
Metal-Based G-Quadruplex Binders for Cancer Theranostics




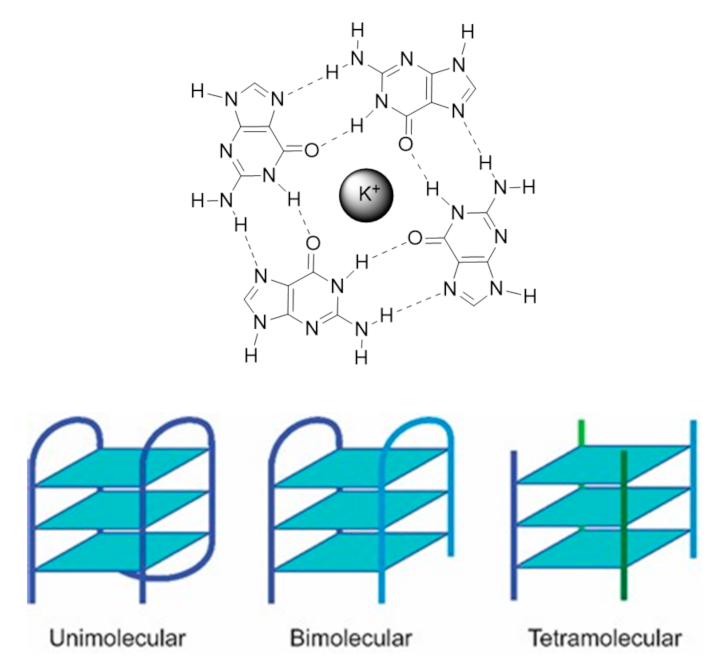{kind=link}
{kind=link}
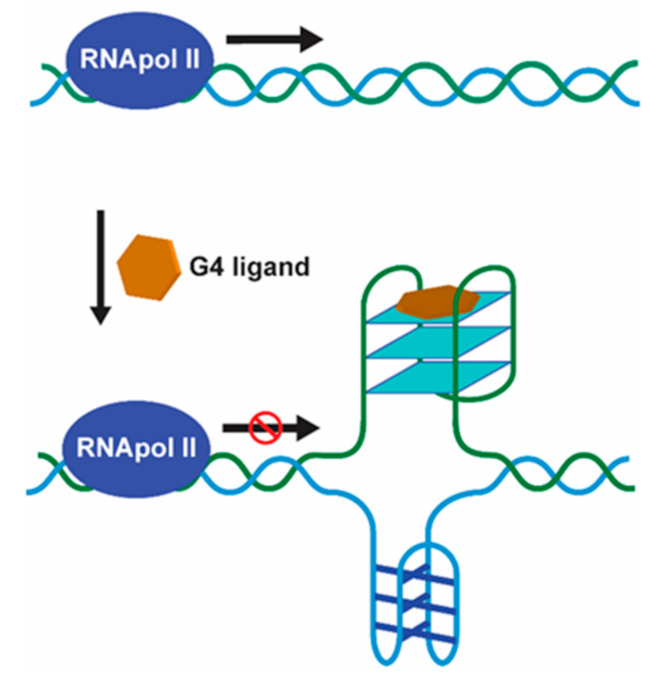{kind=link}
{kind=link}
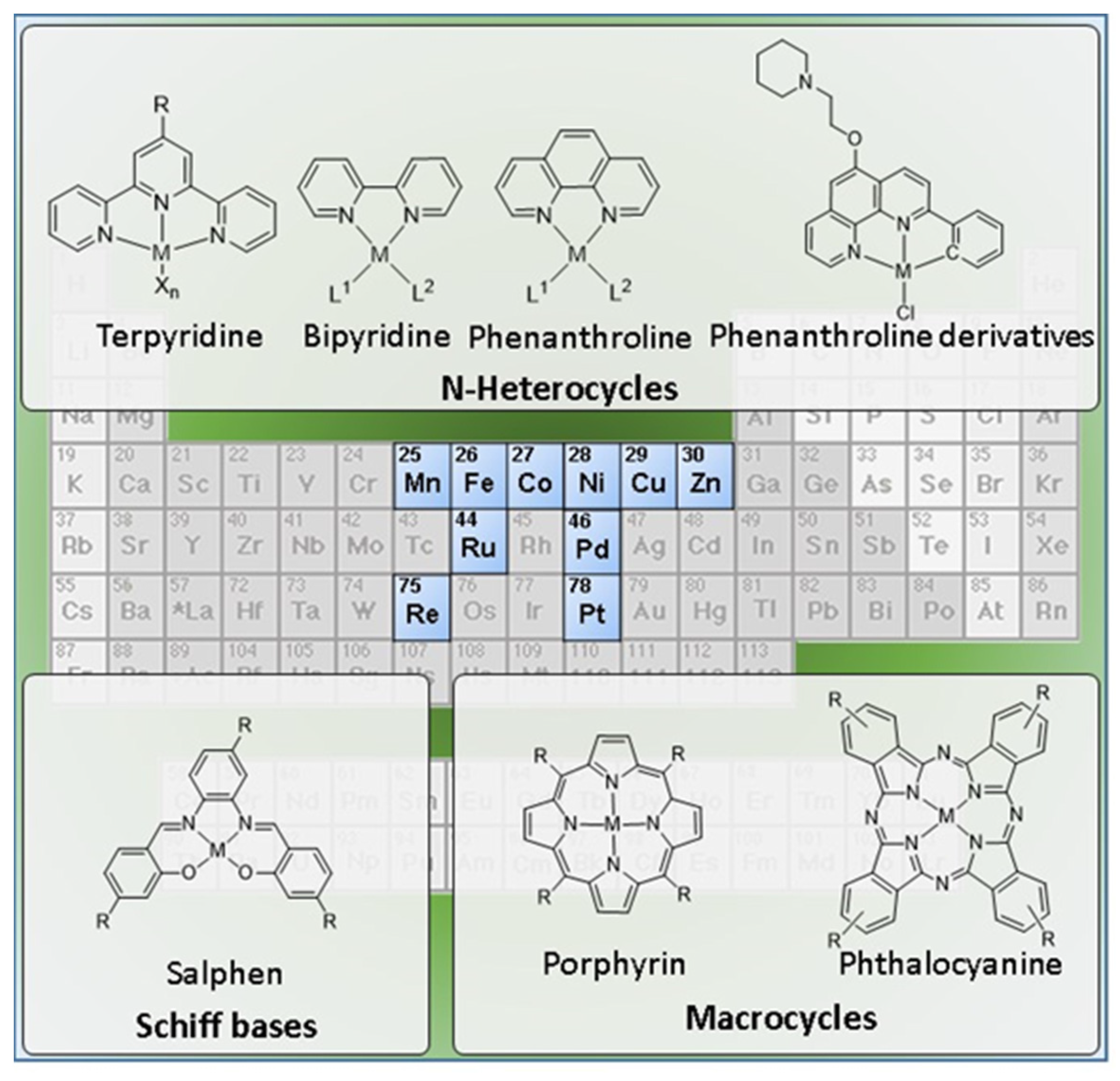{kind=link}
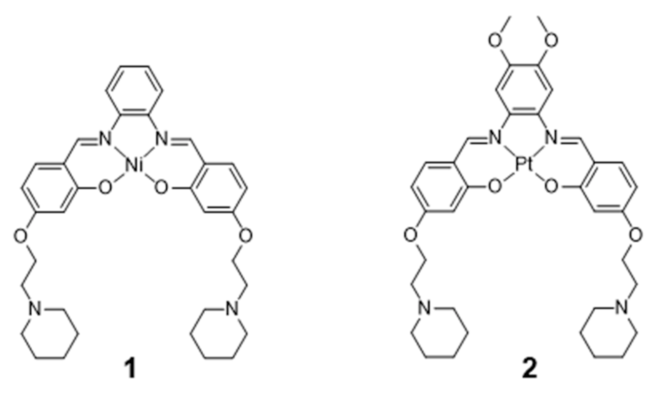{kind=link}
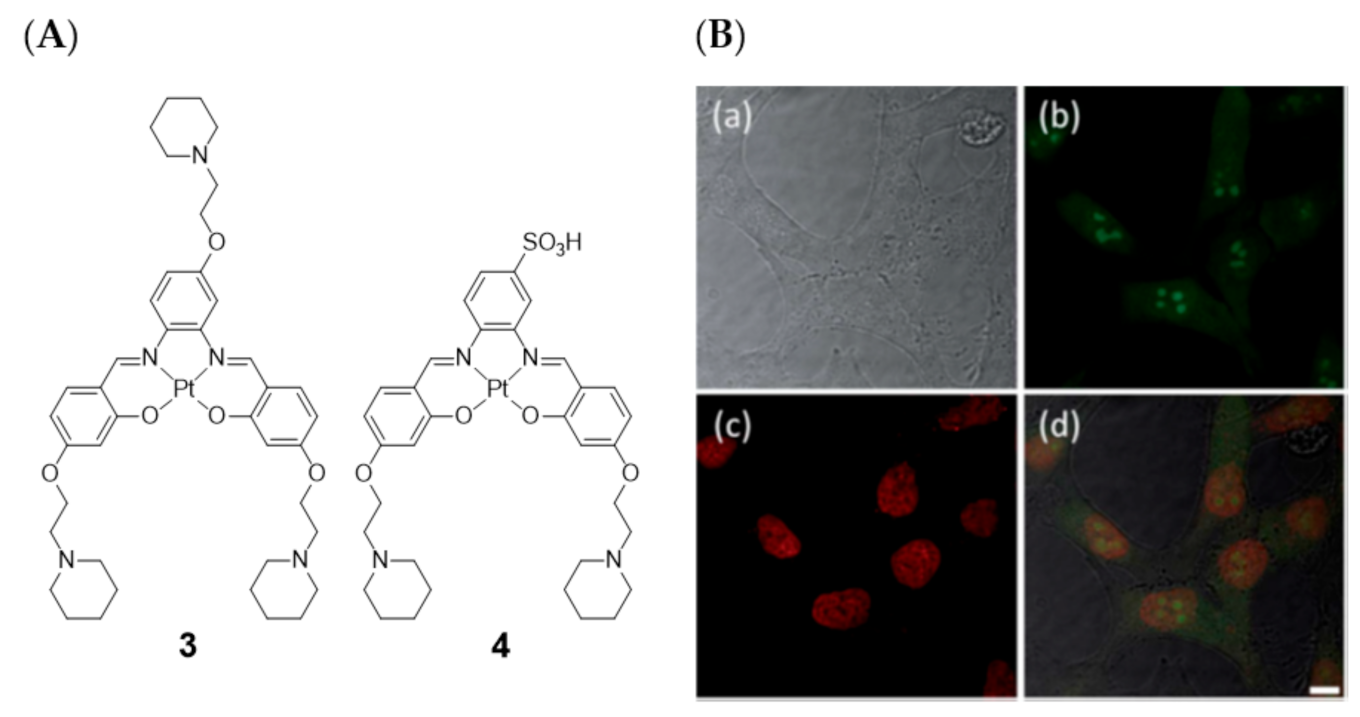{kind=link}
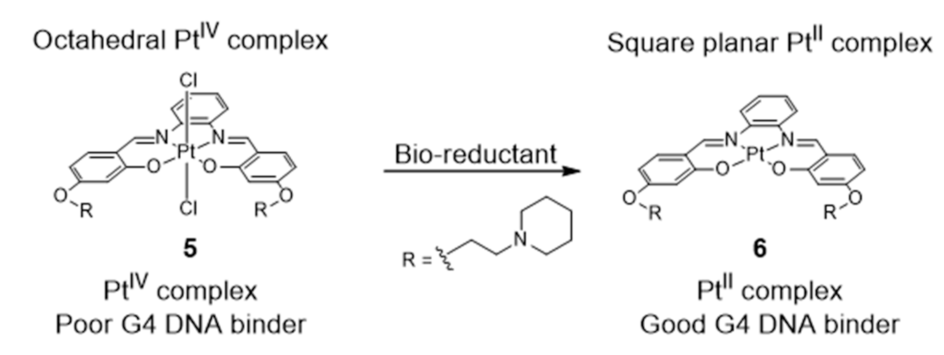{kind=link}
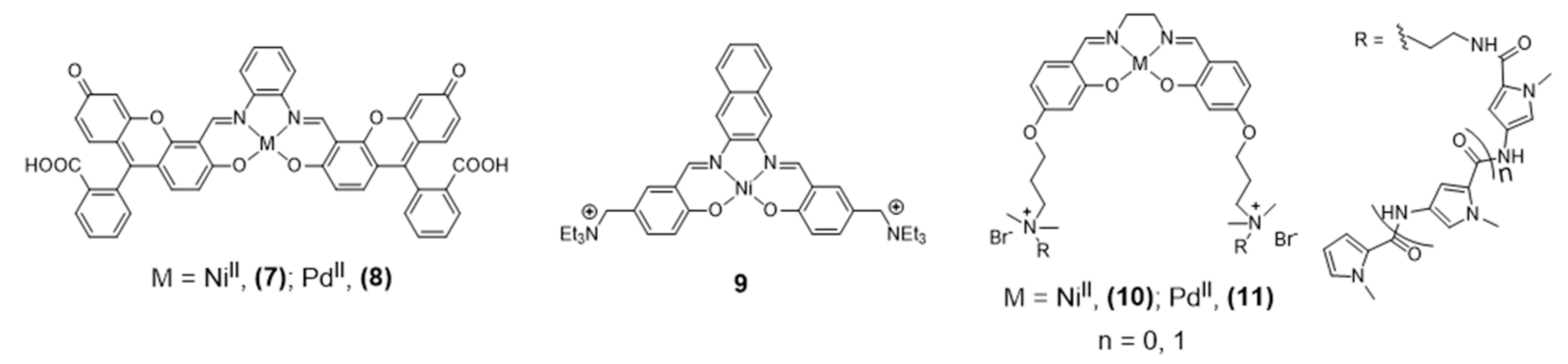{kind=link}
{kind=link}
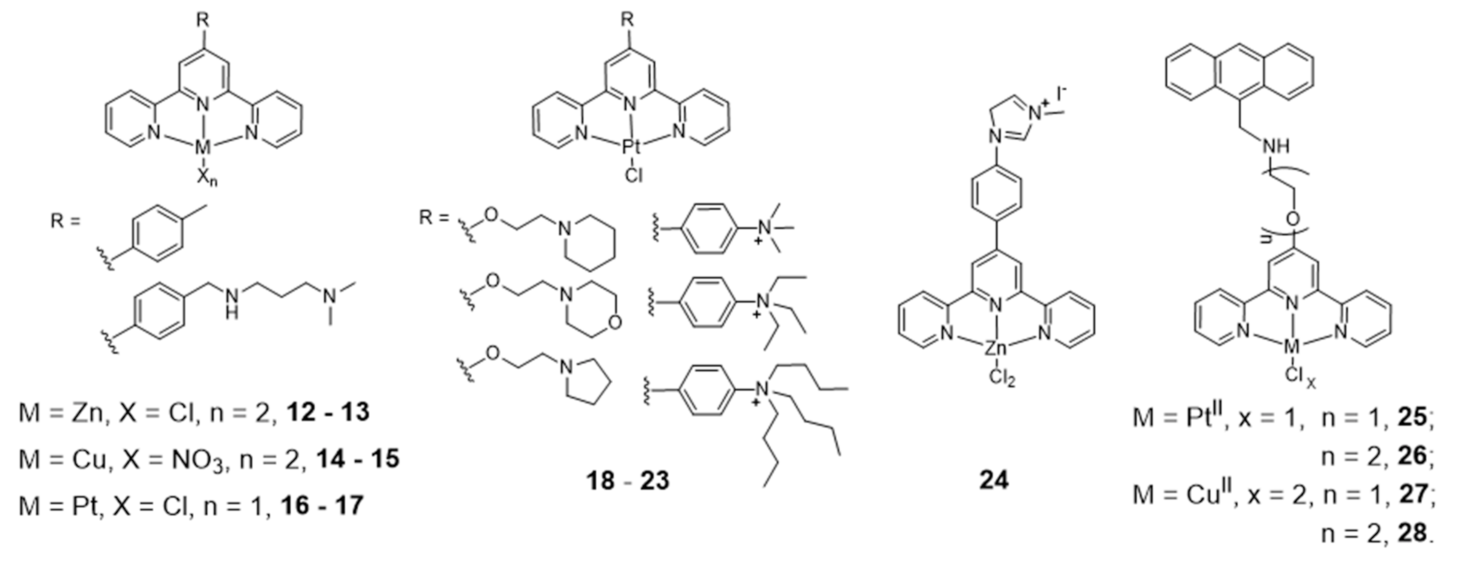{kind=link}
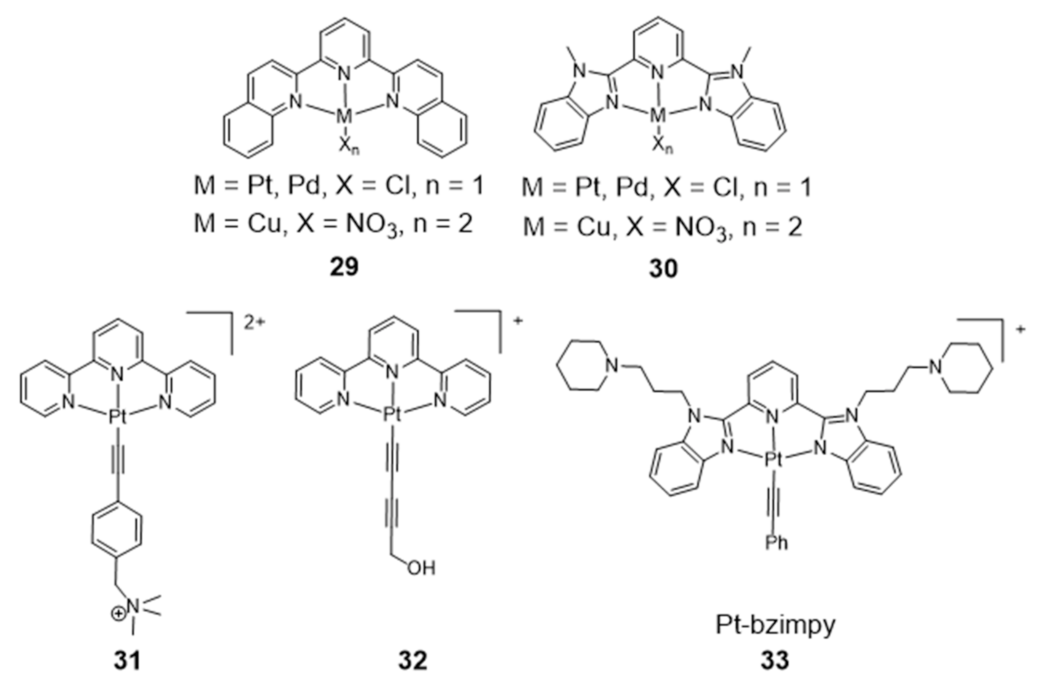{kind=link}
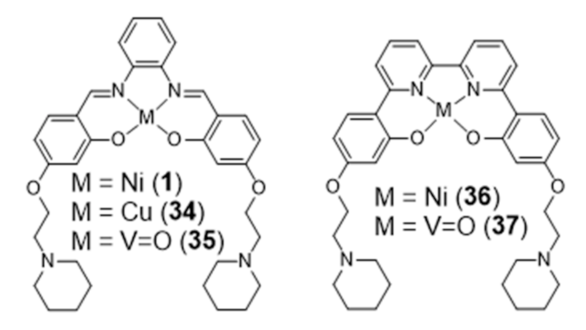{kind=link}
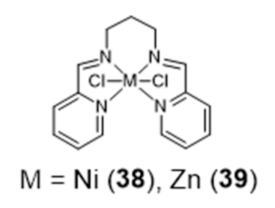{kind=link}
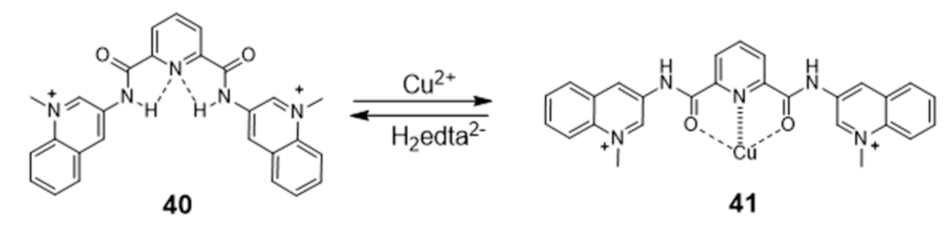{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
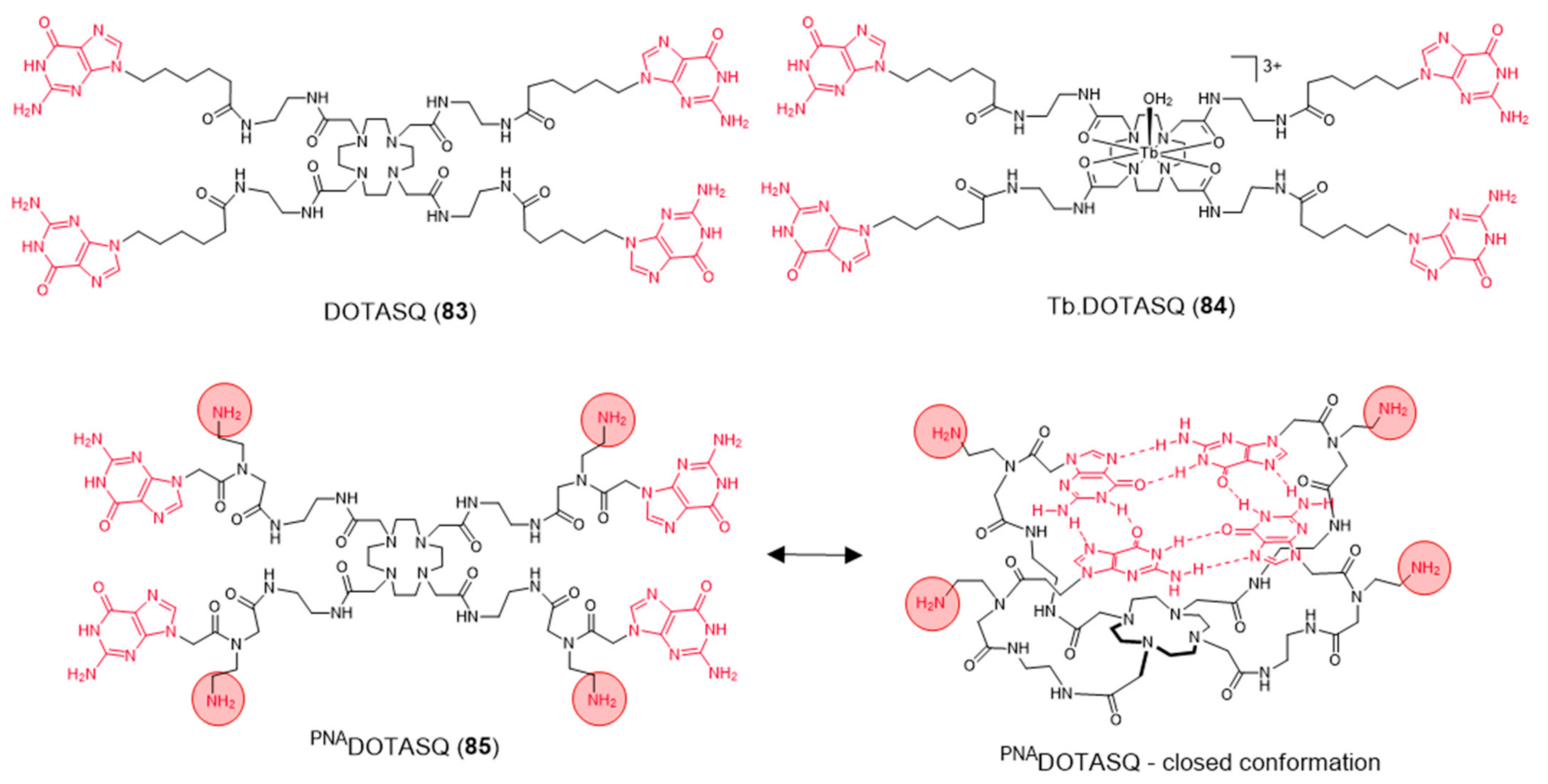{kind=link}
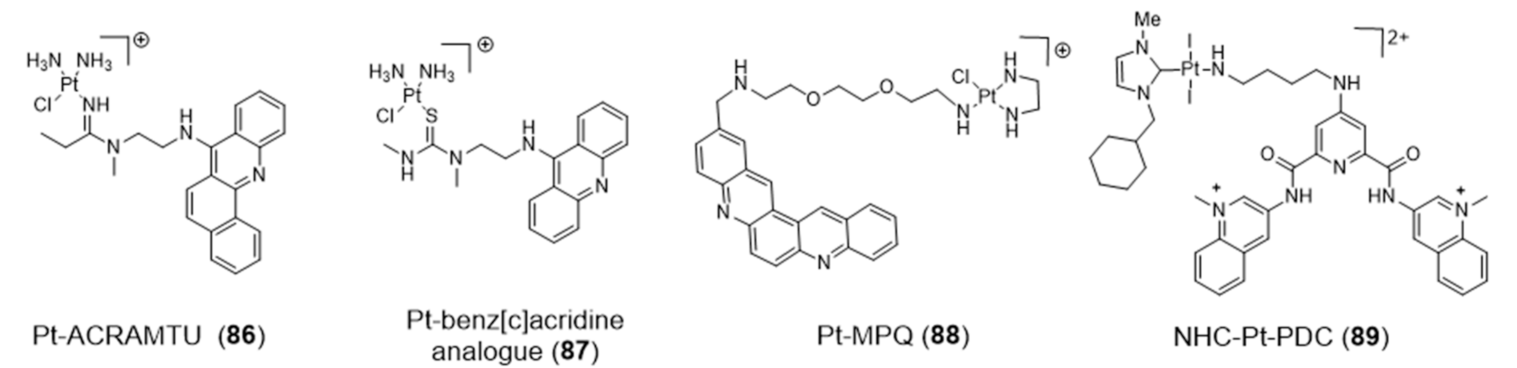{kind=link}
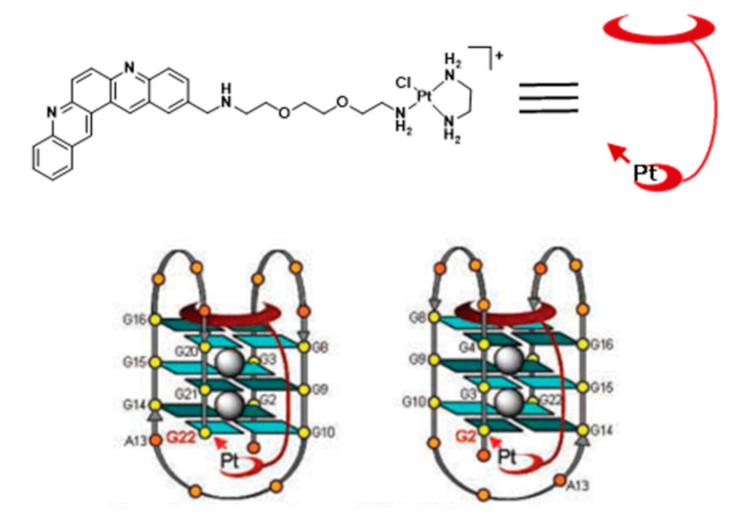{kind=link}
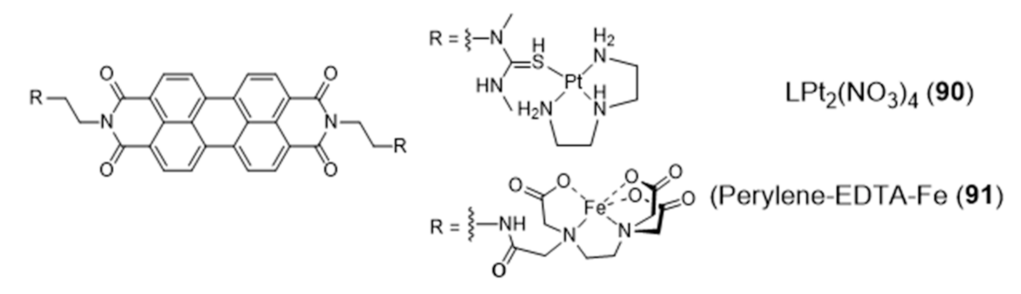{kind=link}
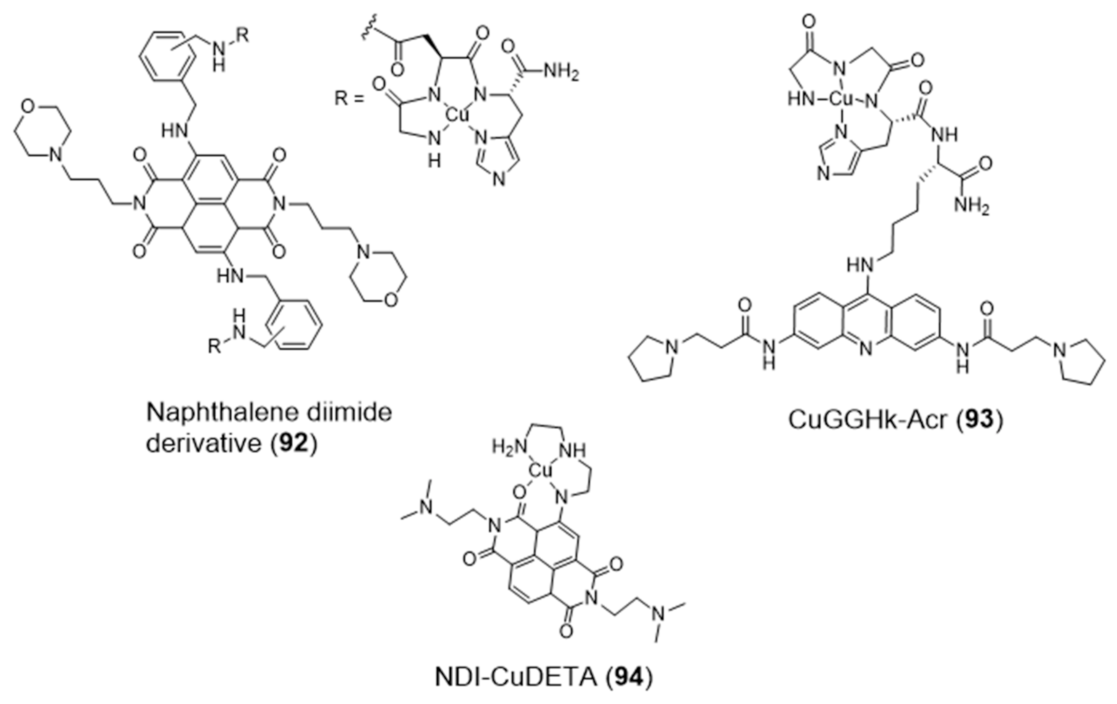{kind=link}
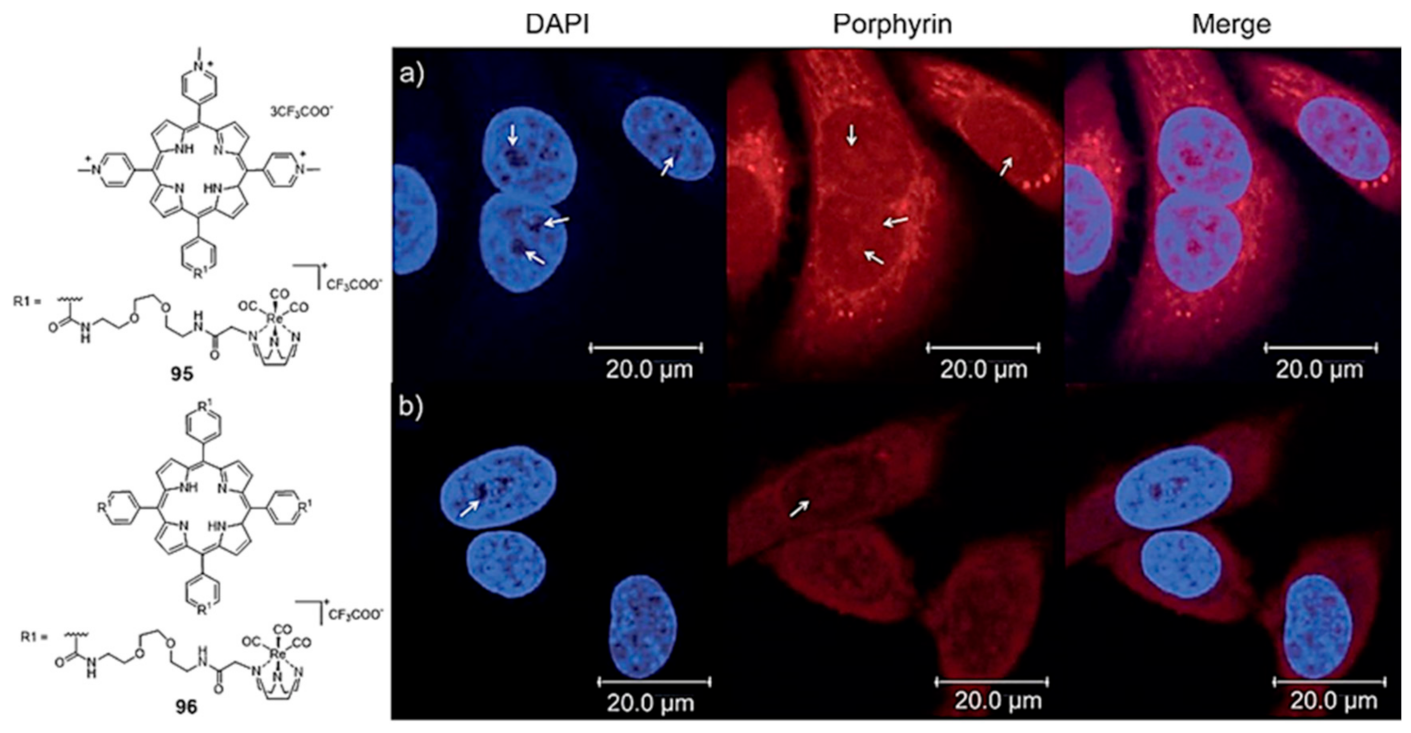{kind=link}
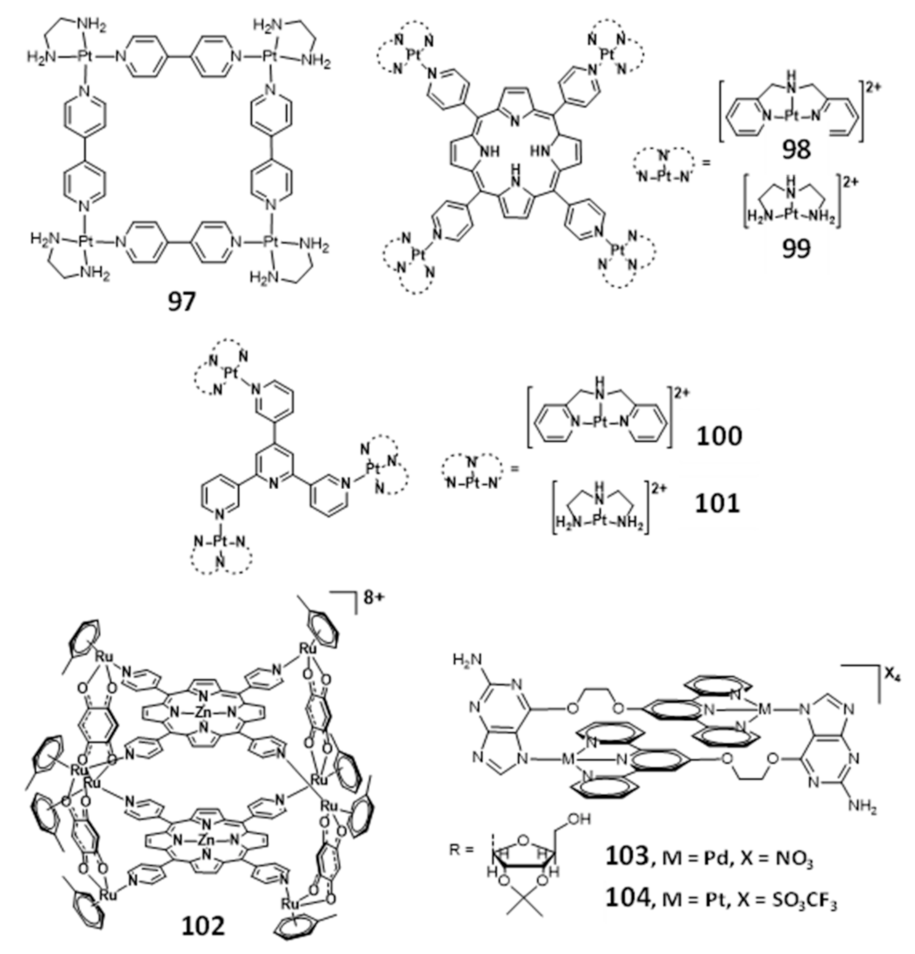{kind=link}
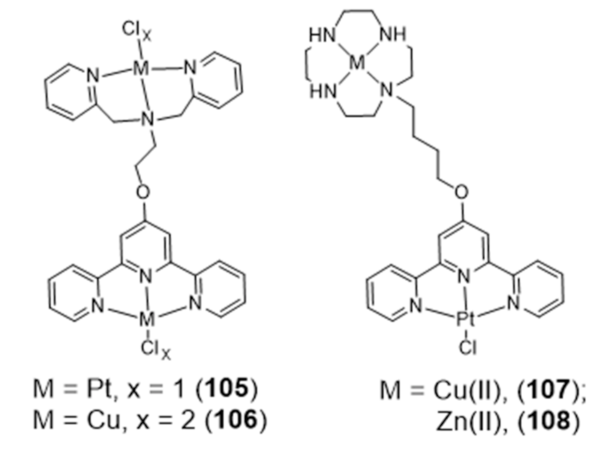{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. G4 Structures: General Aspects and Relevance for Cancer Theranostics
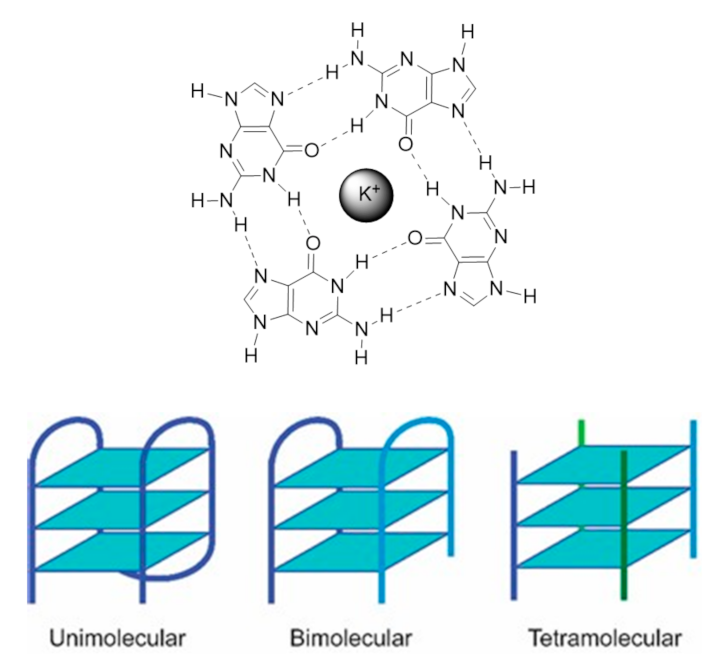
1.1.1. G4 Structures
1.1.2. G4 Targets in Cancer Cells
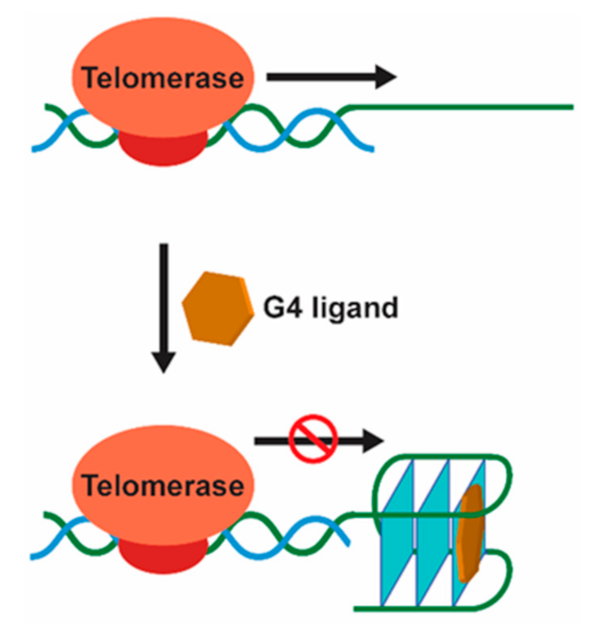
G4-Mediated Targeting of Telomerase and Telomere Maintenance
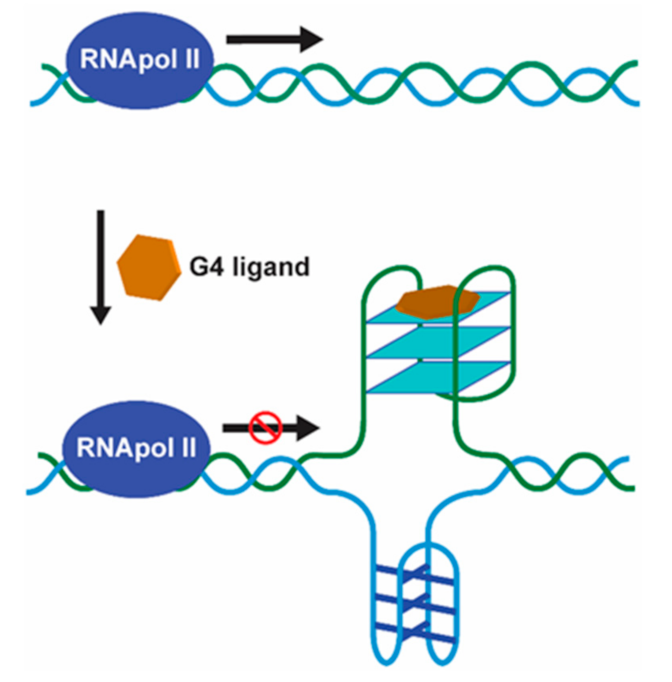
Promoter G4 Targeting
RNA G4 Targeting
1.1.3. Control of G4 Formation and Unwinding
1.1.4. Helicases and Other G4-Binding Proteins
1.1.5. The Therapeutic Relevance of G4s
1.2. Metal Complexes in the Design of G4 Binders
1.3. Scope of the Review
2. Metal-Based G4 Binders for Cancer Theranostics: Fluorescent Probes and Anticancer Agents
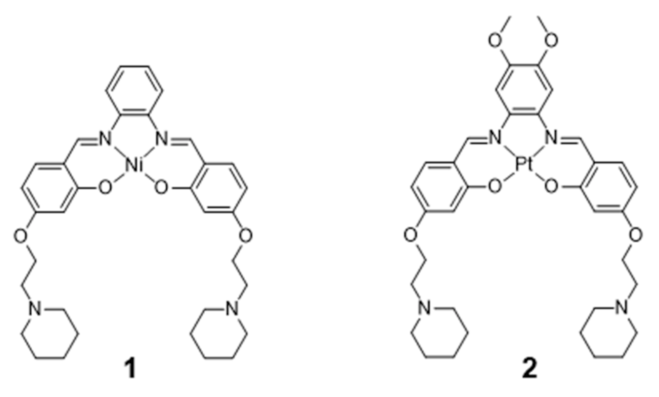
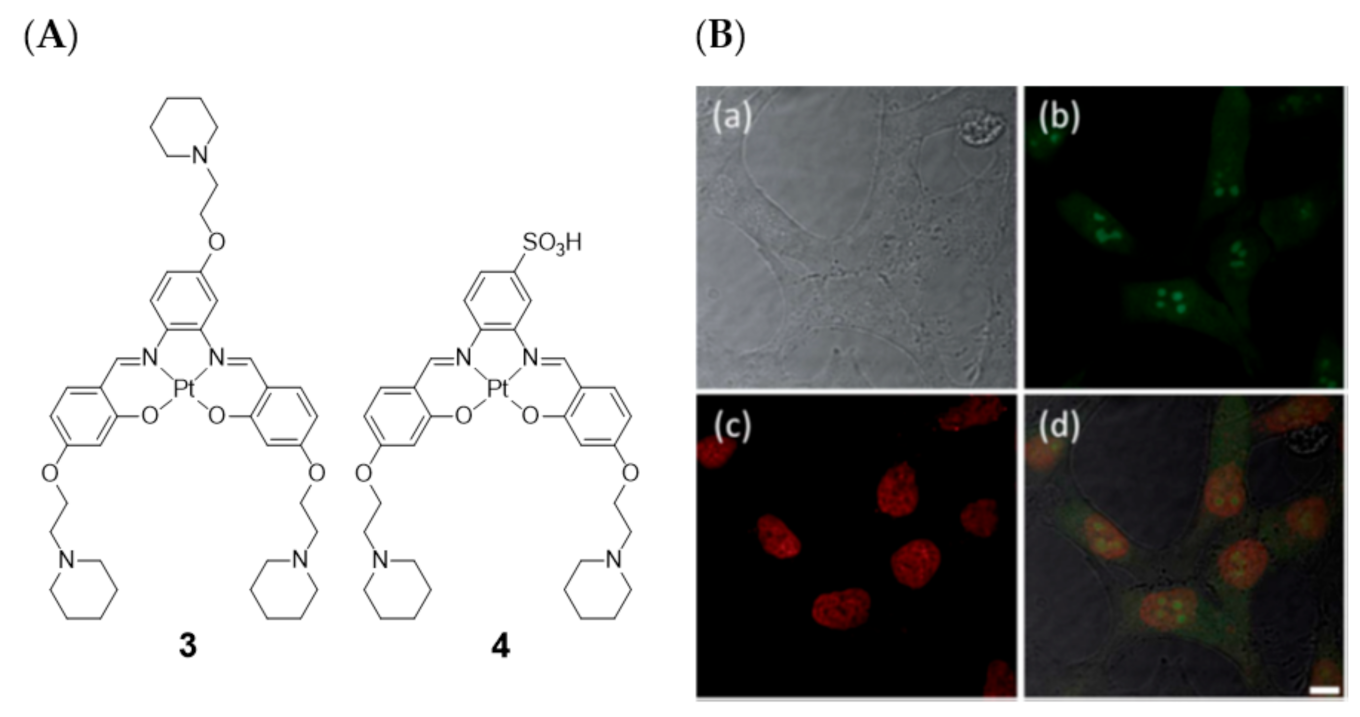
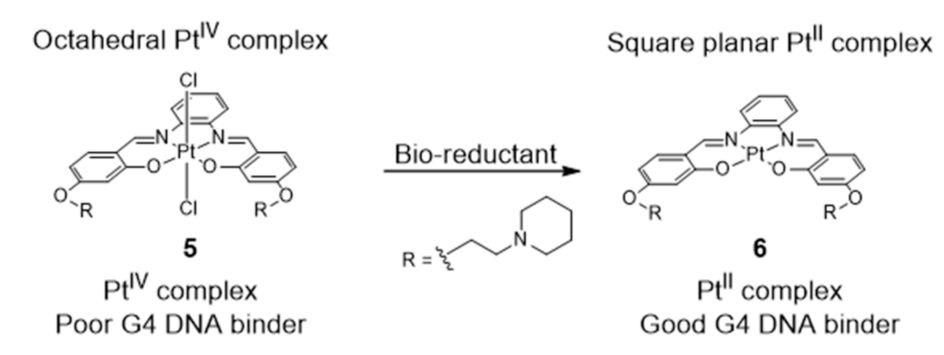
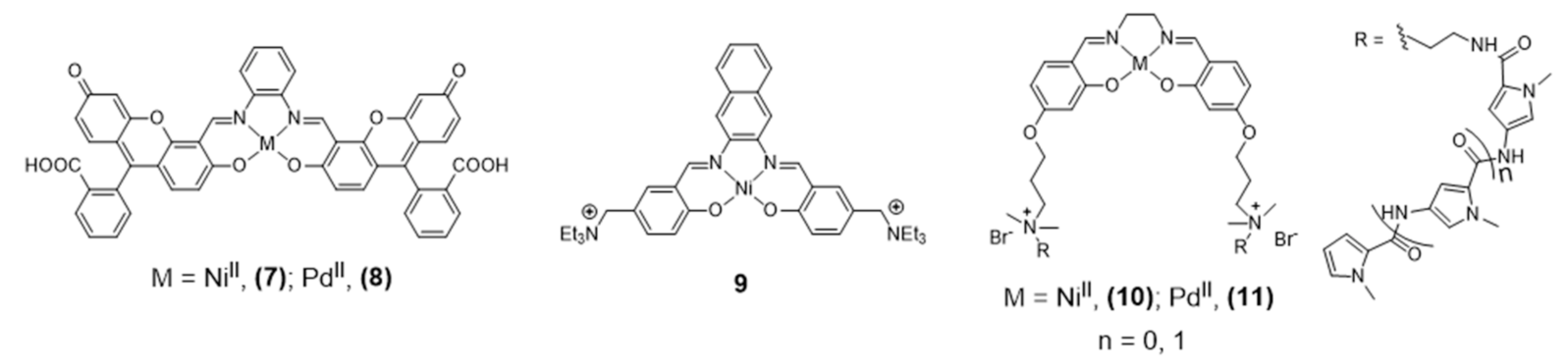
2.1. Complexes with Schiff Bases
2.2. Complexes with N-Heterocyclic Ligands
2.2.1. Terpyridine, Bipyridine and Related Ligands
2.2.2. Phenanthroline and Related Ligands
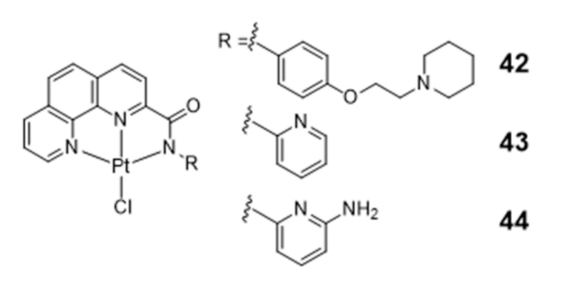
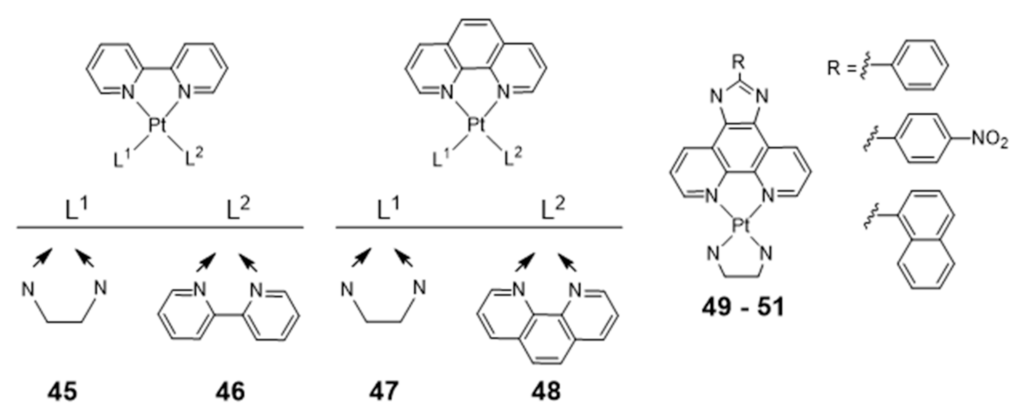
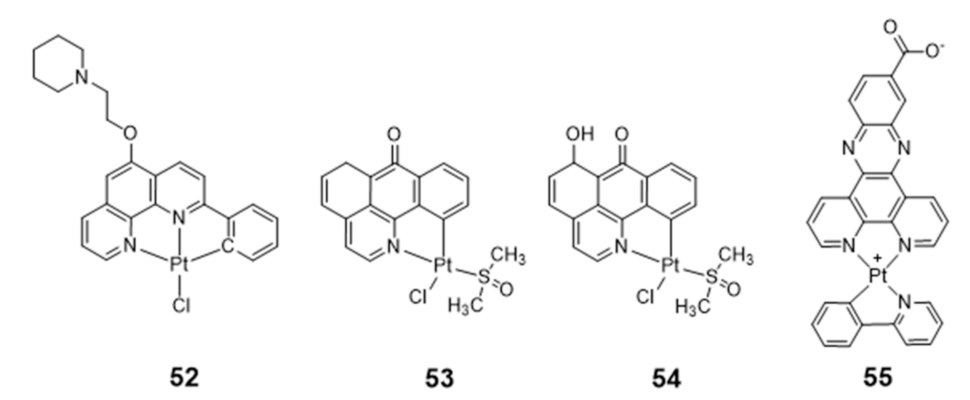
Platinum Complexes
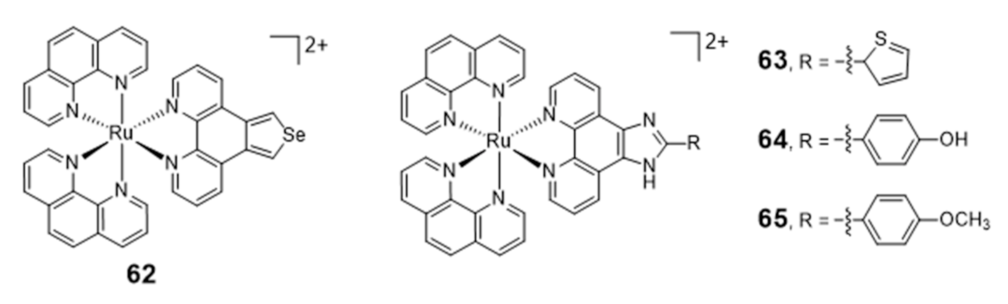
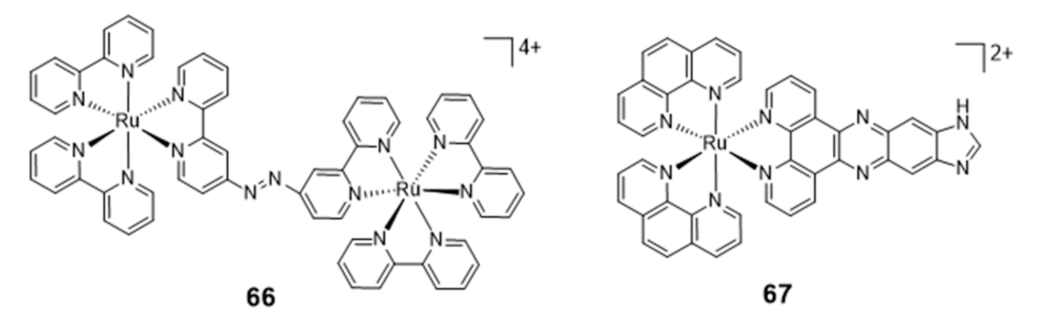
Ruthenium Complexes
2.3. Complexes with Macrocyclic Ligands
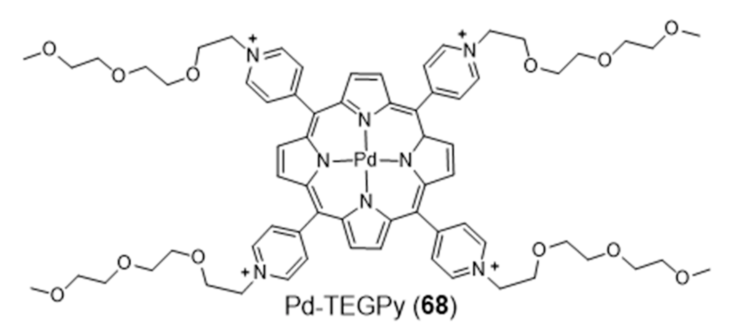
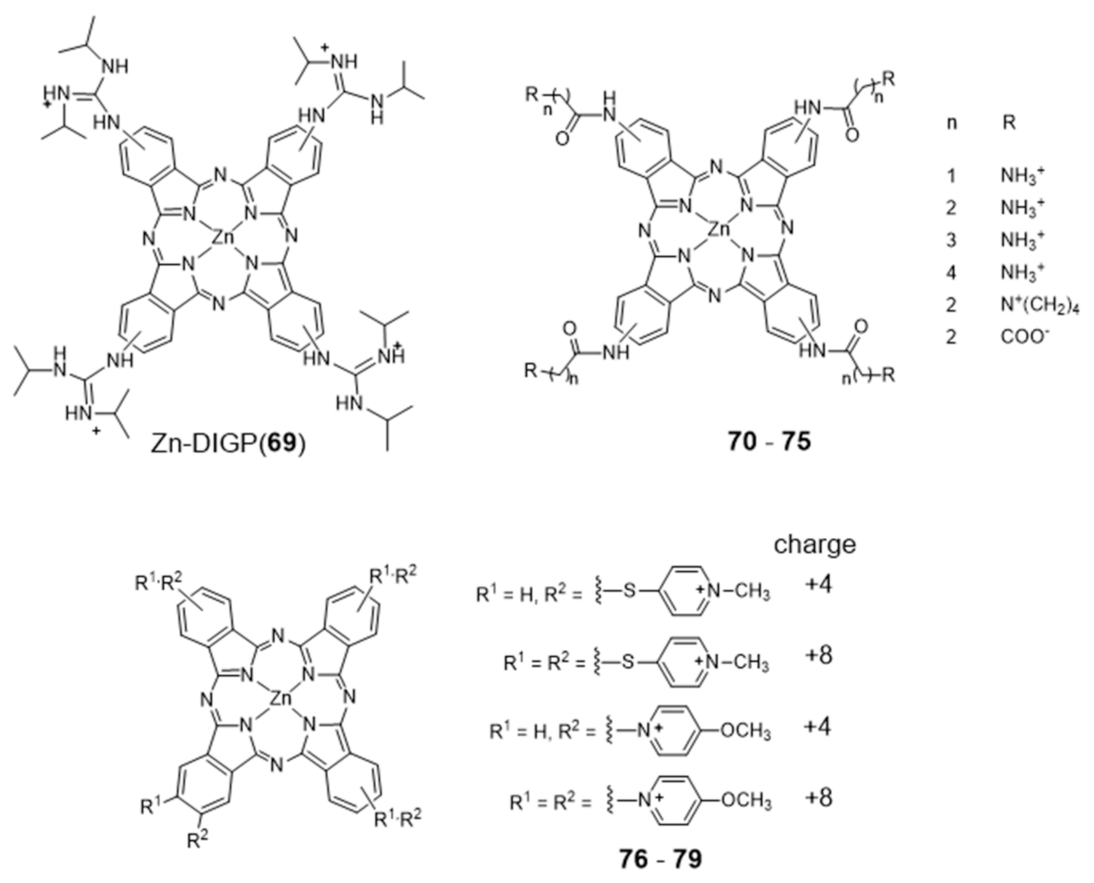
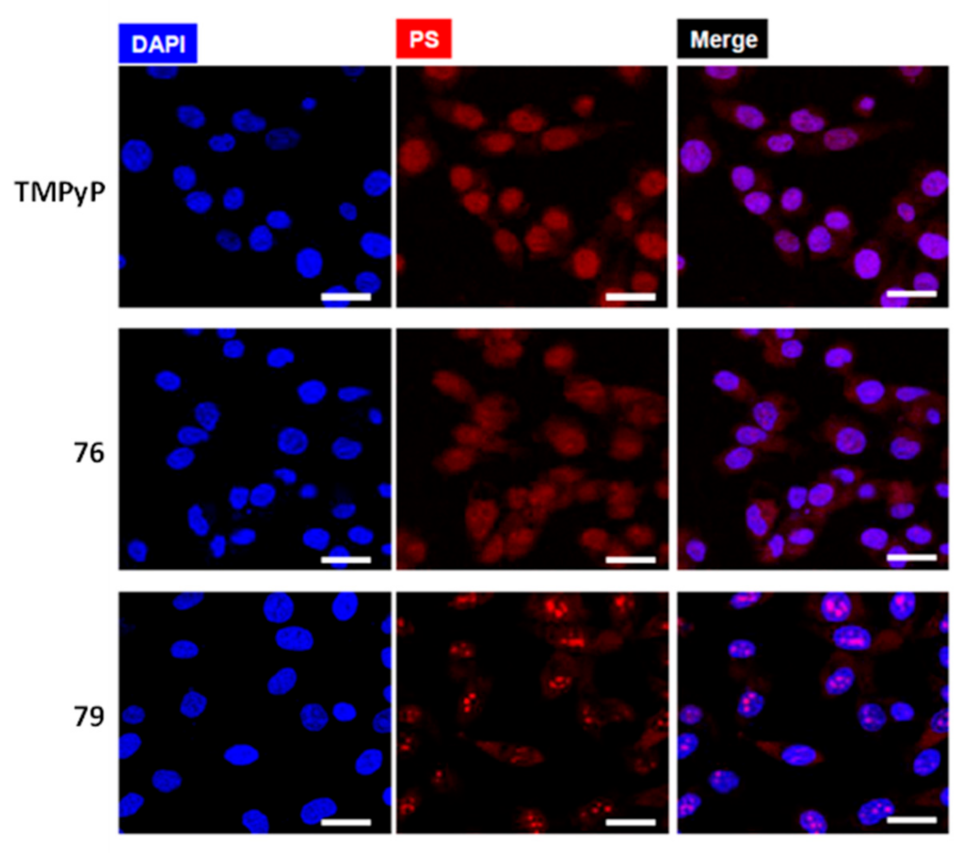
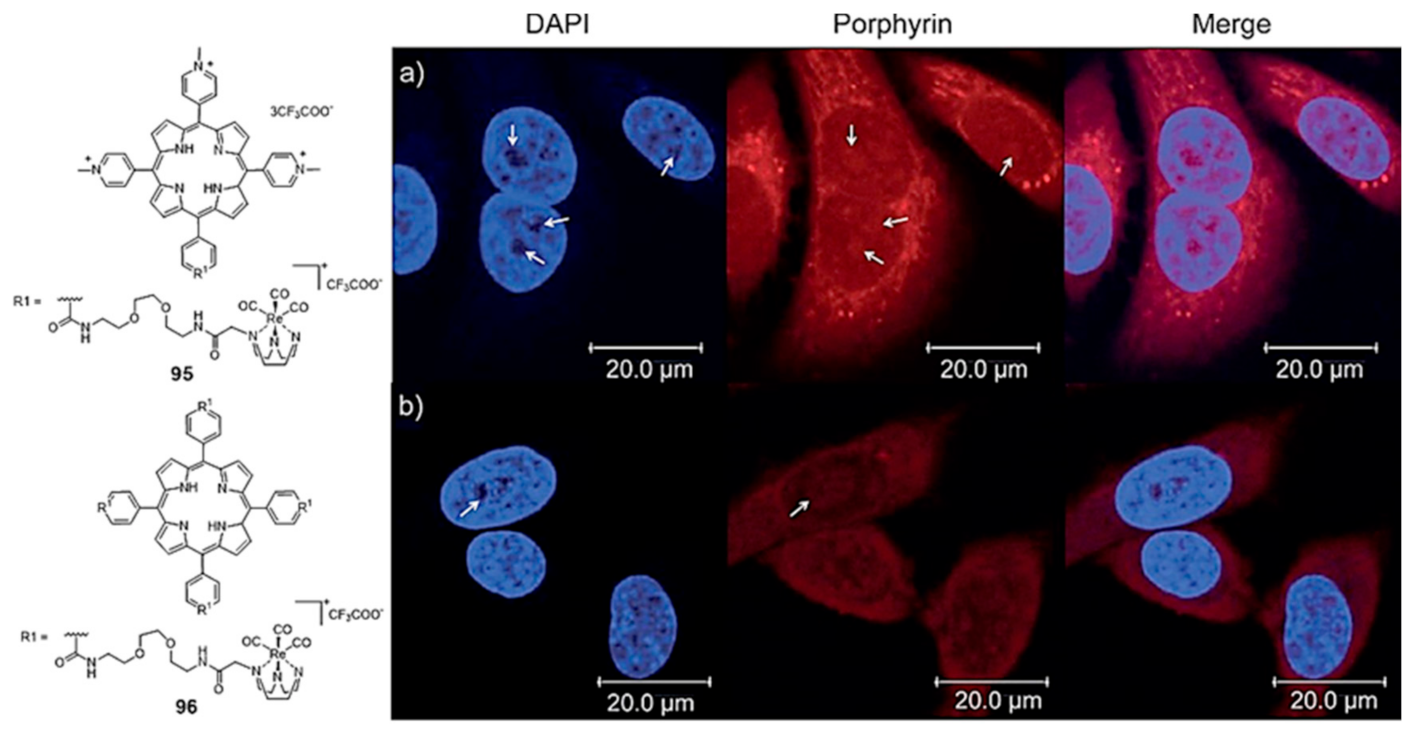
2.3.1. Porphyrin and Phthalocyanine Derivatives
2.3.2. Macrocyclic-Based Metal Complexes with Pendant G4-Binding Motifs
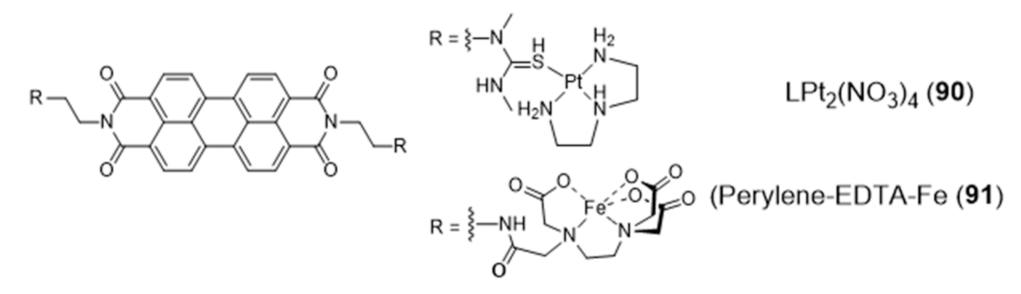
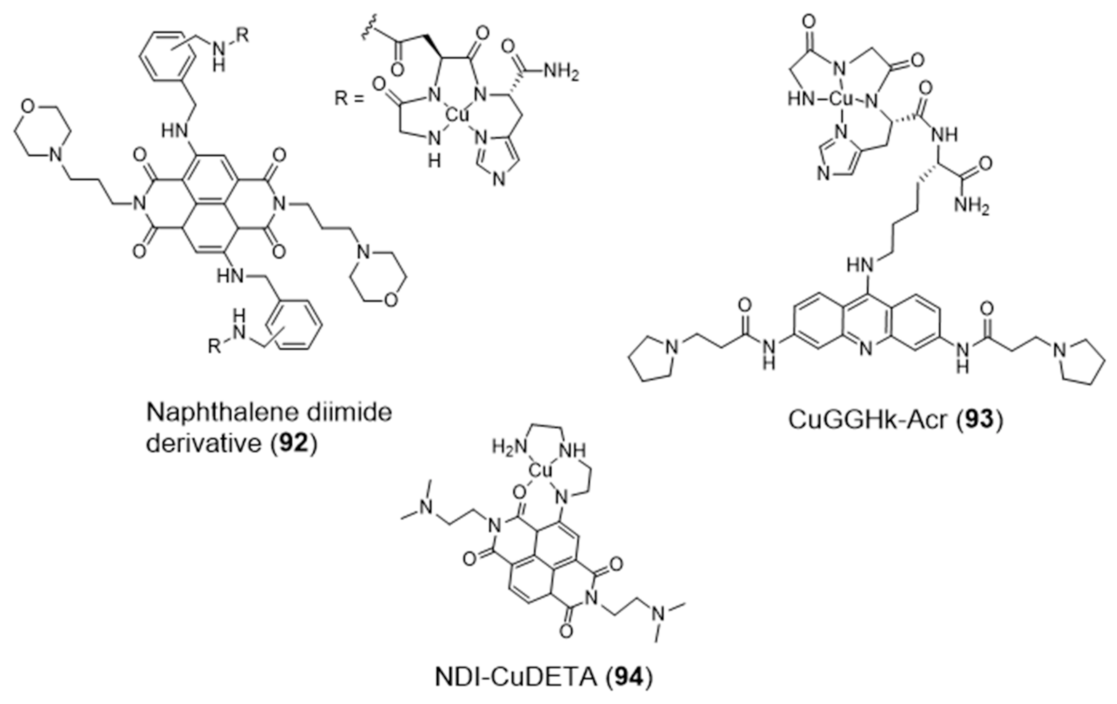
2.4. Other Metal Complexes Covalently Conjugated to G4 Binders
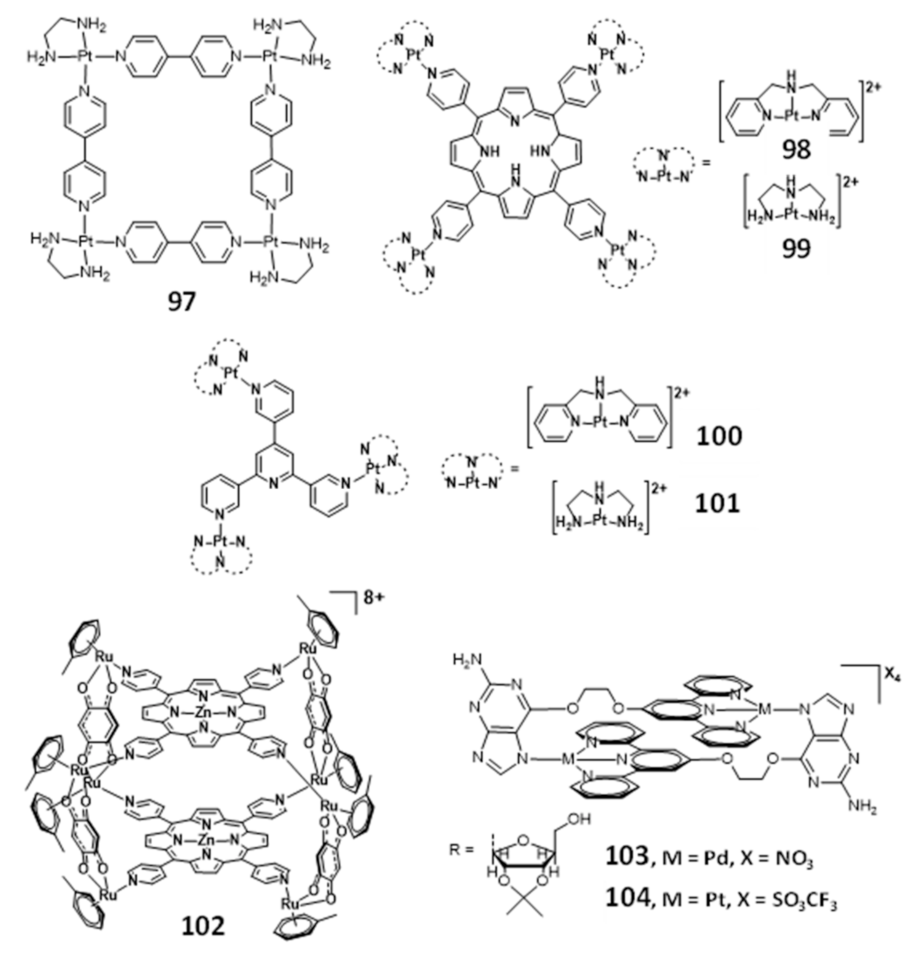
2.5. Multinuclear Metal Assemblies and Dimetallic Complexes
3. DNA-Targeted Radiocomplexes
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H.C. Molecular structure of nucleic acids. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.; BSFrietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Gellert, M.; Lipsett, M.; Davies, D. Helix formation by guanylic acid. Proc. Natl. Acad Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [Green Version]
- Bang, I. Untersuchungen über die Guanylsäure. Biochem. Z. 1910, 26, 293–311. [Google Scholar]
- Carvalho, J.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Reina, C.; Cavalieri, V. Epigenetic modulation of chromatin states and gene expression by G-quadruplex structures. Int. J. Mol. Sci. 2020, 21, 4172. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, S. A triple stranded G-quadruplex formation in the promoter region of human myosin β(Myh7) gene. J. Biomol. Struct. Dyn. 2017, 36, 2773–2786. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, S.; Curtis, E.A. Structure and function of multimeric G-quadruplexes. Molecules 2019, 24, 3074. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wu, Y.; Zhang, W. G-Quadruplex Structures and Their Interaction Diversity with Ligands. ChemMedChem 2014, 9, 899–911. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Bialis, T.; Jones, R.A.; Yang, D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006, 34, 2723–2735. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Perreault, J.-P.; Topisirovic, I.; Richard, S. RNA G-quadruplexes and their potential regulatory roles in translation. Translation 2016, 4, e1244031. [Google Scholar] [CrossRef] [Green Version]
- Bedrat, A.; Lacroix, L.; Mergny, J.-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Wojtyla, A.; Gladych, M.; Rubis, B. Human telomerase activity regulation. Mol. Biol. Rep. 2010, 38, 3339–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Yang, D. Human Telomeric G-Quadruplex Structures and G-Quadruplex-Interactive Compounds. In Telomeres and Telomerase: Methods in Molecular Biology; Songyang, Z., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1587, pp. 171–196. [Google Scholar]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Therapeutic Applications of Quadruplex Nucleic Acids, 1st ed.; Academic Press Inc.: Amsterdam, The Netherlands, 2012. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Guanine residues in d(T2AG3) and d(T2G4) form parallel-stranded potassium cation stabilized G-quadruplexes with anti glycosidic torsion angles in solution. Biochemistry 1992, 31, 8112–8119. [Google Scholar] [CrossRef]
- Zhang, N.; Phan, A.T.; Patel, D.J. (3 + 1) Assembly of Three Human Telomeric Repeats into an Asymmetric Dimeric G-Quadruplex NMR Spectra of Three-Repeat Human Telomeric Se-. J. Am. Chem. Soc. 2005, 127, 17277–17285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of Human Telomerase by a G-Quadruplex-Interactive Compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Basu, S.; Wickstrom, E. Temperature and salt dependence of higher order structure formation by antisense c-myc and c-myb phosphorothioate oligodeoxyribonucleotides containing tetraguanylate tracts. Nucleic Acids Res. 1997, 25, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.-J.; Wu, Y.-L.; Tanaka, Y.; Zhang, W. Small molecules targeting c-myc oncogene: Promising anti-cancer therapeutics. Int. J. Biol. Sci. 2014, 10, 1084–1096. [Google Scholar] [CrossRef]
- Yang, D.; Hurley, L.H. Structure of the biologically relevant G-quadruplex in the c-myc promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution structure of the biologically relevant G-quadruplex element in the human c-myc promoter. Implications for G-quadruplex stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigo, R.; Palumbo, M.; Sissi, C. G-quadruplexes in human promoters: A challenge for therapeutic applications. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 1399–1413. [Google Scholar] [CrossRef]
- Yang, D. G-Quadruplex DNA and RNA. In G-Quadruplex Nucleic Acids: Methods and Protocols; Yang, D., Lin, C., Eds.; Humana Press: New York, NY, USA, 2019; Volume 2035, pp. 1–24. [Google Scholar]
- Xu, Y.; Kaminaga, K.; Komiyama, M. Human telomeric RNA in G-quadruplex structure. Nucleic Acids Symp. Ser. 2008, 52, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Malgowska, M.; Czajczynska, K.; Gudanis, D.; Tworak, A.; Gdaniec, Z. Overview of the RNA G-quadruplex structures. Acta Biochim. Pol. 2017, 63, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Di Antonio, M.; Biffi, G.; Mariani, A.; Raiber, E.-A.; Rodriguez, R.; Balasubramanian, S. Selective RNA versus DNA G-quadruplex targeting by in situ click chemistry. Angew. Chem. Int. Ed. 2012, 51, 11073–11078. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, O.; Bourdoncle, A.; Boulé, J.-B.; Brosh, R.M.; Mergny, J.-L. G-quadruplexes and helicases. Nucleic Acids Res. 2016, 44, 1989–2006. [Google Scholar] [CrossRef] [Green Version]
- Sauer, M.; Paeschke, K. G-quadruplex unwinding helicases and their function in vivo. Biochem. Soc. Trans. 2017, 45, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 2004, 306, 1951–1953. [Google Scholar] [CrossRef]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paeschke, K.; Bochman, M.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nat. Cell Biol. 2013, 497, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, L.T.; Vallur, A.C.; Eddy, J.; Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014, 10, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5’-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef] [Green Version]
- Brázda, V.; Hároníková, L.; Liao, J.C.C.; Fojta, M. DNA and RNA Quadruplex-Binding Proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Bandaria, J.N.; Qureshi, M.H.; Yildiz, A.; Balci, H. G-quadruplex formation in telomeres enhances POT1/TPP1 protection against RPA binding. Proc. Natl. Acad. Sci. USA 2014, 111, 2990–2995. [Google Scholar] [CrossRef] [Green Version]
- Soldatenkov, V.A.; Vetcher, A.A.; Duka, T.; Ladame, S. First Evidence of a Functional Interaction between DNA Quadruplexes and Poly(ADP-ribose) Polymerase-1. ACS Chem. Biol. 2008, 3, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Lago, S.; Tosoni, E.; Nadai, M.; Palumbo, M.; Richter, S.N. The cellular protein nucleolin preferentially binds long-looped G-quadruplex nucleic acids. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 1371–1381. [Google Scholar] [CrossRef]
- González, V.; Guo, K.; Hurley, L.; Sun, D. Identification and Characterization of Nucleolin as a c-myc G-quadruplex-binding Protein. J. Biol. Chem. 2009, 284, 23622–23635. [Google Scholar] [CrossRef] [Green Version]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Haensel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.; Miller, K.M.; Forment, J.; Bradshaw, C.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule–induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Salvati, E.; Scarsella, M.; Porru, M.; Rizzo, A.; Iachettini, S.; Tentori, L.; Graziani, G.; D’Incalci, M.; Stevens, M.F.G.; Orlandi, A.; et al. PARP1 is activated at telomeres upon G4 stabilization: Possible target for telomere-based therapy. Oncogene 2010, 29, 6280–6293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kench, T.; Vilar, R. Metal complexes as G-quadruplex binders. In Annual Reports in Medicinal Chemistry; Neidle, S., Ed.; Academic Press Inc.: Cambridge, MA, USA, 2020; Volume 54, pp. 485–515. [Google Scholar] [CrossRef]
- Georgiades, S.N.; Karim, N.H.A.; Suntharalingam, K.; Vilar, R. Interaction of Metal Complexes with G-Quadruplex DNA. Angew. Chem. Int. Ed. 2010, 49, 4020–4034. [Google Scholar] [CrossRef]
- Vilar, R. Interaction of metal complexes with G-quadruplex DNA. In Advances in Inorganic Chemistry; Sadler, P.J., van Eldik, R., Eds.; Academic Press Inc.: Cambridge, MA, USA, 2020; Volume 75, pp. 425–445. [Google Scholar] [CrossRef]
- Cao, Q.; Li, Y.; Freisinger, E.; Qin, P.Z.; Sigel, R.K.O.; Mao, Z.-W. G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs. Inorg. Chem. Front. 2016, 4, 10–32. [Google Scholar] [CrossRef] [Green Version]
- Erxleben, A. Investigation of non-covalent interactions of metal complexes with DNA in cell-free systems. Chim. Int. J. Chem. 2017, 71, 102–111. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Li, H.; Liu, C.; Xia, J.; Ma, L.; Chu, W.; Zhang, Z.; Chen, C.; Li, S.; et al. Recent Progress and Future Potential for Metal Complexes as Anticancer Drugs Targeting G-quadruplex DNA. Curr. Med. Chem. 2012, 19, 2957–2975. [Google Scholar] [CrossRef]
- Jiang, Y.-L.; Liu, Z.-P. Metallo-organic G-quadruplex ligands in anticancer drug design. Mini Rev. Med. Chem. 2010, 10, 726–736. [Google Scholar] [CrossRef]
- Reed, J.E.; Neidle, S.; Vilar, R. Stabilisation of human telomeric quadruplex DNA and inhibition of telomerase by a platinum–phenanthroline complex. Chem. Commun. 2007, 4366–4368. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.E.; Arola-Arnal, A.; Neidle, S.; Vilar, R. Stabilization of G-quadruplex DNA and inhibition of telomerase activity by square-planar nickel(II) complexes. J. Am. Chem. Soc. 2006, 128, 5992–5993. [Google Scholar] [CrossRef] [PubMed]
- Gama, S.; Rodrigues, I.; Mendes, F.; Santos, I.C.; Gabano, E.; Klejevskaja, B.; Gonzalez-Garcia, J.; Ravera, M.; Vilar, R.; Paulo, A. Anthracene-terpyridine metal complexes as new G-quadruplex DNA binders. J. Inorg. Biochem. 2016, 160, 275–286. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.; Bonsignore, R. Fluorescent metal-based complexes as cancer probes. Bioorganic Med. Chem. Lett. 2020, 30, 127219. [Google Scholar] [CrossRef] [PubMed]
- Kamaladasan, K.; Uttamchandani, M. G-Quadruplex Based Probes for Visual Detection and Sensing. Curr. Pharm. Des. 2012, 18, 2048–2057. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Kudr, J.; Nejdl, L.; Maskova, D.; Kizek, R.; Adam, V. G-quadruplexes as sensing probes. Molecules 2013, 18, 14760–14779. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ding, L. Single-system based discriminative optical sensors: Different strategies and versatile applications. Analyst 2018, 143, 3775–3788. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Granzhan, A.; Hamon, F.; Verga, D.; Teulade-Fichou, M.-P. Visualizing the Quadruplex: From Fluorescent Ligands to Light-Up Probes. Top. Curr. Chem. 2012, 330, 111–177. [Google Scholar] [CrossRef]
- Ma, D.-L.; He, H.-Z.; Leung, K.-H.; Zhong, H.-J.; Chan, D.S.-H.; Leung, C.-H. Label-free luminescent oligonucleotide-based probes. Chem. Soc. Rev. 2013, 42, 3427–3440. [Google Scholar] [CrossRef]
- Ma, D.-L.; He, H.-Z.; Leung, K.-H.; Chan, D.S.-H.; Leung, C.-H. Bioactive luminescent transition-metal complexes for biomedical applications. Angew. Chem. Int. Ed. 2013, 52, 7666–7682. [Google Scholar] [CrossRef]
- Lo, K.K.-W.; Li, S.P.-Y. Utilization of the photophysical and photochemical properties of phosphorescent transition metal complexes in the development of photofunctional cellular sensors, imaging reagents, and cytotoxic agents. RSC Adv. 2014, 4, 10560–10585. [Google Scholar] [CrossRef]
- Pandith, A.; Siddappa, R.G.; Seo, Y.J. Recent developments in novel blue/green/red/NIR small fluorescent probes for in cellulo tracking of RNA/DNA G-quadruplexes. J. Photochem. Photobiol. C Photochem. Rev. 2019, 40, 81–116. [Google Scholar] [CrossRef]
- James, M.L.; Gambhir, S.S. A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications. Physiol. Rev. 2012, 92, 897–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debie, P.; Hernot, S. Emerging fluorescent molecular tracers to guide Intra-operative surgical decision-making. Front. Pharmacol. 2019, 10, 510. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.; Campello, M.P.C.; Paulo, A. Radiolabeled gold nanoparticles for imaging and therapy of cancer. Materials 2020, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Boros, E.; Packard, A.B. Radioactive transition metals for imaging and therapy. Chem. Rev. 2019, 119, 870–901. [Google Scholar] [CrossRef]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy—A review. EJNMMI Radiopharm. Chem. 2019, 4, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Zhong, H.; Chen, Z.-F.; Liu, Y.-C.; Zhang, G.-H.; Qin, Q.-P.; Liang, H. A planar schiff base platinum(II) complex: Crystal structure, cytotoxicity and interaction with DNA. Chem. Pharm. Bull. 2014, 62, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Erxleben, A. Transition metal salen complexes in bioinorganic and medicinal chemistry. Inorganica Chim. Acta 2018, 472, 40–57. [Google Scholar] [CrossRef]
- Yin, H.-Y.; Tang, J.; Zhang, J.-L. Introducing Metallosalens into Biological Studies: The Renaissance of Traditional Coordination Complexes. Eur. J. Inorg. Chem. 2017, 2017, 5085–5093. [Google Scholar] [CrossRef] [Green Version]
- Cametti, M.; Piantanida, I.; Žinić, M.; Cort, A.D.; Mandolini, L.; Marjanović, M.; Kralj, M. Specific sensing of poly G by the aluminum–salophen complex. J. Inorg. Biochem. 2007, 101, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.H.; Karim, N.H.A.; Parkinson, G.N.; Gunaratnam, M.; Petrucci, V.; Todd, A.K.; Vilar, R.; Neidle, S. Molecular basis of structure—activity relationships between salphen metal Complexes and human telomeric DNA quadruplexes. J. Med. Chem. 2011, 55, 209–222. [Google Scholar] [CrossRef]
- Terenzi, A.; Bonsignore, R.; Spinello, A.; Gentile, C.; Martorana, A.; Ducani, C.; Högberg, B.; Almerico, A.M.; Lauria, A.; Barone, G. Selective G-quadruplex stabilizers: Schiff-base metal complexes with anticancer activity. RSC Adv. 2014, 4, 33245–33256. [Google Scholar] [CrossRef]
- Karim, N.H.A.; Mendoza, O.; Shivalingam, A.; Thompson, A.J.; Ghosh, S.; Kuimova, M.K.; Vilar, R. Salphen metal complexes as tunable G-quadruplex binders and optical probes. RSC Adv. 2014, 4, 3355–3363. [Google Scholar] [CrossRef]
- Bandeira, S.; Gonzalez-Garcia, J.; Pensa, E.; Albrecht, T.; Vilar, R. A Redox-Activated G-Quadruplex DNA Binder Based on a Platinum(IV)–Salphen Complex. Angew. Chem. Int. Ed. 2018, 57. [Google Scholar] [CrossRef]
- Wu, P.; Ma, D.-L.; Leung, C.-H.; Yan, S.-C.; Zhu, N.; Abagyan, R.; Che, C.-M. Stabilization of G-Quadruplex DNA with Platinum(II) Schiff Base Complexes: Luminescent Probe and Down-Regulation of c-mycOncogene Expression. Chem. A Eur. J. 2009, 15, 13008–13021. [Google Scholar] [CrossRef]
- Ali, A.; Kamra, M.; Roy, S.; Muniyappa, K.; Bhattacharya, S. Enhanced G-Quadruplex DNA Stabilization and Telomerase Inhibition by Novel Fluorescein Derived Salen and Salphen Based Ni(II) and Pd(II) Complexes. Bioconjugate Chem. 2016, 28, 341–352. [Google Scholar] [CrossRef]
- Ali, A.; Kamra, M.; Roy, S.; Muniyappa, K.; Bhattacharya, S. Novel Oligopyrrole Carboxamide based Nickel(II) and Palladium(II) Salens, Their Targeting of Human G-Quadruplex DNA, and Selective Cancer Cell Toxicity. Chem. Asian J. 2016, 11, 2542–2554. [Google Scholar] [CrossRef]
- Terenzi, A.; Lötsch, D.; Van Schoonhoven, S.; Roller, A.; Kowol, C.R.; Berger, W.; Keppler, B.K.; Barone, G. Another step toward DNA selective targeting: NiII and CuII complexes of a Schiff base ligand able to bind gene promoter G-quadruplexes. Dalton Trans. 2016, 45, 7758–7767. [Google Scholar] [CrossRef] [Green Version]
- Arola-Arnal, A.; Benet-Buchholz, J.; Neidle, S.; Vilar, R. Effects of metal coordination geometry on stabilization of Human telomeric quadruplex DNA by square-planar and square-pyramidal metal complexes. Inorg. Chem. 2008, 47, 11910–11919. [Google Scholar] [CrossRef]
- Lecarme, L.; Prado, E.; De Rache, A.; Nicolau-Travers, M.-L.; Bonnet, R.; Van Der Heyden, A.; Philouze, C.; Gomez, D.; Mergny, J.-L.; Jamet, H.; et al. Interaction of Polycationic Ni(II)-Salophen Complexes with G-Quadruplex DNA. Inorg. Chem. 2014, 53, 12519–12531. [Google Scholar] [CrossRef]
- Summers, P.A.; Lewis, B.W.; Gonzalez-Garcia, J.; Porreca, R.M.; Lim, A.H.M.; Cadinu, P.; Martin-Pintado, N.; Mann, D.J.; Edel, J.B.; Vannier, J.B.; et al. Visualising G-quadruplex DNA dynamics in live cells by fluorescence lifetime imaging microscopy. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Bertrand, H.; Monchaud, D.; De Cian, A.; Guillot, R.; Mergny, J.-L.; Teulade-Fichou, M.-P. The importance of metal geometry in the recognition of G-quadruplex-DNA by metal-terpyridine complexes. Org. Biomol. Chem. 2007, 5, 2555–2559. [Google Scholar] [CrossRef]
- Wang, J.-T.; Li, Y.; Tan, J.-H.; Ji, L.-N.; Mao, Z.-W. Platinum(ii)-triarylpyridines complexes with electropositive pendants as efficient G-quadruplex binders. Dalton Trans. 2011, 40, 564–566. [Google Scholar] [CrossRef]
- Suntharalingam, K.; White, A.J.P.; Vilar, R. Synthesis, structural characterization, and quadruplex DNA binding studies of platinum(II)-terpyridine complexes. Inorg. Chem. 2009, 48, 9427–9435. [Google Scholar] [CrossRef]
- Busto, N.; Carrión, M.C.; Montanaro, S.; de Greñu, B.D.; Biver, T.; Jalon, F.A.; Manzano, B.R.; García, B. Targeting G-quadruplex structures with Zn(ii) terpyridine derivatives: A SAR study. Dalton Trans. 2020, 49, 13372–13385. [Google Scholar] [CrossRef]
- Largy, E.; Hamon, F.; Rosu, F.; Gabelica, V.; De Pauw, E.; Guédin, A.; Mergny, J.-L.; Teulade-Fichou, M. Tridentate N-donor palladium(II) complexes as efficient coordinating quadruplex DNA binders. Chem. A Eur. J. 2011, 17, 13274–13283. [Google Scholar] [CrossRef]
- Label-Free Optical Sensing and Characterization of Biomolecules by D8 or D10 Metal Complexes. 2008. Available online: http://hub.hku.hk/handle/10722/176960 (accessed on 22 May 2021).
- Yu, C.; Chan, K.H.-Y.; Wong, K.M.-C.; Yam, V.W.-W. Nucleic acid-induced self-assembly of a platinum(ii) terpyridyl complex: Detection of G-quadruplex formation and nuclease activity. Chem. Commun. 2009, 3756–3758. [Google Scholar] [CrossRef]
- Wang, P.; Leung, C.-H.; Ma, D.-L.; Yan, S.-C.; Che, C.-M. Structure-Based design of Platinum(II) Complexes as c-myc oncogene down-regulators and luminescent probes for G-quadruplex DNA. Chem. A Eur. J. 2010, 16, 6900–6911. [Google Scholar] [CrossRef]
- Łęczkowska, A.; Gonzalez-Garcia, J.; Perez-Arnaiz, C.; Garcia, B.; White, A.J.P.; Vilar, R. Binding Studies of Metal–Salphen and Metal–Bipyridine Complexes towards G-Quadruplex DNA. Chem. A Eur. J. 2018, 24, 11785–11794. [Google Scholar] [CrossRef]
- Butkus, J.M.; Pytko, K.G.; Stead, C.E.; Basu, S. Binding of quadruplex DNA to nickel and zinc complexes monitored by surface-enhanced raman and fluorescence spectroscopy. J. Photochem. Photobiol. A Chem. 2020, 397, 112513. [Google Scholar] [CrossRef]
- Monchaud, D.; Yang, P.; Lacroix, L.; Teulade-Fichou, M.-P.; Mergny, J.-L. A metal-mediated conformational switch controls G-quadruplex binding affinity. Angew. Chem. Int. Ed. 2008, 47, 4858–4861. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.E.; White, A.J.P.; Neidle, S.; Vilar, R. Effect of metal coordination on the interaction of substituted phenanthroline and pyridine ligands with quadruplex DNA. Dalton Trans. 2009, 2558. [Google Scholar] [CrossRef]
- Wang, J.-T.; Zheng, X.-H.; Xia, Q.; Mao, Z.-W.; Ji, L.-N.; Wang, K. 1,10-Phenanthroline platinum(ii) complex: A simple molecule for efficient G-quadruplex stabilization. Dalton Trans. 2010, 39, 7214–7216. [Google Scholar] [CrossRef]
- Talib, J.; Green, C.; Davis, K.J.; Urathamakul, T.; Beck, J.L.; Aldrich-Wright, J.R.; Ralph, S.F. A comparison of the binding of metal complexes to duplex and quadruplex DNA. Dalton Trans. 2008, 1018–1026. [Google Scholar] [CrossRef]
- Kieltyka, R.; Fakhoury, J.; Moitessier, N.; Sleiman, H.F. Platinum phenanthroimidazole complexes as G-quadruplex DNA selective binders. Chem. A Eur. J. 2008, 14, 1145–1154. [Google Scholar] [CrossRef]
- Luo, X.J.; Qin, Q.P.; Li, Y.L.; Liu, Y.C.; Liang, H. Synthesis, antitumor activity and G-quadruplex DNA/ct-DNA binding prop-erty of a cationic platinum(II) complex of 2-(4-nitro)-imidazo-[5,6-f][1,10]-phenanthroline. Indian J. Chem. Sect. A Inorg. Phys. Theor. Anal. Chem. 2014, 53, 787–792. [Google Scholar]
- Castor, K.J.; Mancini, J.; Fakhoury, J.; Weill, N.; Kieltyka, R.; Englebienne, P.; Avakyan, N.; Mittermaier, A.; Autexier, C.; Moitessier, N.; et al. Platinum(II) phenanthroimidazoles for targeting telomeric G-quadruplexes. ChemMedChem 2011, 7, 85–94. [Google Scholar] [CrossRef]
- Pierce, S.E.; Kieltyka, R.; Sleiman, H.; Brodbelt, J.S. Evaluation of binding selectivities and affinities of platinum-based quadruplex interactive complexes by electrospray ionization mass spectrometry. Biopolymers 2009, 91, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Suntharalingam, K.; Łęczkowska, A.; Furrer, M.A.; Wu, Y.; Kuimova, M.K.; Therrien, B.; White, A.J.P.; Vilar, R. A Cyclometallated platinum complex as a selective optical switch for quadruplex DNA. Chem. A Eur. J. 2012, 18, 16277–16282. [Google Scholar] [CrossRef]
- Chen, Z.-F.; Qin, Q.-P.; Qin, J.-L.; Liu, Y.-C.; Huang, K.-B.; Li, Y.-L.; Meng, T.; Zhang, G.-H.; Peng, Y.; Luo, X.-J.; et al. Stabilization of G-quadruplex DNA, inhibition of telomerase activity, and tumor cell apoptosis by organoplatinum(II) complexes with oxoisoaporphine. J. Med. Chem. 2015, 58, 2159–2179. [Google Scholar] [CrossRef]
- Ma, D.-L.; Che, C.M.; Yan, S.-C. Platinum(II) complexes with dipyridophenazine ligands as human telomerase inhibitors and luminescent probes for G-quadruplex DNA. J. Am. Chem. Soc. 2009, 131, 1835–1846. [Google Scholar] [CrossRef]
- Li, G.; Sun, L.; Ji, L.; Chao, H. Ruthenium(ii) complexes with dppz: From molecular photoswitch to biological applications. Dalton Trans. 2016, 45, 13261–13276. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhao, J.; Geng, X.; Yao, T.; Huang, H.; Liu, T.; Zheng, L.; Li, Z.; Yang, D.; Ji, L. Molecular “light switch” for G-quadruplexes and i-motif of human telomeric DNA: [Ru(phen)2(dppz)]2+. Dalton Trans. 2010, 39, 2490–2493. [Google Scholar] [CrossRef]
- Shi, S.; Geng, X.; Zhao, J.; Yao, T.; Wang, C.; Yang, D.; Zheng, L.; Ji, L. Interaction of [Ru(bpy)2(dppz)]2+ with human telomeric DNA: Preferential binding to G-quadruplexes over i-motif. Biochimie 2010, 92, 370–377. [Google Scholar] [CrossRef]
- Sun, J.; An, Y.; Zhang, L.; Chen, H.-Y.; Wang, Y.-J.; Mao, Z.-W.; Ji, L.-N. Studies on synthesis, characterization, and G-quadruplex binding of Ru(II) complexes containing two dppz ligands. J. Inorg. Biochem. 2011, 105, 149–154. [Google Scholar] [CrossRef]
- Rajput, C.; Rutkaite, R.; Swanson, L.; Haq, I.; Thomas, J.A. Dinuclear monointercalating ruii complexes that display high affinity binding to duplex and quadruplex DNA. Chem. A Eur. J. 2006, 12, 4611–4619. [Google Scholar] [CrossRef]
- Gill, M.R.; Garcia-Lara, J.; Foster, S.J.; Smythe, C.; Battaglia, G.; Thomas, J.A. A ruthenium(II) polypyridyl complex for direct imaging of DNA structure in living cells. Nat. Chem. 2009, 1, 662–667. [Google Scholar] [CrossRef]
- Sun, D.; Liu, Y.; Liu, D.; Zhang, R.; Yang, X.; Liu, J. Stabilization of G-quadruplex DNA, inhibition of telomerase activity and live cell imaging studies of chiral ruthenium(II) complexes. Chem. A Eur. J. 2012, 18, 4285–4295. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, Y.; Wang, C.; Sun, D.; Yang, X.; Liu, Y.; Liu, J. Chiral Ruthenium(II) Polypyridyl Complexes: Stabilization of G-Quadruplex DNA, Inhibition of Telomerase Activity and Cellular Uptake. PLoS ONE 2012, 7, e50902. [Google Scholar] [CrossRef]
- Li, Q.; Sun, N.; Zhou, Y.; Liu, D.; Zhang, Q.; Liu, J. Anticancer activity of novel ruthenium complex with 1,10-phenanthrolineselenazole as potent telomeric G-quadruplex inhibitor. Inorg. Chem. Commun. 2012, 20, 142–146. [Google Scholar] [CrossRef]
- Liu, D.; Liu, Y.; Wang, C.; Shi, S.; Sun, D.; Gao, F.; Zhang, Q.; Liu, J. Polypyridyl complexes of ruthenium(II): Stabilization of G-quadruplex DNA and inhibition of telomerase activity. Chem. Plus. Chem. 2012, 77, 551–562. [Google Scholar] [CrossRef]
- Chen, Z.-F.; Qin, Q.-P.; Qin, J.-L.; Zhou, J.; Li, Y.-L.; Li, N.; Liu, Y.-C.; Liang, H. Water-Soluble Ruthenium(II) Complexes with Chiral 4-(2,3-Dihydroxypropyl)-formamide Oxoaporphine (FOA): In Vitro and in Vivo Anticancer Activity by Stabilization of G-Quadruplex DNA, Inhibition of Telomerase Activity, and Induction of Tumor Cell Apoptosis. J. Med. Chem. 2015, 58, 4771–4789. [Google Scholar] [CrossRef]
- Ruthenium (II)-polypyridine Complex, and Preparation Method and Application Thereof. Google Patents CN102464676A, 23 May 2021. Available online: https://patents.google.com/patent/CN102464676A/en (accessed on 23 May 2021).
- Thomas, J.A. Colorimetric Sensing and/or Detection of DNA Sequences Using Ruthenium or Osmium Bipyridine Complexes. WO2009050509. 2009. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009050509 (accessed on 23 May 2021).
- Diabate, P.D.; Laguerre, A.; Pirrotta, M.; Desbois, N.; Boudon, J.; Gros, C.P.; Monchaud, D. DNA structure-specific sensitization of a metalloporphyrin leads to an efficient in vitro quadruplex detection molecular tool. New J. Chem. 2016, 40, 5683–5689. [Google Scholar] [CrossRef]
- Pattanayak, R.; Barua, A.; Das, A.; Chatterjee, T.; Pathak, A.; Choudhury, P.; Sen, S.; Saha, P.; Bhattacharyya, M. Porphyrins to restrict progression of pancreatic cancer by stabilizing KRAS G-quadruplex: In silico, in vitro and in vivo validation of anticancer strategy. Eur. J. Pharm. Sci. 2018, 125, 39–53. [Google Scholar] [CrossRef]
- Alzeer, J.; Vummidi, B.; Roth, P.; Luedtke, N. Guanidinium-modified phthalocyanines as high-affinity G-quadruplex fluorescent probes and transcriptional regulators. Angew. Chem. Int. Ed. 2009, 48, 9362–9365. [Google Scholar] [CrossRef]
- Membrino, A.; Paramasivam, M.; Cogoi, S.; Alzeer, J.; Luedtke, N.W.; Xodo, L. Cellular uptake and binding of guanidine-modified phthalocyanines to KRAS/HRASG-quadruplexes. Chem. Commun. 2010, 46, 625–627. [Google Scholar] [CrossRef] [Green Version]
- Luedtke, N.W.; Carmichael, P.; Tor, Y. Cellular uptake of aminoglycosides, guanidinoglycosides, and poly-arginine. J. Am. Chem. Soc. 2003, 125, 12374–12375. [Google Scholar] [CrossRef]
- Luedtke, N.W.; Baker, T.; Goodman, M.; Tor, Y. Guanidinoglycosides: A novel family of RNA ligands. J. Am. Chem. Soc. 2000, 122, 12035–12036. [Google Scholar] [CrossRef]
- Alzeer, J.; Luedtke, N.W. pH-mediated fluorescence and G-quadruplex binding of amido phthalocyanines. Biochemistry 2010, 49, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.I.V.; Almeida, S.P.; Lourenço, L.M.O.; Pereira, P.M.R.; Fernandes, R.; Faustino, M.A.F.; Tomé, J.P.C.; Carvalho, J.; Cruz, C.; Neves, M.G.P.M.S. Multicharged Phthalocyanines as Selective Ligands for G-Quadruplex DNA Structures. Molecules 2019, 24, 733. [Google Scholar] [CrossRef] [Green Version]
- You, D.; Liu, L.; Yang, Q.; Wu, X.; Li, S.; Li, A. A far-red aza-crown ether fluorescent probe for selective G-quadruplex DNA targeting. Dye. Pigment. 2020, 176, 108222. [Google Scholar] [CrossRef]
- Siters, K.E.; Sander, S.A.; Devlin, J.R.; Morrow, J.R. Bifunctional Zn( ii ) complexes for recognition of non-canonical thymines in DNA bulges and G-quadruplexes. Dalton Trans. 2014, 44, 3708–3716. [Google Scholar] [CrossRef]
- Del Mundo, I.M.A.; Siters, K.E.; Fountain, M.A.; Morrow, J.R. Structural basis for bifunctional zinc(ii) macrocyclic complex recognition of thymine bulges in DNA. Inorg. Chem. 2012, 51, 5444–5457. [Google Scholar] [CrossRef]
- Siters, K.E.; Fountain, M.A.; Morrow, J.R. Selective binding of Zn2+ complexes to human telomeric G-quadruplex DNA. Inorg. Chem. 2014, 53, 11540–11551. [Google Scholar] [CrossRef]
- Haudecoeur, R.; Stefan, L.; Denat, F.; Monchaud, D. A Model of smart G-quadruplex ligand. J. Am. Chem. Soc. 2012, 135, 550–553. [Google Scholar] [CrossRef]
- Pickard, A.; Liu, F.; Bartenstein, T.F.; Haines, L.G.; Levine, K.; Kucera, G.L.; Bierbach, U. Redesigning the DNA-targeted chromophore in platinum-acridine anticancer agents: A structure-activity relationship study. Chem. A Eur. J. 2014, 20, 16174–16187. [Google Scholar] [CrossRef]
- Rao, L.; Bierbach, U. Kinetically favored platination of adenine in the G-rich human telomeric repeat. J. Am. Chem. Soc. 2007, 129, 15764–15765. [Google Scholar] [CrossRef] [Green Version]
- Baruah, H. Unusual intercalation of acridin-9-ylthiourea into the 5’-GA/TC DNA base step from the minor groove: Implications for the covalent DNA adduct profile of a novel platinum-intercalator conjugate. Nucleic Acids Res. 2003, 31, 4138–4146. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, H.; Bombard, S.; Monchaud, D.; Teulade-Fichou, M.-P. New platinum(II) complexes targeting the loops of the human telomeric G-quadruplex. Nucleic Acids Symp. Ser. 2008, 52, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Charif, R.; Granotier-Beckers, C.; Bertrand, H.C.; Poupon, J.; Ségal-Bendirdjian, E.; Teulade-Fichou, M.-P.; Boussin, F.; Bombard, S. Association of a Platinum Complex to a G-Quadruplex Ligand Enhances Telomere Disruption. Chem. Res. Toxicol. 2017, 30, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Betzer, J.-F.; Nuter, F.; Chtchigrovsky, M.; Hamon, F.; Kellermann, G.; Ali, S.; Calméjane, M.-A.; Roque, S.; Poupon, J.; Cresteil, T.; et al. Linking of antitumor trans NHC-Pt(II) complexes to G-quadruplex DNA ligand for telomeric targeting. Bioconjugate Chem. 2016, 27, 1456–1470. [Google Scholar] [CrossRef]
- Rao, L.; Dworkin, J.D.; Nell, W.E.; Bierbach, U. Interactions of a platinum-modified perylene derivative with the human telomeric G-quadruplex. J. Phys. Chem. B 2011, 115, 13701–13712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuntiwechapikul, W.; Salazar, M. Cleavage of telomeric G-quadruplex DNA with perylene-EDTA·Fe(II). Biochemistry 2001, 40, 13652–13658. [Google Scholar] [CrossRef] [PubMed]
- Tuntiwechapikul, W.; Lee, J.T.; Salazar, M. Design and synthesis of the G-quadruplex-specific cleaving reagent perylene-EDTA·iron(II). J. Am. Chem. Soc. 2001, 123, 5606–5607. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Han, M.; Cowan, J.A. Toward the design of a catalytic metallodrug: Selective cleavage of G-quadruplex telomeric DNA by an anticancer copper-acridine-ATCUN complex. Angew. Chem. Int. Ed. 2015, 54, 1901–1905. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Fenk, K.D.; Huang, D.; Sen, S.; Cowan, J.A. Rapid Telomere Reduction in Cancer Cells Induced by G-Quadruplex-Targeting Copper Complexes. J. Med. Chem. 2019, 62, 5040–5048. [Google Scholar] [CrossRef] [PubMed]
- Nadai, M.; Doria, F.; Scalabrin, M.; Pirota, V.; Grande, V.; Bergamaschi, G.; Amendola, V.; Winnerdy, F.R.; Phan, A.T.; Richter, S.N.; et al. A Catalytic and Selective Scissoring Molecular Tool for Quadruplex Nucleic Acids. J. Am. Chem. Soc. 2018, 140, 14528–14532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mion, G.; Gianferrara, T.; Bergamo, A.; Gasser, G.; Pierroz, V.; Rubbiani, R.; Vilar, R.; Leczkowska, A.; Alessio, E. Phototoxic activity and DNA interactions of water-soluble porphyrins and their rhenium(I) conjugates. ChemMedChem 2015, 10, 1901–1914. [Google Scholar] [CrossRef] [PubMed]
- Kieltyka, R.; Englebienne, P.; Fakhoury, J.; Autexier, C.; Moitessier, N.; Sleiman, H. A Platinum Supramolecular Square as an Effective G-Quadruplex Binder and Telomerase Inhibitor. J. Am. Chem. Soc. 2008, 130, 10040–10041. [Google Scholar] [CrossRef]
- Zheng, X.-H.; Chen, H.-Y.; Tong, M.-L.; Ji, L.-N.; Mao, Z.-W. Platinum squares with high selectivity and affinity for human telomeric G-quadruplexes. Chem. Commun. 2012, 48, 7607–7609. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-H.; Zhong, Y.-F.; Tan, C.-P.; Ji, L.-N.; Mao, Z.-W. Pt(ii) squares as selective and effective human telomeric G-quadruplex binders and potential cancer therapeutics. Dalton Trans. 2012, 41, 11807–11812. [Google Scholar] [CrossRef]
- Garci, A.; Castor, K.J.; Fakhoury, J.; Do, J.-L.; Di Trani, J.; Chidchob, P.; Stein, R.S.; Mittermaier, A.K.; Friščić, T.; Sleiman, H. Efficient and Rapid Mechanochemical Assembly of Platinum(II) Squares for Guanine Quadruplex Targeting. J. Am. Chem. Soc. 2017, 139, 16913–16922. [Google Scholar] [CrossRef]
- Domarco, O.; Lötsch, D.; Schreiber, J.; Dinhof, C.; Van Schoonhoven, S.; Garcia, M.D.; Peinador, C.; Keppler, B.K.; Berger, W.; Terenzi, A. Self-assembled Pt2L2 boxes strongly bind G-quadruplex DNA and influence gene expression in cancer cells. Dalton Trans. 2016, 46, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.-H.; Mu, G.; Zhong, Y.-F.; Zhang, T.-P.; Cao, Q.; Ji, L.-N.; Zhao, Y.; Mao, Z.-W. Trigeminal star-like platinum complexes induce cancer cell senescence through quadruplex-mediated telomere dysfunction. Chem. Commun. 2016, 52, 14101–14104. [Google Scholar] [CrossRef]
- Xu, C.-X.; Shen, Y.; Hu, Q.; Zheng, Y.-X.; Cao, Q.; Qin, P.Z.; Zhao, Y.; Ji, L.-N.; Mao, Z.-W. Stabilization of human telomeric G-quadruplex and inhibition of telomerase activity by propeller-shaped trinuclear PtII complexes. Chem. Asian J. 2014, 9, 2519–2526. [Google Scholar] [CrossRef]
- Zheng, X.-H.; Cao, Q.; Ding, Y.-L.; Zhong, Y.-F.; Mu, G.; Qin, P.Z.; Ji, L.-N.; Mao, Z.-W. Platinum(ii) clovers targeting G-quadruplexes and their anticancer activities. Dalton Trans. 2014, 44, 50–53. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, H.; Liu, W.; Zheng, X.; Zhou, Y.; Cao, Q.; Shen, Y.; Zhao, Y.; Qin, P.Z.; Ji, L.; et al. A Platinum(II)-based Photosensitive Tripod as an Effective Photodynamic Anticancer Agent through DNA Damage. Chem. A Eur. J. 2017, 23, 16442–16446. [Google Scholar] [CrossRef] [PubMed]
- Barry, N.; Karim, N.H.A.; Vilar, R.; Therrien, B. Interactions of ruthenium coordination cubes with DNA. Dalton Trans. 2009, 10717–10719. [Google Scholar] [CrossRef]
- Ghosh, S.; Mendoza, O.; Cubo, L.; Rosu, F.; Gabelica, V.; White, A.J.P.; Vilar, R. Assembly of palladium(II) and platinum(II) metallo-rectangles with a guanosine-substituted terpyridine and study of their interactions with quadruplex DNA. Chem. A Eur. J. 2014, 20, 4772–4779. [Google Scholar] [CrossRef] [Green Version]
- Stafford, V.S.; Suntharalingam, K.; Shivalingam, A.; White, A.J.P.; Mann, D.J.; Vilar, R. Syntheses of polypyridyl metal complexes and studies of their interaction with quadruplex DNA. Dalton Trans. 2014, 44, 3686–3700. [Google Scholar] [CrossRef] [Green Version]
- Khosravifarsani, M.; Ait-mohand, S.; GuÃ, B. In-vitro cytotoxic effect of 64Cu/NOTA-terpyridine platinum conjugate, as a novel theranostic agent. J. Nucl. Med. 2019, 60 (Suppl. 1), 1615. [Google Scholar]
- Vultos, F.; Fernandes, C.; Mendes, F.; Marques, F.; Correia, J.D.G.; Santos, I.; Gano, L. A Multifunctional Radiotheranostic Agent for Dual Targeting of Breast Cancer Cells. ChemMedChem 2017, 12, 1103–1107. [Google Scholar] [CrossRef]
- Reissig, F.; Mamat, C.; Steinbach, J.; Pietzsch, H.-J.; Freudenberg, R.; Navarro-Retamal, C.; Caballero, J.; Kotzerke, J.; Wunderlich, G. Direct and auger electron-induced, single- and double-strand breaks on plasmid DNA caused by 99mTc-labeled pyrene derivatives and the effect of bonding distance. PLoS ONE 2016, 11, e0161973. [Google Scholar] [CrossRef]
- Balagurumoorthy, P.; Xu, X.; Wang, K.; Adelstein, S.J.; Kassis, A.I. Effect of distance between decaying125I and DNA on Auger-electron induced double-strand break yield. Int. J. Radiat. Biol. 2012, 88, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Imstepf, S.; Pierroz, V.; Raposinho, P.; Bauwens, M.; Felber, M.; Fox, T.; Shapiro, A.B.; Freudenberg, R.; Fernandes, C.; Gama, S.; et al. Nuclear targeting with an auger electron emitter potentiates the action of a widely used antineoplastic drug. Bioconjugate Chem. 2015, 26, 2397–2407. [Google Scholar] [CrossRef]
- Chung, W.-J.; Cui, Y.; Huang, F.-Y.J.; Tu, T.-H.; Yang, T.-S.; Lo, J.-M.; Chiang, C.-S.; Hsu, I.C. 99mTc pyrene derivative complex causes double-strand breaks in dsDNA mainly through cluster-mediated indirect effect in aqueous solution. PLoS ONE 2014, 9, e108162. [Google Scholar] [CrossRef]
- Haefliger, P.; Agorastos, N.; Renard, A.; Giambonini-Brugnoli, G.; Marty, C.; Alberto, R. Cell uptake and radiotoxicity studies of an nuclear localization signal peptide−intercalator conjugate labeled with [99mTc(CO)3]+. Bioconjugate Chem. 2005, 16, 582–587. [Google Scholar] [CrossRef]
- Gardette, M.; Papon, J.; Bonnet, M.; Desbois, N.; Labarre, P.; Wu, T.-D.; Miot-Noirault, E.; Madelmont, J.-C.; Guerquin-Kern, J.-L.; Chezal, J.-M.; et al. Evaluation of new iodinated acridine derivatives for targeted radionuclide therapy of melanoma using 125I, an Auger electron emitter. Investig. New Drugs 2010, 29, 1253–1263. [Google Scholar] [CrossRef]
- Gill, M.R.; Walker, M.G.; Able, S.; Tietz, O.; Lakshminarayanan, A.; Anderson, R.; Chalk, R.; El-Sagheer, A.H.; Brown, T.; Thomas, J.A.; et al. An 111In-labelled bis-ruthenium(ii) dipyridophenazine theranostic complex: Mismatch DNA binding and selective radiotoxicity towards MMR-deficient cancer cells. Chem. Sci. 2020, 11, 8936–8944. [Google Scholar] [CrossRef]
- Gama, S.; Mendes, F.; Esteves, T.; Marques, F.; Matos, A.; Rino, J.; Coimbra, J.; Ravera, M.; Gabano, E.; Santos, I.; et al. Synthesis and Biological Studies of Pyrazolyl-Diamine PtIIComplexes Containing Polyaromatic DNA-Binding Groups. ChemBioChem 2012, 13, 2352–2362. [Google Scholar] [CrossRef]
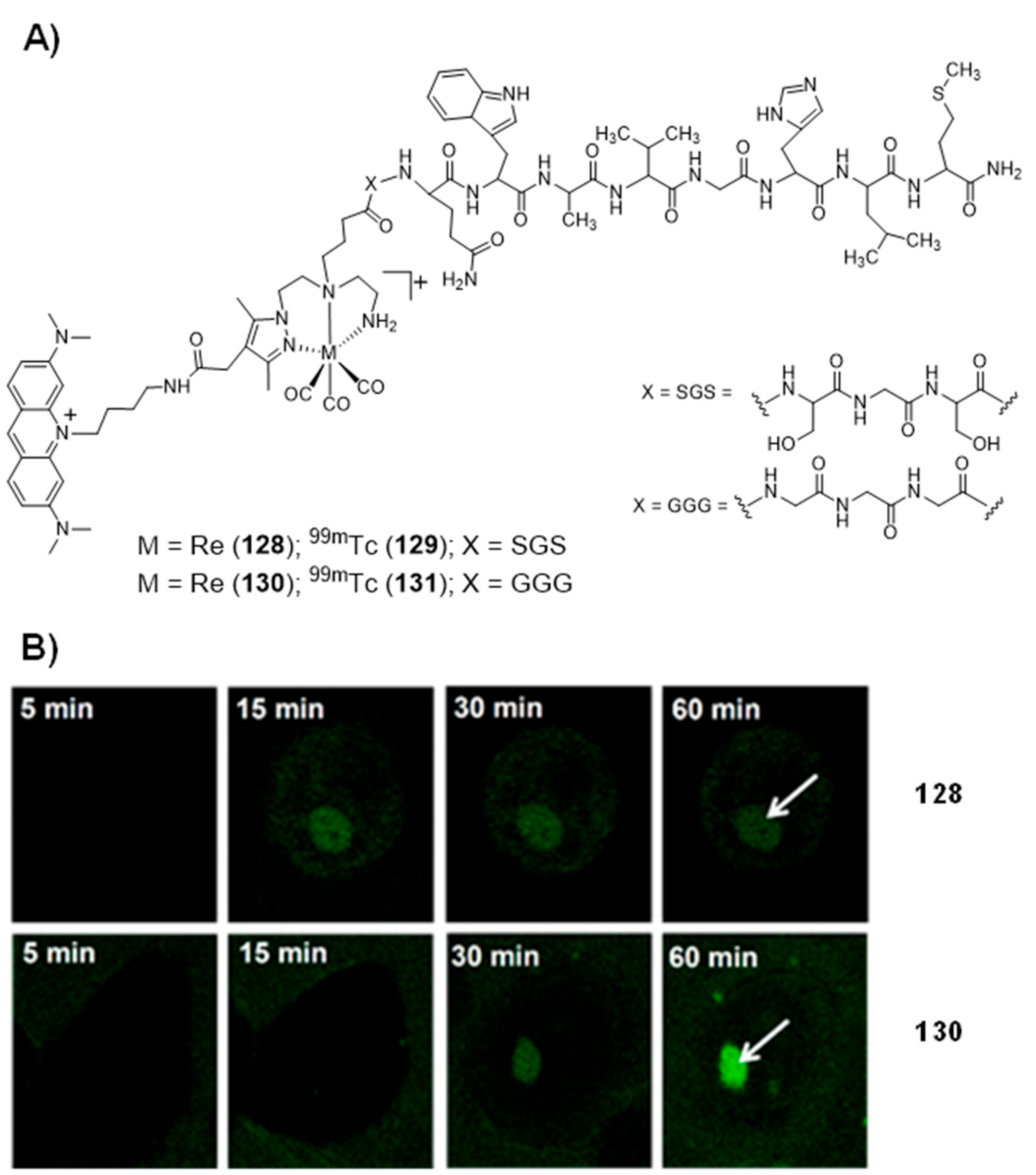
- Esteves, T.; Marques, F.; Paulo, A.; Rino, J.; Nanda, P.; Smith, C.J.; Santos, I. Nuclear targeting with cell-specific multifunctional tricarbonyl M(I) (M is Re, 99mTc) complexes: Synthesis, characterization, and cell studies. JBIC J. Biol. Inorg. Chem. 2011, 16, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Esteves, T.; Xavier, C.; Gama, S.; Mendes, F.; Raposinho, P.; Marques, F.; Paulo, A.; Pessoa, J.C.; Rino, J.; Viola, G.; et al. Tricarbonyl M(I) (M = Re, 99mTc) complexes bearing acridine fluorophores: Synthesis, characterization, DNA interaction studies and nuclear targeting. Org. Biomol. Chem. 2010, 8, 4104–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, E.; Quental, L.D.; Palma, E.; Oliveira, M.C.; Mendes, F.; Raposinho, P.; Correia, I.; Lavrado, J.; Di Maria, S.; Belchior, A.; et al. Evaluation of Acridine Orange Derivatives as DNA-Targeted Radiopharmaceuticals for Auger Therapy: Influence of the Radionuclide and Distance to DNA. Sci. Rep. 2017, 7, srep42544. [Google Scholar] [CrossRef] [PubMed]
- Belchior, A.; Di Maria, S.; Fernandes, C.; Vaz, P.; Paulo, A.; Raposinho, P. Radiobiological and dosimetric assessment of DNA-intercalated 99mTc-complexes bearing acridine orange derivatives. EJNMMI Res. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Carvalho, J.; Pereira, E.; Marquevielle, J.; Campello, M.P.C.; Mergny, J.-L.; Paulo, A.; Salgado, G.F.; Queiroz, J.; Cruz, C. Fluorescent light-up acridine orange derivatives bind and stabilize KRAS-22RT G-quadruplex. Biochimie 2018, 144, 144–152. [Google Scholar] [CrossRef]
- Carvalho, J.; Lopes-Nunes, J.; Campello, M.P.C.; Paulo, A.; Milici, J.; Meyers, C.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. Human Papillomavirus G-Rich Regions as Potential Antiviral Drug Targets. Nucleic Acid Ther. 2021, 31, 68–81. [Google Scholar] [CrossRef]
- Carvalho, J.; Paiva, A.; Campello, M.P.C.; Paulo, A.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.; Cruz, C. Aptamer-based Targeted Delivery of a G-quadruplex Ligand in Cervical Cancer Cells. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, J.; Nunes, J.L.; Lopes, A.C.; Campello, M.P.C.; Paulo, A.R.; Queiroz, J.A.; Cruz, C. Aptamer-guided acridine derivatives for cervical cancer. Org. Biomol. Chem. 2019, 17, 2992–3002. [Google Scholar] [CrossRef]
- Figueiredo, J.P.R.; Nunes, J.; Carvalho, J.; Antunes, F.; Ribeiro, M.; Campello, M.P.C.; Paulo, A.; Paiva, A.; Salgado, G.F.; Queiroz, J.; et al. AS1411 derivatives as carriers of G-quadruplex ligands for cervical cancer cells. Int. J. Pharm. 2019, 568, 118511. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.; Pereira, P.; Campello, M.P.C.; Paulo, A.; Queiroz, J.; Cabrita, E.; Cruz, C. RNA G-quadruplex as supramolecular carrier for cancer-selective delivery. Eur. J. Pharm. Biopharm. 2019, 142, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, D.; Fernandes, C.; Silva, F.; Palma, E.; Raposinho, P.; Belchior, A.; Vaz, P.; Paulo, A. Synthesis and Biological Evaluation of 99mTc(I) Tricarbonyl Complexes Dual-Targeted at Tumoral Mitochondria. Molecules 2021, 26, 441. [Google Scholar] [CrossRef] [PubMed]






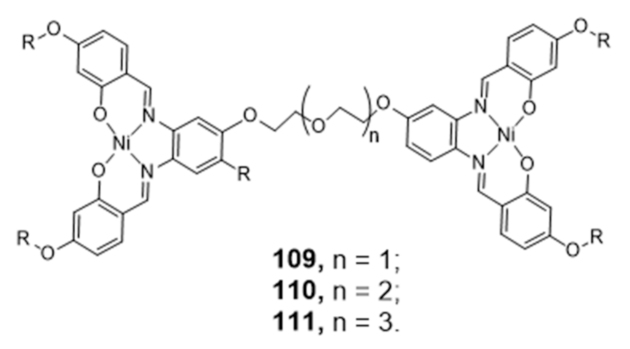

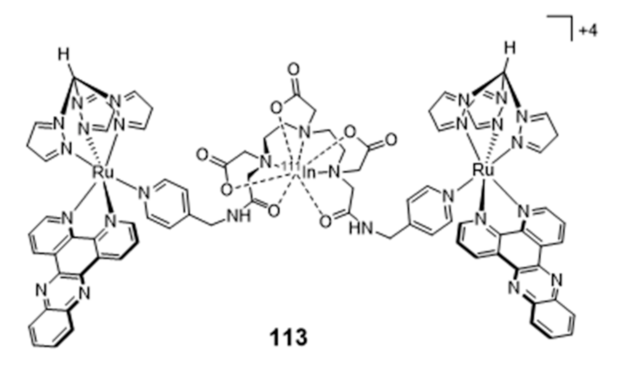
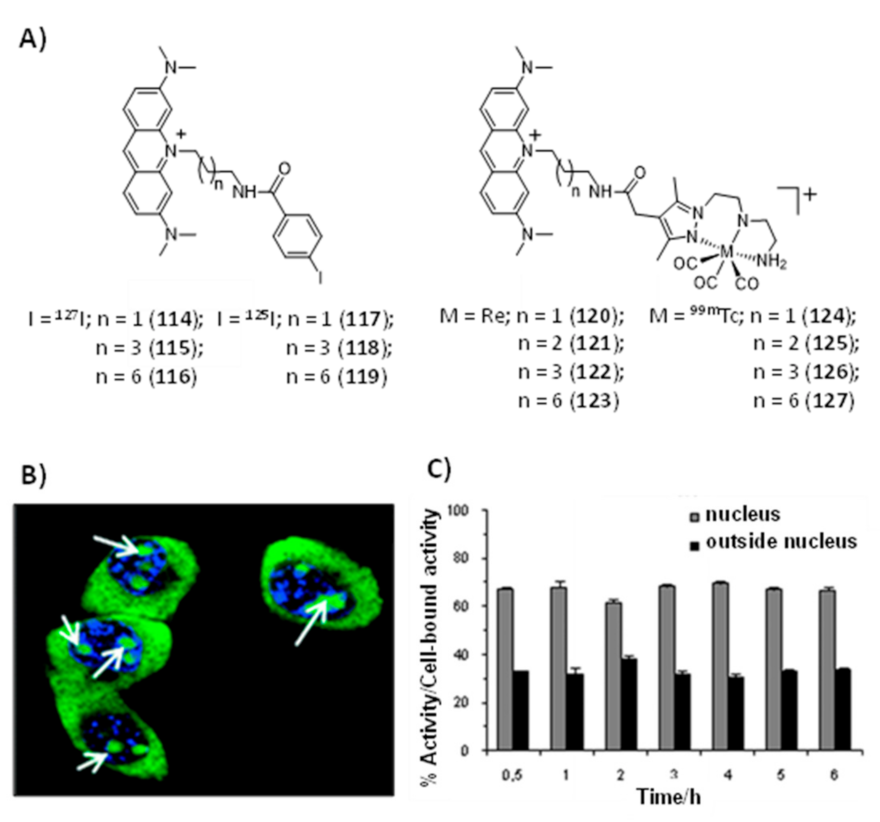
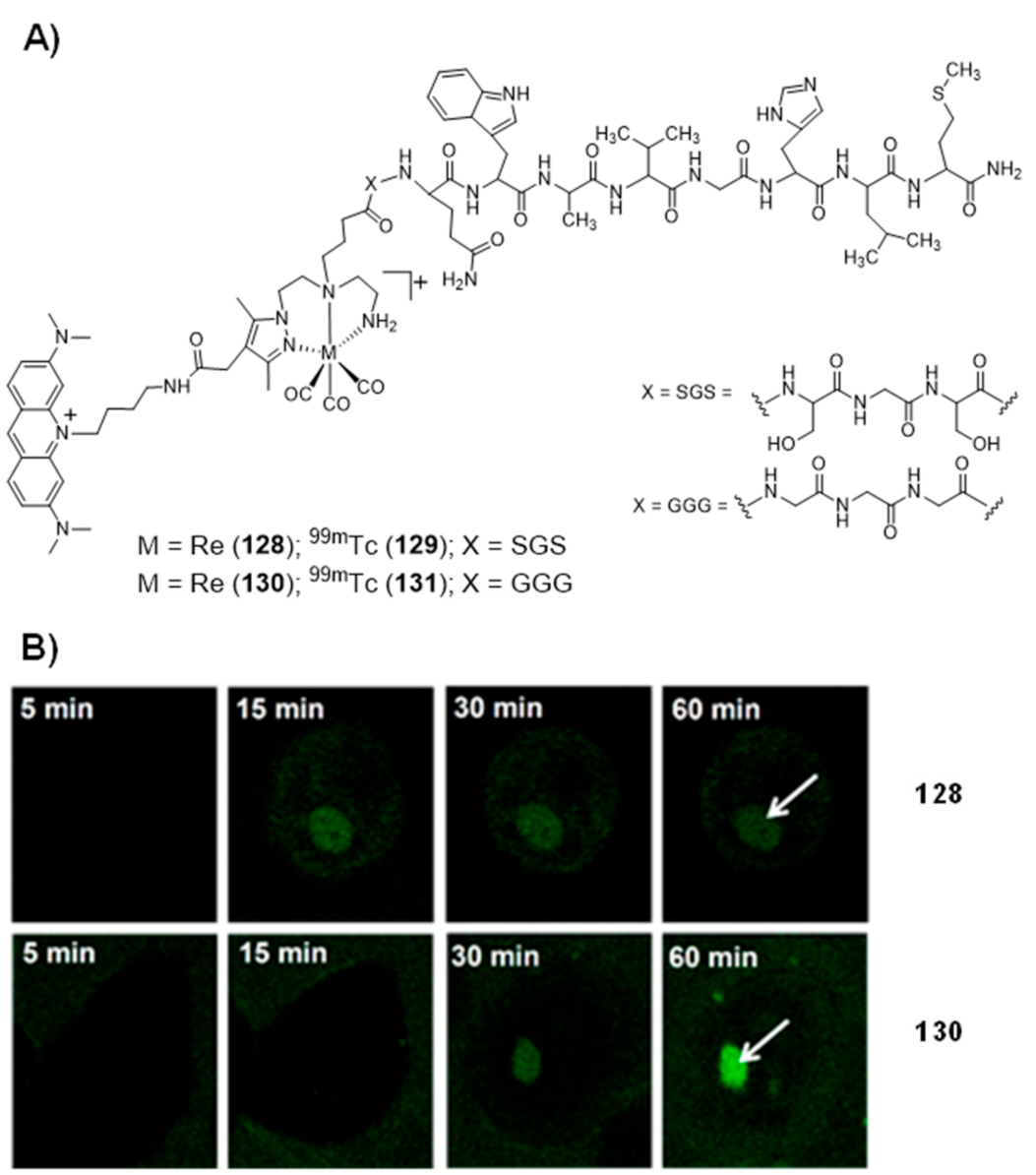
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma, E.; Carvalho, J.; Cruz, C.; Paulo, A. Metal-Based G-Quadruplex Binders for Cancer Theranostics. Pharmaceuticals 2021, 14, 605. https://doi.org/10.3390/ph14070605
Palma E, Carvalho J, Cruz C, Paulo A. Metal-Based G-Quadruplex Binders for Cancer Theranostics. Pharmaceuticals. 2021; 14(7):605. https://doi.org/10.3390/ph14070605
Chicago/Turabian StylePalma, Elisa, Josué Carvalho, Carla Cruz, and António Paulo. 2021. "Metal-Based G-Quadruplex Binders for Cancer Theranostics" Pharmaceuticals 14, no. 7: 605. https://doi.org/10.3390/ph14070605
APA StylePalma, E., Carvalho, J., Cruz, C., & Paulo, A. (2021). Metal-Based G-Quadruplex Binders for Cancer Theranostics. Pharmaceuticals, 14(7), 605. https://doi.org/10.3390/ph14070605