 57, 6668–6678. [Google Scholar] [CrossRef]
- Hayden, F.G.; Shindo, N. Influenza virus polymerase inhibitors in clinical development. Curr. Opin. Infect. Dis. 2019, 32, 176–186. [Google Scholar] [CrossRef]
- Kim, M.; Yim, J.H.; Kim, S.Y.; Kim, H.S.; Lee, W.G.; Kim, S.J.; Kang, P.S.; Lee, C.K. In vitro inhibition of influenza A virus infection by marine microalga-derived sulfated polysaccharide p-KG03. Antivir. Res. 2012, 93, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Shin, J.S.; Yoon, Y.S.; Go, Y.Y.; Lee, H.W.; Kwon, O.S.; Park, S.; Park, M.S.; Kim, M. Salinomycin Inhibits Influenza Virus Infection by Disrupting Endosomal Acidification and Viral Matrix Protein 2 Function. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.J.; Achary, R.; Lee, H.W.; Lee, H.J.; Lee, C.K.; Han, S.B.; Jung, Y.S.; Kang, N.S.; Kim, P.; Kim, M. Synthesis and anti-influenza virus activity of 4-oxo- or thioxo-4,5-dihydrofuro[3,4-c]pyridin-3(1H)-ones. Antivir. Res. 2014, 107, 66–75. [Google Scholar] [CrossRef]
- Murtaugh, W.; Mahaman, L.; Healey, B.; Peters, H.; Anderson, B.; Tran, M.; Ziese, M.; Carlos, M.P. Evaluation of three influenza neuraminidase inhibition assays for use in a public health laboratory setting during the 2011–2012 influenza season. Public Health Rep. 2013, 128 (Suppl. 2), 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).