
Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase

 ,
,  ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results
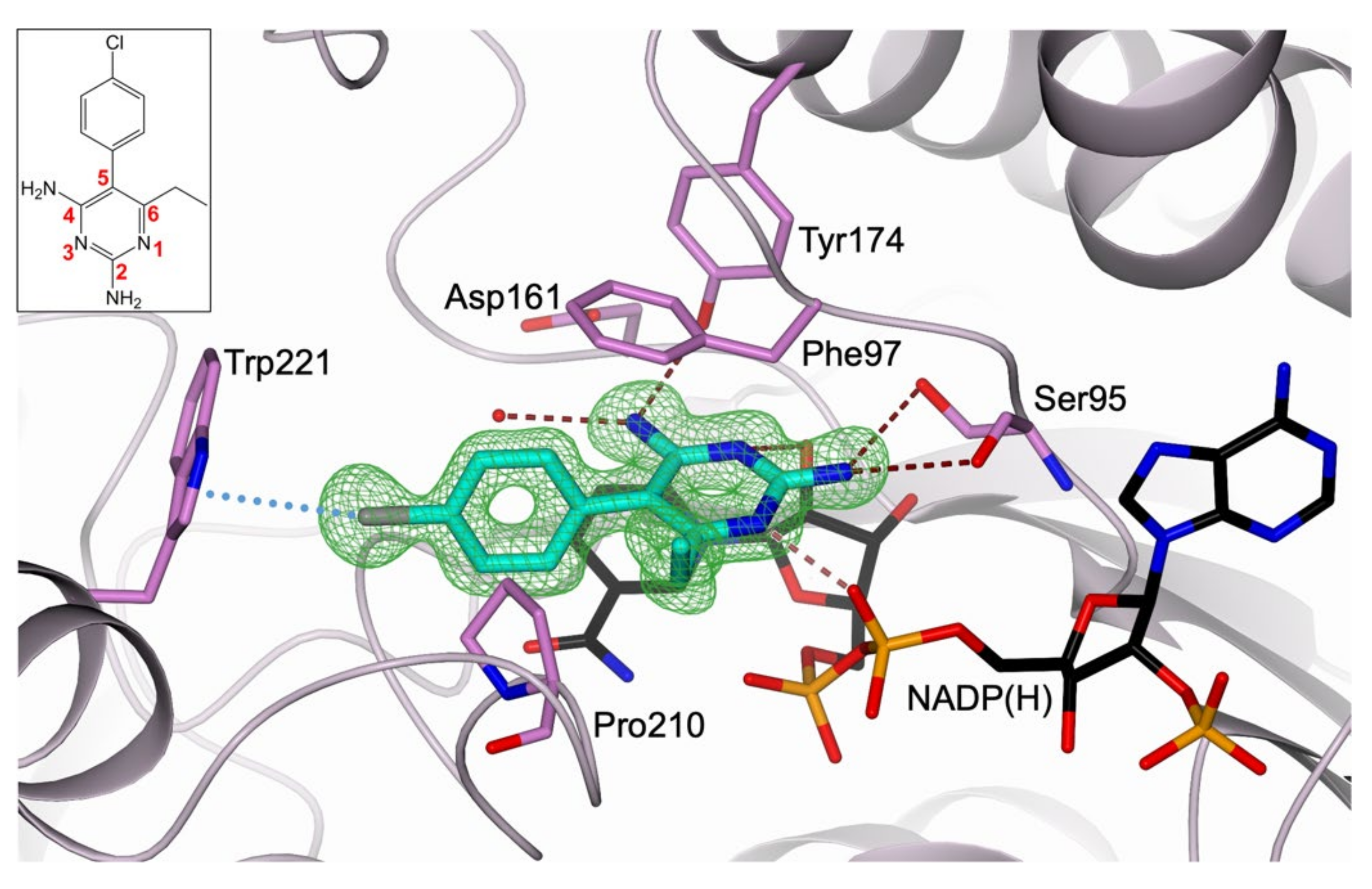
2.1. Structural Characterization of the TbPTR1:NADP(H):PYR Complex
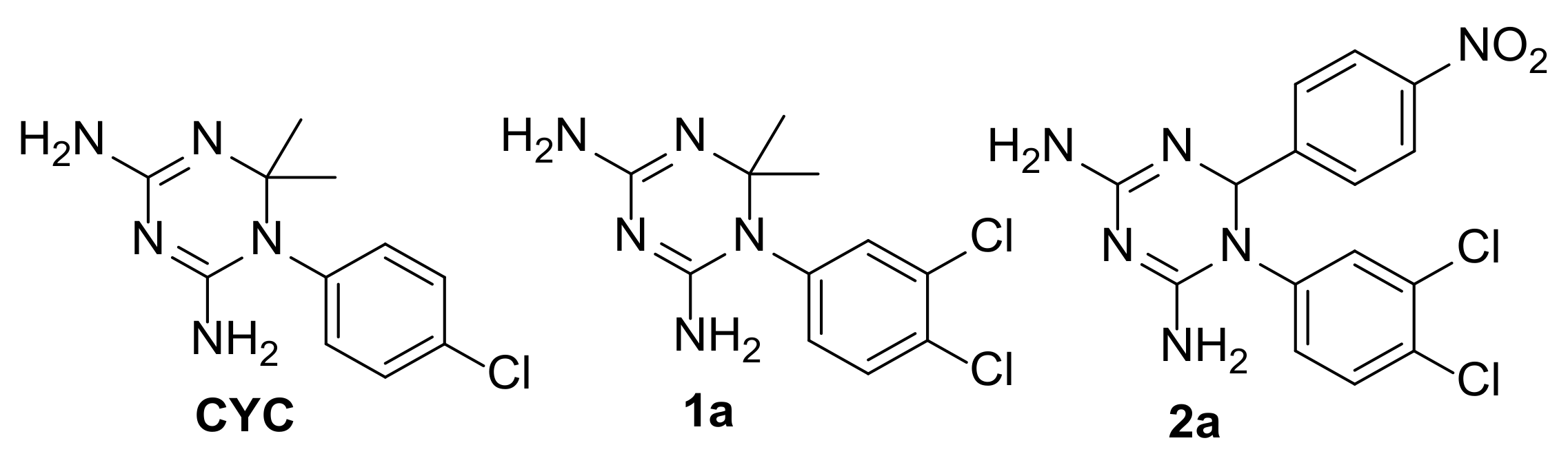
2.2. Selection of the Small Library of CYC Derivatives
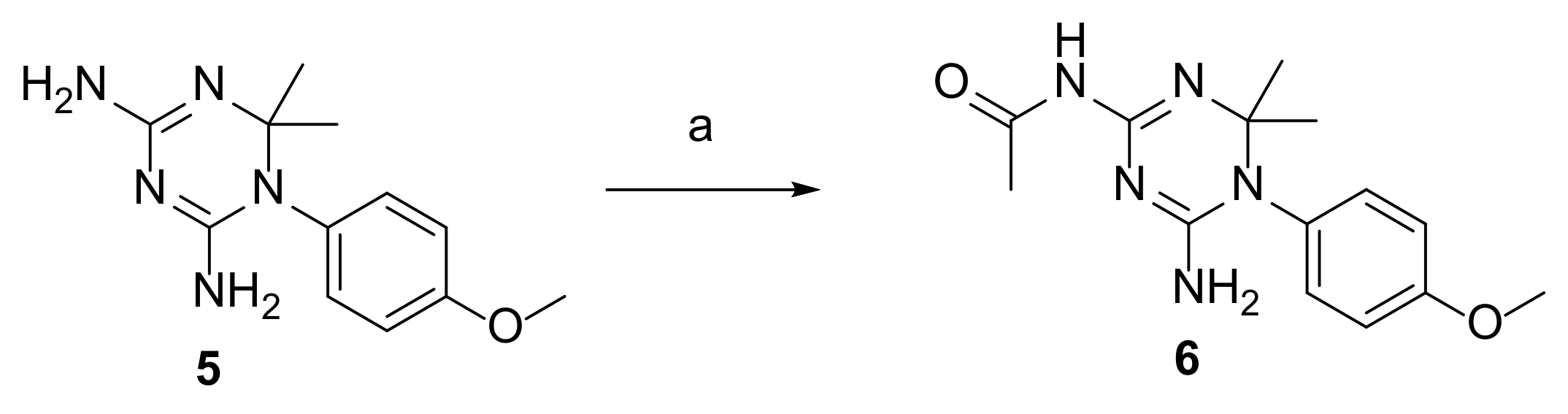
2.3. Synthesis of CYC Derivatives
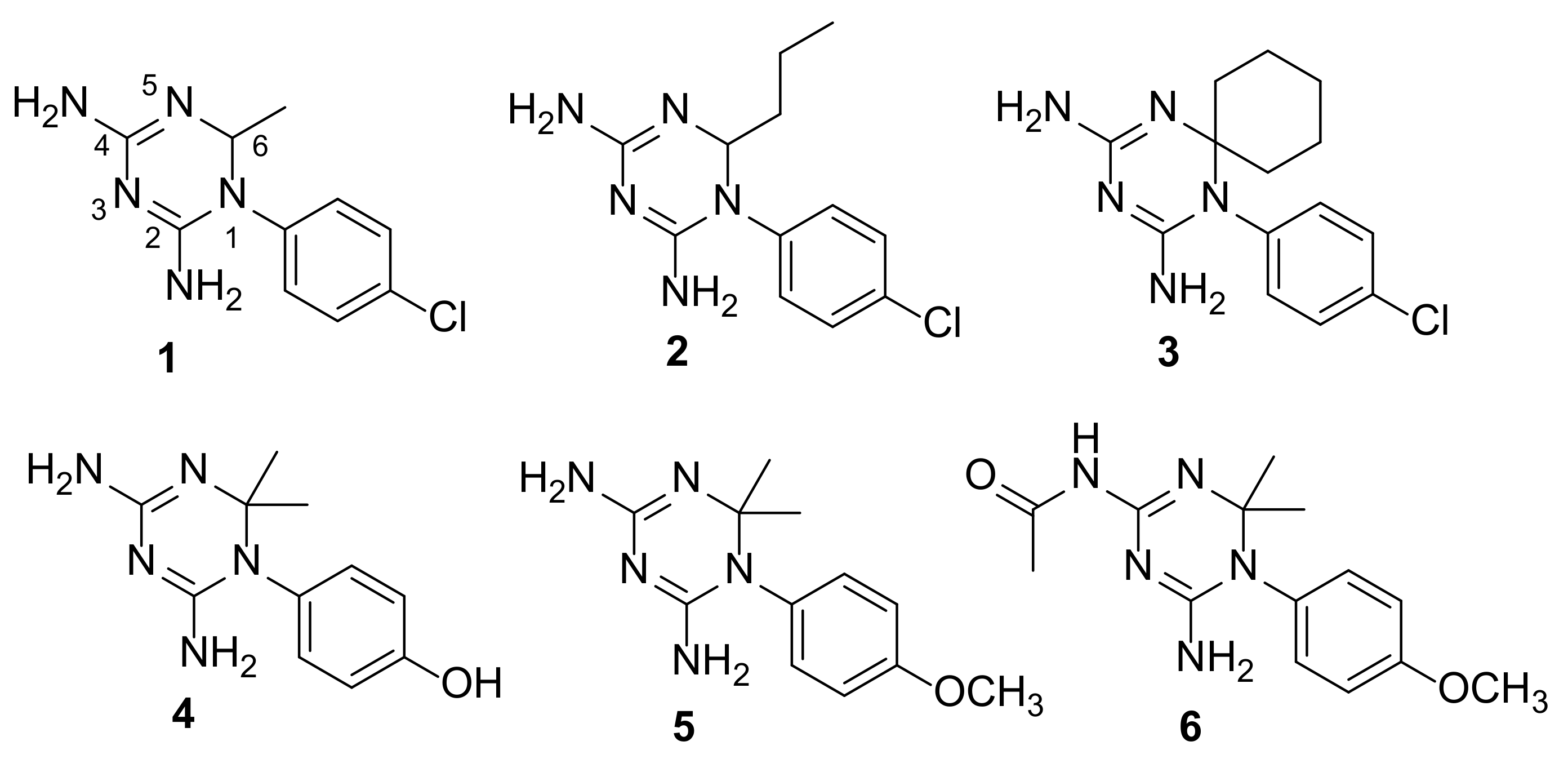
2.4. Structure–Activity Relationship Studies of CYC Analogues against Parasitic and Human Enzymes
2.5. Compounds’ Selectivity between the PTR1 and DHFRs Enzymes
3. Discussion
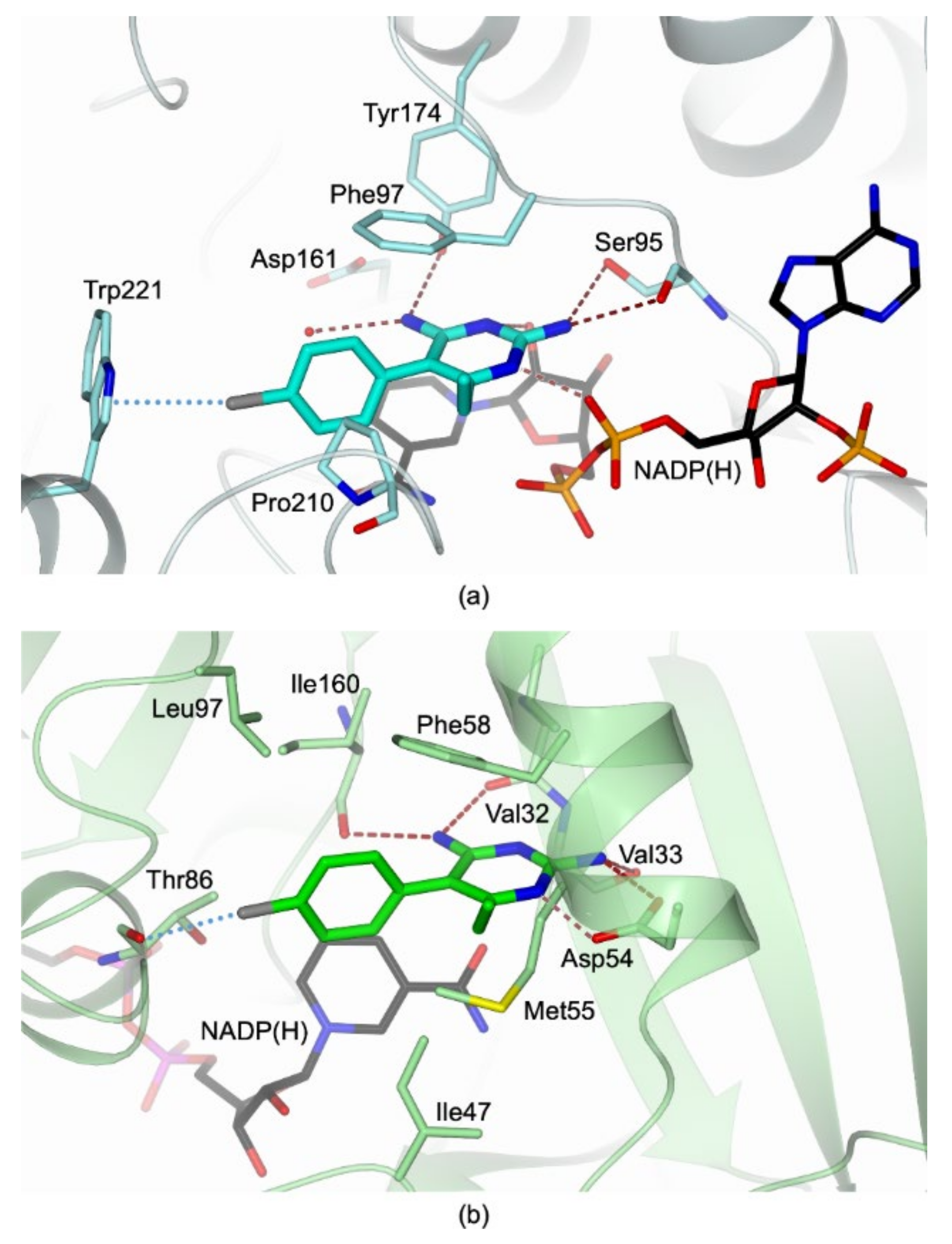
3.1. Comparative Analysis of CYC versus PYR TbPTR1 Structure
3.2. Structural Basis for Developing Dual Inhibitors of TbPTR1 and TbDHFR
4. Materials and Methods
4.1. Synthesis of CYC Analogues
4.2. TbPTR1 and TbDHFR-TS Expression, Purification, Kinetics, and Inhibition Assays
4.3. TbPTR1 and TbDHFR-TS Crystallization
4.4. Data Collection, Structure Solution, and Refinement
4.5. Stability of Compound 6 in Solution at Different pH
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castillo, E.; Dea-Ayuela, M.A.; Bolás-Fernández, F.; Rangel, M.; González-Rosende, M.E. The Kinetoplastid Chemotherapy Revisited: Current Drugs, Recent Advances and Future Perspectives. Curr. Med. Chem. 2012, 17, 4027–4051. [Google Scholar] [CrossRef] [PubMed]
- Mackey, T.K.; Liang, B.A.; Cuomo, R.; Hafen, R.; Brouwer, K.C.; Lee, D.E. Emerging and Reemerging Neglected Tropical Diseases: A Review of Key Characteristics, Risk Factors, and the Policy and Innovation Environment. Clin. Microbiol. Rev. 2014, 27, 949–979. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gürtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related Protozoan Pathogens, Different Diseases. J. Clin. Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Executive summary. Ending the neglect to attain the Sustainable Development Goals: A road map for neglected tropical diseases 2021–2030. Available online: http://www.who.int/publications/i/item/WHO-UCN-NTD-2020.01 (accessed on 28 January 2020).
- Barrett, M.P.; Croft, S.L. Management of Trypanosomiasis and Leishmaniasis. Br. Med Bull. 2012, 104, 175–196. [Google Scholar] [CrossRef]
- Alirol, E.; Schrumpf, D.; Amici Heradi, J.; Riedel, A.; de Patoul, C.; Quere, M.; Chappuis, F. Nifurtimox-Eflornithine Combination Therapy for Second-Stage Gambiense Human African Trypanosomiasis: Médecins Sans Frontières Experience in the Democratic Republic of the Congo. Clin. Infect. Dis. 2012, 56, 195–203. [Google Scholar] [CrossRef]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazué, G.; Bray, M.A.; Pécoul, B. Fexinidazole–a New Oral Nitroimidazole Drug Candidate Entering Clinical Development for the Treatment of Sleeping Sickness. PLoS Negl. Trop. Dis. 2010, 4, e923. [Google Scholar] [CrossRef]
- Maxmen, A. Sleeping Sickness Can Now Be Cured with Pills. Nature 2017, 550, 441. [Google Scholar] [CrossRef][Green Version]
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Capela, R.; Moreira, R.; Lopes, F. An Overview of Drug Resistance in Protozoal Diseases. Int. J. Mol. Sci. 2019, 20, 5748. [Google Scholar] [CrossRef]
- Barrett, M.P.; Kyle, D.E.; Sibley, L.D.; Radke, J.B.; Tarleton, R.L. Protozoan Persister-like Cells and Drug Treatment Failure. Nat. Rev. Microbiol. 2019, 17, 607–620. [Google Scholar] [CrossRef]
- Dawidowski, M.; Emmanouilidis, L.; Kalel, V.C.; Tripsianes, K.; Schorpp, K.; Hadian, K.; Kaiser, M.; Mäser, P.; Kolonko, M.; Tanghe, S.; et al. Inhibitors of PEX14 Disrupt Protein Import into Glycosomes and Kill Trypanosoma Parasites. Science 2017, 355, 1416–1420. [Google Scholar] [CrossRef]
- Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate Reductase Inhibitors as Antibacterial Agents. Biochem. Pharmacol. 2006, 71, 941–948. [Google Scholar] [CrossRef]
- Yuthavong, Y.; Yuvaniyama, J.; Chitnumsub, P.; Vanichtanankul, J.; Chusacultanachai, S.; Tarnchompoo, B.; Vilaivan, T.; Kamchonwongpaisan, S. Malarial (Plasmodium Falciparum) Dihydrofolate Reductase-Thymidylate Synthase: Structural Basis for Antifolate Resistance and Development of Effective Inhibitors. Parasitology 2005, 130, 249–259. [Google Scholar] [CrossRef]
- Borsari, C.; Luciani, R.; Pozzi, C.; Poehner, I.; Henrich, S.; Trande, M.; Cordeiro-da-Silva, A.; Santarem, N.; Baptista, C.; Tait, A.; et al. Profiling of Flavonol Derivatives for the Development of Antitrypanosomatidic Drugs. J. Med. Chem. 2016, 59, 7598–7616. [Google Scholar] [CrossRef]
- Di Pisa, F.; Landi, G.; Dello Iacono, L.; Pozzi, C.; Borsari, C.; Ferrari, S.; Santucci, M.; Santarem, N.; Cordeiro-da-Silva, A.; Moraes, C.B.; et al. Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity. Molecules 2017, 22, 426. [Google Scholar] [CrossRef]
- Linciano, P.; Dawson, A.; Pöhner, I.; Costa, D.M.; Sá, M.S.; Cordeiro-da-Silva, A.; Luciani, R.; Gul, S.; Witt, G.; Ellinger, B.; et al. Exploiting the 2-Amino-1,3,4-Thiadiazole Scaffold To Inhibit Trypanosoma Brucei Pteridine Reductase in Support of Early-Stage Drug Discovery. ACS Omega 2017, 2, 5666–5683. [Google Scholar] [CrossRef]
- Tulloch, L.B.; Martini, V.P.; Iulek, J.; Huggan, J.K.; Lee, J.H.; Gibson, C.L.; Smith, T.K.; Suckling, C.J.; Hunter, W.N. Structure-Based Design of Pteridine Reductase Inhibitors Targeting African Sleeping Sickness and the Leishmaniases. J. Med. Chem. 2010, 53, 221–229. [Google Scholar] [CrossRef]
- Linciano, P.; Pozzi, C.; Iacono, L.D.; di Pisa, F.; Landi, G.; Bonucci, A.; Gul, S.; Kuzikov, M.; Ellinger, B.; Witt, G.; et al. Enhancement of Benzothiazoles as Pteridine Reductase-1 Inhibitors for the Treatment of Trypanosomatidic Infections. J. Med. Chem. 2019, 62, 3989–4012. [Google Scholar] [CrossRef]
- Landi, G.; Linciano, P.; Borsari, C.; Bertolacini, C.P.; Moraes, C.B.; Cordeiro-da-Silva, A.; Gul, S.; Witt, G.; Kuzikov, M.; Costi, M.P.; et al. Structural Insights into the Development of Cycloguanil Derivatives as Trypanosoma Brucei Pteridine-Reductase-1 Inhibitors. ACS Infect. Dis. 2019, 5, 1105–1114. [Google Scholar] [CrossRef]
- Landi, G.; Linciano, P.; Tassone, G.; Costi, M.P.; Mangani, S.; Pozzi, C. High-Resolution Crystal Structure of Trypanosoma Brucei Pteridine Reductase 1 in Complex with an Innovative Tricyclic-Based Inhibitor. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 558–564. [Google Scholar] [CrossRef]
- Dawson, A.; Gibellini, F.; Sienkiewicz, N.; Tulloch, L.B.; Fyfe, P.K.; McLuskey, K.; Fairlamb, A.H.; Hunter, W.N. Structure and Reactivity of Trypanosoma Brucei Pteridine Reductase: Inhibition by the Archetypal Antifolate Methotrexate. Mol. Microbiol. 2006, 61, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; Tassone, G.; Mangani, S. Chapter Five-X-ray Crystallography Contributions to Drug Discovery Against Parasite. In Annual Reports in Medicinal Chemistry; Botta, M., Ed.; Neglected Diseases: Extensive Space for Modern Drug Discovery; Academic Press: Cambridge, MA, USA, 2018; Volume 51, pp. 175–230. [Google Scholar]
- Ouellette, M.; Drummelsmith, J.; El Fadili, A.; Kündig, C.; Richard, D.; Roy, G. Pterin Transport and Metabolism in Leishmania and Related Trypanosomatid Parasites. Int. J. Parasitol. 2002, 32, 385–398. [Google Scholar] [CrossRef]
- Barrett, M.P.; Gilbert, I.H. Targeting of Toxic Compounds to the Trypanosome’s Interior. Adv. Parasitol. 2006, 63, 125–183. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.R.; Nare, B.; Freedman, D.; Hardy, L.; Beverley, S.M. PTR1: A Reductase Mediating Salvage of Oxidized Pteridines and Methotrexate Resistance in the Protozoan Parasite Leishmania Major. Proc. Natl. Acad. Sci. USA 1994, 91, 11442–11446. [Google Scholar] [CrossRef] [PubMed]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The Genome of the African Trypanosome Trypanosoma Brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef]
- Zuccotto, F.; Martin, A.C.; Laskowski, R.A.; Thornton, J.M.; Gilbert, I.H. Dihydrofolate Reductase: A Potential Drug Target in Trypanosomes and Leishmania. J. Comput. Mol. Des. 1998, 12, 241–257. [Google Scholar] [CrossRef]
- Sienkiewicz, N.; Jarosławski, S.; Wyllie, S.; Fairlamb, A.H. Chemical and Genetic Validation of Dihydrofolate Reductase-Thymidylate Synthase as a Drug Target in African Trypanosomes. Mol. Microbiol. 2008, 69, 520–533. [Google Scholar] [CrossRef]
- Nare, B.; Hardy, L.W.; Beverley, S.M. The Roles of Pteridine Reductase 1 and Dihydrofolate Reductase-Thymidylate Synthase in Pteridine Metabolism in the Protozoan Parasite Leishmania Major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef]
- Nare, B.; Luba, J.; Hardy, L.W.; Beverley, S. New Approaches to Leishmania Chemotherapy: Pteridine Reductase 1 (PTR1) as a Target and Modulator of Antifolate Sensitivity. Parasitology 1997, 114, 101–110. [Google Scholar] [CrossRef]
- Hardy, L.W.; Matthews, W.; Nare, B.; Beverley, S.M. Biochemical and Genetic Tests for Inhibitors of Leishmania Pteridine Pathways. Exp. Parasitol. 1997, 87, 158–170. [Google Scholar] [CrossRef]
- Papadopoulou, B.; Roy, G.; Ouellette, M. A Novel Antifolate Resistance Gene on the Amplified H Circle of Leishmania. EMBO J. 1992, 11, 3601–3608. [Google Scholar] [CrossRef]
- Robello, C.; Navarro, P.; Castanys, S.; Gamarro, F. A Pteridine Reductase Gene Ptr1 Contiguous to a P-Glycoprotein Confers Resistance to Antifolates in Trypanosoma Cruzi. Mol. Biochem. Parasitol. 1997, 90, 525–535. [Google Scholar] [CrossRef]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine Reductase Mechanism Correlates Pterin Metabolism with Drug Resistance in Trypanosomatid Parasites. Nat. Genet. 2001, 8, 521–525. [Google Scholar] [CrossRef]
- Ong, H.B.; Sienkiewicz, N.; Wyllie, S.; Fairlamb, A.H. Dissecting the Metabolic Roles of Pteridine Reductase 1 in Trypanosoma Brucei and Leishmania Major. J. Biol. Chem. 2011, 286, 10429–10438. [Google Scholar] [CrossRef]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosomal Dihydrofolate Reductase Reveals Natural Antifolate Resistance. ACS Chem. Biol. 2011, 6, 905–911. [Google Scholar] [CrossRef]
- Tonelli, M.; Naesens, L.; Gazzarrini, S.; Santucci, M.; Cichero, E.; Tasso, B.; Moroni, A.; Costi, M.P.; Loddo, R. Host Dihydrofolate Reductase (DHFR)-Directed Cycloguanil Analogues Endowed with Activity against Influenza Virus and Respiratory Syncytial Virus. Eur. J. Med. Chem. 2017, 135, 467–478. [Google Scholar] [CrossRef]
- Panecka-Hofman, J.; Pöhner, I.; Spyrakis, F.; Zeppelin, T.; Di Pisa, F.; Dello Iacono, L.; Bonucci, A.; Quotadamo, A.; Venturelli, A.; Mangani, S.; et al. Comparative Mapping of On-Targets and off-Targets for the Discovery of Anti-Trypanosomatid Folate Pathway Inhibitors. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3215–3230. [Google Scholar] [CrossRef]
- Chan, D.C.M.; Anderson, A.C. Towards Species-Specific Antifolates. Curr. Med. Chem. 2006, 13, 377–398. [Google Scholar] [CrossRef]
- Gangjee, A.; Kurup, S.; Namjoshi, O. Dihydrofolate Reductase as a Target for Chemotherapy in Parasites. Available online: http://www.eurekaselect.com/58895/article (accessed on 9 April 2018).
- Sharma, V.K.; Abbat, S.; Bharatam, P.V. Pharmacoinformatic Study on the Selective Inhibition of the Protozoan Dihydrofolate Reductase Enzymes. Mol. Inform. 2017, 36, 1600156. [Google Scholar] [CrossRef]
- Modest, E.J.; Levine, P. Chemical and Biological Studies on 1,2-Dihydro-s-Triazines. III. Two-Component Synthesis. J. Org. Chem. 1956, 21, 14–20. [Google Scholar] [CrossRef]
- Modest, E.J. Chemical and Biological Studies on 1,2-Dihydro-s-Triazines. II. Three-Component Synthesis. J. Org. Chem. 1956, 21, 1–13. [Google Scholar] [CrossRef]
- Hansch, C.; Dietrich, S.W.; Fukunaga, J.Y. Inhibition of Bovine and Rat Liver Dihydrofolate Reductase by 4,6-Diamino-1,2-Dihydro-2,2-Dimethyl-1-(4-Substituted-Phenyl)-s-Triazines. J. Med. Chem. 1981, 24, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Bami, H. Studies in dihydrotriazines: 1-aryl-2,4-diamino-1,6-dihydro-6,6-dialkyl-1,3,5-triazines. J. Sci. Ind. Res. 1955, 14(C), 231–236. [Google Scholar]
- Šícho, M.; Stork, C.; Mazzolari, A.; de Bruyn Kops, C.; Pedretti, A.; Testa, B.; Vistoli, G.; Svozil, D.; Kirchmair, J. FAME 3: Predicting the Sites of Metabolism in Synthetic Compounds and Natural Products for Phase 1 and Phase 2 Metabolic Enzymes. J. Chem. Inf. Model. 2019, 59, 3400–3412. [Google Scholar] [CrossRef]
- Stork, C.; Embruch, G.; Šícho, M.; de Bruyn Kops, C.; Chen, Y.; Svozil, D.; Kirchmair, J. NERDD: A Web Portal Providing Access to in Silico Tools for Drug Discovery. Bioinformatics 2020, 36, 1291–1292. [Google Scholar] [CrossRef]
- Sramek, M.; Neradil, J.; Veselska, R. Much More than You Expected: The Non-DHFR-Mediated Effects of Methotrexate. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 499–503. [Google Scholar] [CrossRef]
- GraphPad Prism 9.1.0 Version, Free Demo Licence. Available online: https://www.graphpad.com/support/faq/prism-910-release-notes/ (accessed on 10 April 2021).
- OriginLab Corporation. Origin(Pro), Version 2021; OriginLab Corporation: Northampton, MA, USA, 2021. [Google Scholar]
- Benvenuti, M.; Mangani, S. Crystallization of Soluble Proteins in Vapor Diffusion for X-Ray Crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P.R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular Replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 22–25. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated Macromolecular Model Building for X-Ray Crystallography Using ARP/WARP Version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Potterton, L.; McNicholas, S.; Krissinel, E.; Gruber, J.; Cowtan, K.; Emsley, P.; Murshudov, G.N.; Cohen, S.; Perrakis, A.; Noble, M. Developments in the CCP4 Molecular-Graphics Project. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2288–2294. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | TbPTR1 (IC50 µM) | TbDHFR-TS (IC50 µM) | hDHFR (IC50 µM) | Selectivity IC50 TbDHFR-TS /IC50 TbPTR1 | Selectivity IC50 hDHFR /IC50 TbDHFR-TS |
|---|---|---|---|---|---|
| PYR | 0.090 | Ki = 0.024 [37] | Ki = 0.47 [38] | c.a. 0.27 a | 19.6 |
| CYC | 31.6 | Ki = 0.256 [37] | Ki = 0.41 [38] | c.a. 0.002 a | 1.6 |
| MTX | 0.793 | 0.040 | 0.014 | 0.05 | 0.035 |
| 1 (f) | 6.02 (±1.10) | 103 (±14) | 337 (±53) | 16.9 | 3.27 |
| 2 (a) | 27.5 (±4.54) | 7.26 (±2.35) | 66.7 (±18.5) | 0.26 | 9.19 |
| 3 (b) | 1.22 (±0.24) | 17.6 (±4.69) | 65.6 (±9.3) | 14.4 | 3.73 |
| 4 (e) | 4.10 (±0.75) | 89.0 (±9.9) | 284 (±77.4) | 21.7 | 3.19 |
| 5 (c) | 0.67 (±0.18) | 33.2 (±9.64) | 413 (±102) | 49.6 | 12.4 |
| 6 (d) | 0.83 (±0.07) | 18.9 (±5.17) | 62.5 (±9.1) | 22.8 | 3.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tassone, G.; Landi, G.; Linciano, P.; Francesconi, V.; Tonelli, M.; Tagliazucchi, L.; Costi, M.P.; Mangani, S.; Pozzi, C. Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase. Pharmaceuticals 2021, 14, 636. https://doi.org/10.3390/ph14070636
Tassone G, Landi G, Linciano P, Francesconi V, Tonelli M, Tagliazucchi L, Costi MP, Mangani S, Pozzi C. Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase. Pharmaceuticals. 2021; 14(7):636. https://doi.org/10.3390/ph14070636
Chicago/Turabian StyleTassone, Giusy, Giacomo Landi, Pasquale Linciano, Valeria Francesconi, Michele Tonelli, Lorenzo Tagliazucchi, Maria Paola Costi, Stefano Mangani, and Cecilia Pozzi. 2021. "Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase" Pharmaceuticals 14, no. 7: 636. https://doi.org/10.3390/ph14070636
APA StyleTassone, G., Landi, G., Linciano, P., Francesconi, V., Tonelli, M., Tagliazucchi, L., Costi, M. P., Mangani, S., & Pozzi, C. (2021). Evidence of Pyrimethamine and Cycloguanil Analogues as Dual Inhibitors of Trypanosoma brucei Pteridine Reductase and Dihydrofolate Reductase. Pharmaceuticals, 14(7), 636. https://doi.org/10.3390/ph14070636