
A Radiobrominated Tyrosine Kinase Inhibitor for EGFR with L858R/T790M Mutations in Lung Carcinoma

 ,
,  ,
, 

Abstract
1. Introduction
2. Results
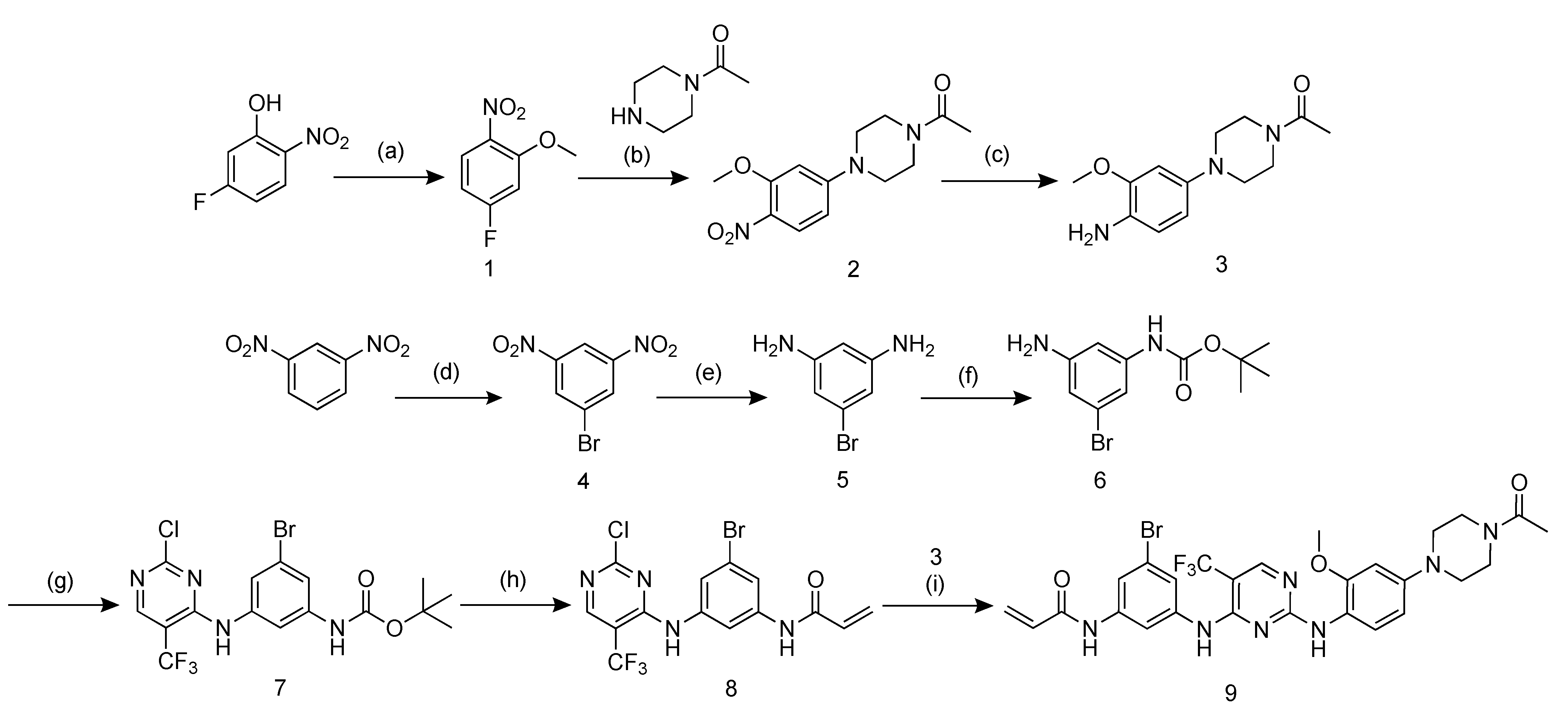
2.1. Probe Design and Synthesis
2.2. Cytotoxicity Assays
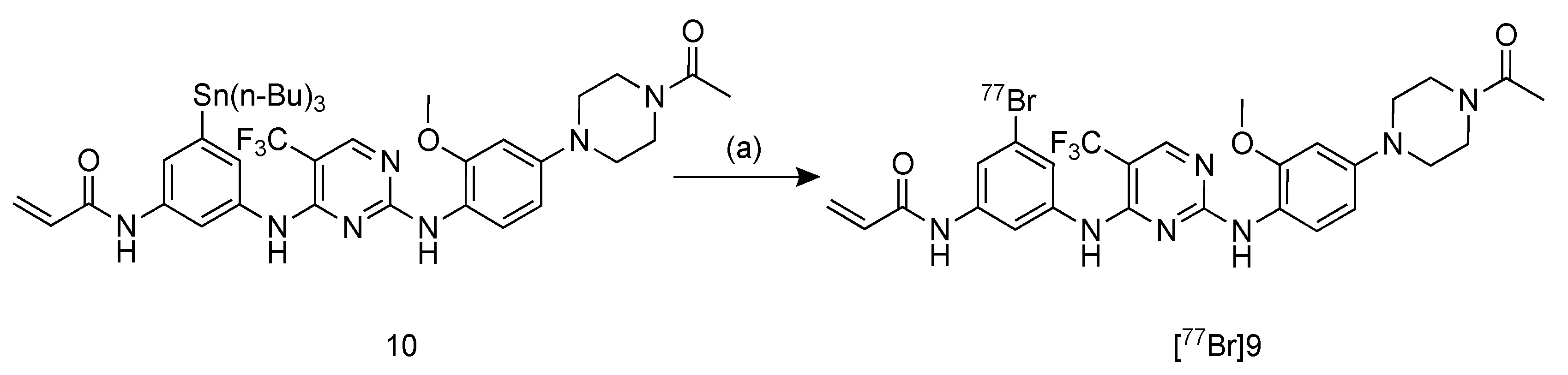
2.3. Radiosynthesis of [77Br]BrCO1686 ([77Br]9)
2.4. Determination of Partition Coefficient
2.5. Stability Assessment
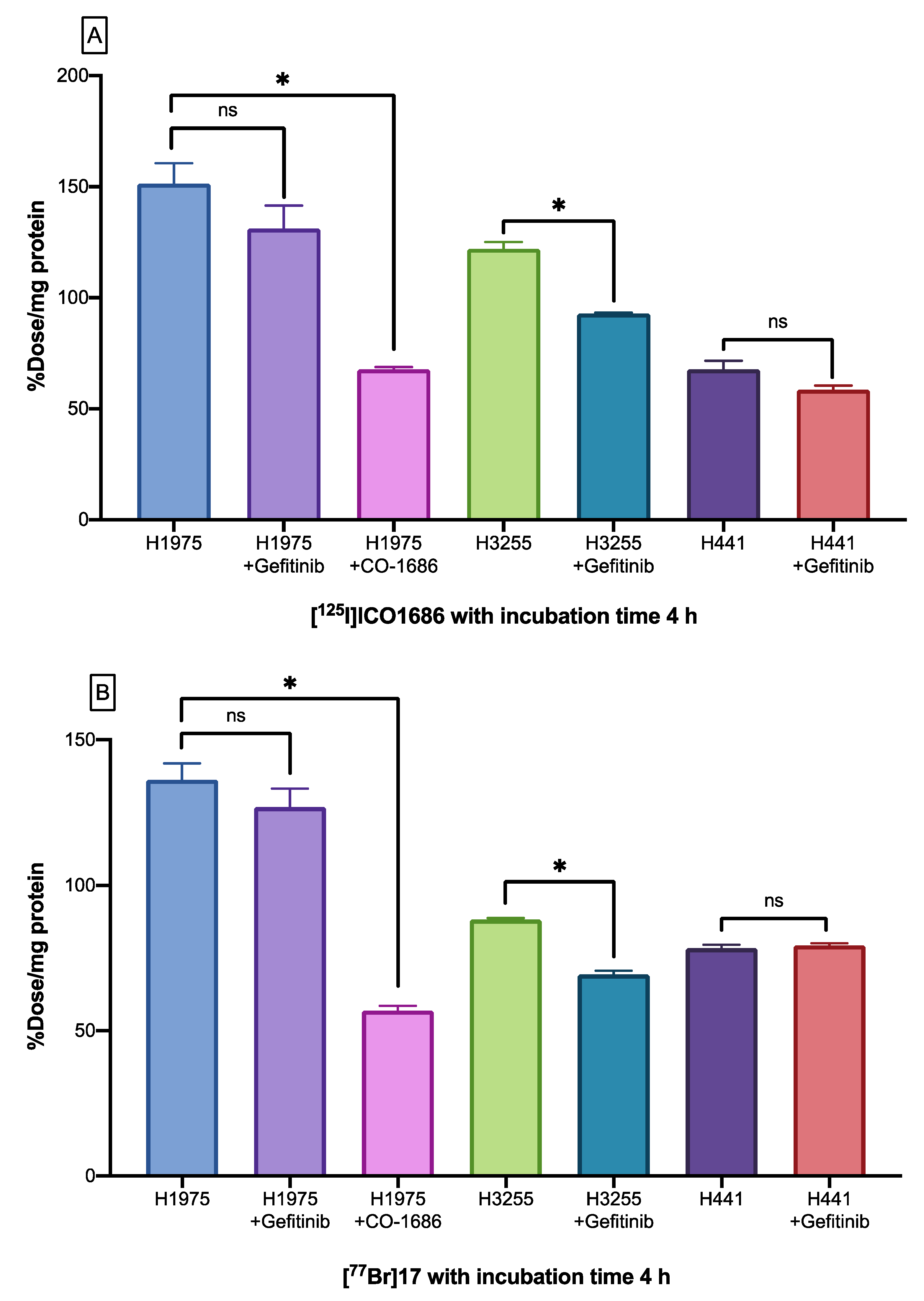
2.6. Cellular Uptake Studies
2.7. Biodistribution Studies
3. Discussion
4. Material and Methods
4.1. General Chemistry
4.2. Probe Design and Synthesis
4.2.1. Synthesis of 1-bromo-3,5-dinitrobenzene (4)
4.2.2. Synthesis of 5-bromobenzene-1,3-diamine (5)
4.2.3. Synthesis of tert-butyl (3-amino-5-bromophenyl)carbamate (6)
4.2.4. Synthesis of tert-butyl(3-{[2-chloro-5-(trifluoromethyl)pyrimidin-4-yl]amino}-5- bromophenyl)carbamate (7)
4.2.5. Synthesis of N-(3-{[2-chloro-5-(trifluoromethyl)pyrimidin-4-yl]amino}-5- bromophenyl)acrylamide (8)
4.2.6. Synthesis of N-(3-{[2-({4-[4-acetylpiperazin-1-yl]-2-methoxyphenyl}amino)-5- (trifluoromethyl)pyrimidin-4-yl]amino}-5-bromophenyl)acrylamide (9)
4.3. Cytotoxicity Assays
4.4. Production of Bromine-77
4.5. Radiosynthesis of [77Br]BrCO1686 ([77Br]9)
4.6. Determination of Partition Coefficient
4.7. Stability Assessment
4.8. Cellular Uptake Studies
4.9. Tumor Model
4.10. Biodistribution Studies
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, R.; Dastane, A.M.; Forozan, F.; Riley-Portuguez, A.; Chung, F.; Lopategui, J.; Marchevsky, A.M. Evaluation of EGFR abnormalities in patients with pulmonary adenocarcinoma: The need to test neoplasms with more than one method. Mod. Pathol. 2009, 22, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Altine, B.; Gai, Y.; Han, N.; Jiang, Y.; Ji, H.; Fang, H.; Niyonkuru, A.; Bakari, K.H.; Rajab Arnous, M.M.; Liu, Q.; et al. Preclinical Evaluation of a Fluorine-18 18F-Labeled Phosphatidylinositol 3-Kinase Inhibitor for Breast Cancer Imaging. Mol. Pharm. 2019, 16, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Ogawa, K.; Balatoni, J.; Mukhapadhyay, U.; Pal, A.; Gonzalez-Lepera, C.; Shavrin, A.; Soghomonyan, S.; Flores, L., 2nd; Young, D.; et al. Molecular imaging of active mutant L858R EGF receptor (EGFR) kinase-expressing nonsmall cell lung carcinomas using PET/CT. Proc. Natl. Acad. Sci. USA 2011, 108, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Balatoni, J.A.; Mukhopadhyay, U.; Ogawa, K.; Gonzalez-Lepera, C.; Shavrin, A.; Volgin, A.; Tong, W.; Alauddin, M.M.; Gelovani, J.G. Radiosynthesis and initial in vitro evaluation of [18F]F-PEG6-IPQA--a novel PET radiotracer for imaging EGFR expression-activity in lung carcinomas. Mol. Imaging Biol. 2011, 13, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, I.; Planchard, D. Next-Generation EGFR Tyrosine Kinase Inhibitors for Treating EGFR-Mutant Lung Cancer beyond First Line. Front. Med. (Lausanne) 2016, 3, 76. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Jadhav, H.R. Targeting non-small cell lung cancer with small-molecule EGFR tyrosine kinase inhibitors. Drug Discov. Today 2018, 23, 745–753. [Google Scholar] [CrossRef]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [PubMed]
- Fawwaz, M.; Mishiro, K.; Nishii, R.; Sawazaki, I.; Shiba, K.; Kinuya, S.; Ogawa, K. Synthesis and Fundamental Evaluation of Radioiodinated Rociletinib (CO-1686) as a Probe to Lung Cancer with L858R/T790M Mutations of Epidermal Growth Factor Receptor (EGFR). Molecules 2020, 25, 2914. [Google Scholar] [CrossRef]
- Zhou, D.; Kim, S.H.; Chu, W.; Voller, T.; Katzenellenbogen, J.A. Evaluation of aromatic radiobromination by nucleophilic substitution using diaryliodonium salt precursors. J. Label. Comp. Radiopharm. 2017, 60, 450–456. [Google Scholar] [CrossRef]
- Yan, X.E.; Zhu, S.J.; Liang, L.; Zhao, P.; Choi, H.G.; Yun, C.H. Structural basis of mutant-selectivity and drug-resistance related to CO-1686. Oncotarget 2017, 8, 53508–53517. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xiao, Z.; Wang, K.; Wang, X.; Zhang, C.; Fang, F.; Sun, X.; Shen, B. Development and Evaluation of 18F-IRS for Molecular Imaging Mutant EGF Receptors in NSCLC. Sci. Rep. 2017, 7, 3121. [Google Scholar] [CrossRef]
- Chen, W.; Shen, B.; Sun, X. Analysis of Progress and Challenges of EGFR-Targeted Molecular Imaging in Cancer With a Focus on Affibody Molecules. Mol. Imaging 2019, 18, 1536012118823473. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, H.; Ohshima, Y.; Suzuki, Y.; Yamaguchi, A.; Watanabe, S.; Uehara, T.; Nagamori, S.; Kanai, Y.; Ishioka, N.S.; Tsushima, Y.; et al. Development of a Widely Usable Amino Acid Tracer: 76Br-alpha-Methyl-Phenylalanine for Tumor PET Imaging. J. Nucl. Med. 2015, 56, 791–797. [Google Scholar] [CrossRef]
- Su, H.; Seimbille, Y.; Ferl, G.Z.; Bodenstein, C.; Fueger, B.; Kim, K.J.; Hsu, Y.T.; Dubinett, S.M.; Phelps, M.E.; Czernin, J.; et al. Evaluation of [18F]gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1089–1099. [Google Scholar] [CrossRef]
- Guo, A.; Villen, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci. USA 2008, 105, 692–697. [Google Scholar] [CrossRef]
- Bruehlmeier, M.; Roelcke, U.; Bläuenstein, P.; Missimer, J.; Schubiger, P.A.; Locher, J.T.; Pellikka, R.; Ametamey, S.M. Measurement of the extracellular space in brain tumors using 76Br-bromide and PET. J. Nucl. Med. 2003, 44, 1210–1218. [Google Scholar] [PubMed]
- Rossin, R.; Berndorff, D.; Friebe, M.; Dinkelborg, L.M.; Welch, M.J. Small-animal PET of tumor angiogenesis using a (76)Br-labeled human recombinant antibody fragment to the ED-B domain of fibronectin. J. Nucl. Med. 2007, 48, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Finck, B.N.; Jones, L.A.; Welch, M.J.; Mach, R.H. Synthesis and evaluation of a bromine-76-labeled PPARgamma antagonist 2-bromo-5-nitro-N-phenylbenzamide. Nucl. Med. Biol. 2006, 33, 847–854. [Google Scholar] [CrossRef]
- Ogawa, K.; Masuda, R.; Mizuno, Y.; Makino, A.; Kozaka, T.; Kitamura, Y.; Kiyono, Y.; Shiba, K.; Odani, A. Development of a novel radiobromine-labeled sigma-1 receptor imaging probe. Nucl. Med. Biol. 2018, 61, 28–35. [Google Scholar] [CrossRef]
- Ohshima, Y.; Hanaoka, H.; Watanabe, S.; Sugo, Y.; Watanabe, S.; Tominaga, H.; Oriuchi, N.; Endo, K.; Ishioka, N.S. Preparation and biological evaluation of 3-[76Br]bromo-α-methyl-l-tyrosine, a novel tyrosine analog for positron emission tomography imaging of tumors. Nucl. Med. Biol. 2011, 38, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Windhorst, A.D.; Walsum, M.S.-v.; Schuit, R.C.; Smit, E.F.; Niessen, H.G.; Solca, F.; Stehle, G.; van Dongen, G.A.M.S.; Poot, A.J. Development of [18F]afatinib as new TKI-PET tracer for EGFR positive tumors. Nucl. Med. Biol. 2014, 41, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Takeda, T.; Yokokawa, M.; Yu, J.; Makino, A.; Kiyono, Y.; Shiba, K.; Kinuya, S.; Odani, A. Comparison of Radioiodine- or Radiobromine-Labeled RGD Peptides between Direct and Indirect Labeling Methods. Chem. Pharm. Bull. (Tokyo) 2018, 66, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yuan, L.; Yin, L.; Jiang, Y.; Gai, Y.; Liu, Q.; Wang, Y.; Zhang, Y.; Lan, X. Synthesis and Preclinical Evaluation of 18F-PEG3-FPN for the Detection of Metastatic Pigmented Melanoma. Mol. Pharm. 2017, 14, 3896–3905. [Google Scholar] [CrossRef]
- Lux, J.; Chan, M.; Elst, L.V.; Schopf, E.; Mahmoud, E.; Laurent, S.; Almutairi, A. Metal Chelating Crosslinkers Form Nanogels with High Chelation Stability. J. Mater. Chem. B 2013, 1, 6359–6364. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Koura, M.; Yamaguchi, Y.; Kurobuchi, S.; Sumida, H.; Watanabe, Y.; Enomoto, T.; Matsuda, T.; Okuda, A.; Koshizawa, T.; Matsumoto, Y.; et al. Discovery of a 2-hydroxyacetophenone derivative as an outstanding linker to enhance potency and β-selectivity of liver X receptor agonist. Bioorganic Med. Chem. 2016, 24, 3436–3446. [Google Scholar] [CrossRef]
- Fischer, C.; Sparr, C. Synthesis of 1,5-bifunctional organolithium reagents by a double directed ortho-metalation: Direct transformation of esters into 1,8-dimethoxy-acridinium salts. Tetrahedron 2018, 74, 5486–5493. [Google Scholar] [CrossRef]
- Michael, D.A.; Brian, M.A.; Kenneth, L.A.; Sathesh, B.; Jason, B.; Kaleen, K.C.; Bernard, C.; Maria, E. Aminopyrimidines as syk inhibitors. U.S. Patent 2011/0245205 A1, 6 October 2011. [Google Scholar]
- Ogawa, K.; Kanbara, H.; Kiyono, Y.; Kitamura, Y.; Kiwada, T.; Kozaka, T.; Kitamura, M.; Mori, T.; Shiba, K.; Odani, A. Development and evaluation of a radiobromine-labeled sigma ligand for tumor imaging. Nucl. Med. Biol. 2013, 40, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Shiba, K.; Akhter, N.; Yoshimoto, M.; Washiyama, K.; Kinuya, S.; Kawai, K.; Mori, H. Evaluation of radioiodinated vesamicol analogs for sigma receptor imaging in tumor and radionuclide receptor therapy. Cancer Sci. 2009, 100, 2188–2192. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cells Lines | Mutation Status | IC50 (μM) | |||
|---|---|---|---|---|---|
| 9 | ICO1686 * | CO-1686 * | Gefitinib * | ||
| H1975 | L858R/T790M | 0.18 ± 0.06 | 0.20 ± 0.05 | 0.14 ± 0.05 | >10 |
| H3255 | L858R | 0.20 ± 0.01 | 0.50 ± 0.21 | 0.15 ± 0.02 | 0.02 ± 0.02 |
| H441 | Wild type | 0.64 ± 0.04 | 1.84 ± 0.44 | 0.26 ± 0.04 | >10 |
| Tissues | Time after Injection | |||||||
|---|---|---|---|---|---|---|---|---|
| 10 min | 1 h | 4 h | 24 h | |||||
| [77Br]9 | ||||||||
| Blood | 3.57 | (0.21) | 3.24 | (0.30) | 3.19 | (0.18) | 1.99 | (0.28) |
| Liver | 11.04 | (0.55) | 3.83 | (0.38) | 3.72 | (0.30) | 1.04 | (0.21) |
| Kidney | 11.68 | (0.94) | 4.21 | (0.67) | 2.81 | (1.17) | 1.33 | (0.25) |
| Small intestine | 25.38 | (5.45) | 26.49 | (2.77) | 5.23 | (1.06) | 1.00 | (0.15) |
| Large intestine | 2.83 | (0.57) | 3.18 | (0.19) | 47.73 | (7.88) | 0.98 | (0.11) |
| Spleen | 5.11 | (3.46) | 4.83 | (1.82) | 2.92 | (2.02 | 1.14 | (0.46) |
| Pancreas | 3.78 | (2.53) | 3.58 | (0.10) | 3.04 | (0.28) | 1.13 | (0.09) |
| Lung | 8.72 | (1.25) | 4.97 | (1.97) | 5.08 | (0.28) | 1.62 | (0.22) |
| Heart | 4.57 | (0.89) | 2.93 | (0.24) | 1.87 | (1.25) | 0.80 | (0.21) |
| Stomach ‡ | 6.17 | (1.45) | 7.00 | (0.58) | 6.68 | (1.02) | 2.21 | (0.33) |
| Bone | 2.76 | (0.83) | 1.66 | (1.18) | 1.54 | (0.62) | 0.89 | (0.93) |
| Muscle | 3.18 | (0.55) | 2.02 | (0.55) | 1.13 | (0.78) | 0.47 | (0.20) |
| Brain | 0.45 | (0.30) | 0.95 | (0.14) | 0.74 | (0.50) | 0.42 | (0.09) |
| Urine | - | - | - | 13.48 | (1.28) | |||
| Feces | - | - | - | 35.01 | (6.85) | |||
| [125I]ICO1686 | ||||||||
| Blood | 1.41 | (0.38) | 0.17 | (0.02) | 0.06 | (0.01) | 0.13 | (0.06) |
| Liver | 19.21 | (2.04) | 7.40 | (1.01) | 6.45 | (1.21) | 1.40 | (0.17) |
| Kidney | 9.66 | (1.19) | 2.40 | (0.56) | 0.75 | (0.18) | 0.54 | (0.10) |
| Small intestine | 36.32 | (12.00) | 40.99 | (4.13) | 6.08 | (1.99) | 0.30 | (0.11) |
| Large intestine | 0.94 | (0.18) | 1.65 | (0.34) | 81.54 | (10.19) | 0.52 | (0.55) |
| Spleen | 2.73 | (0.56) | 0.55 | (0.09) | 0.20 | (0.07) | 0.13 | (0.04) |
| Pancreas | 2.03 | (0.20) | 0.86 | (0.15) | 0.12 | (0.04) | 0.05 | (0.01) |
| Lung | 3.61 | (0.40) | 0.59 | (0.25) | 0.16 | (0.03) | 0.14 | (0.02) |
| Heart | 2.24 | (0.49) | 0.30 | (0.08) | 0.08 | (0.04) | 0.20 | (0.25) |
| Stomach ‡ | 2.60 | (1.23) | 1.64 | (0.71) | 0.76 | (0.61) | 0.05 | (0.02) |
| Bone | 0.74 | (0.19) | 0.16 | (0.03) | 0.05 | (0.08) | 0.09 | (0.05) |
| Muscle | 1.20 | (0.14) | 0.23 | (0.09) | 0.04 | (0.01) | 0.04 | (0.02) |
| Brain | 0.08 | (0.01) | 0.01 | (0.01) | 0.01 | (0.00) | 0.00 | (0.00) |
| Urine ‡ | - | - | - | 1.63 | (0.24) | |||
| Feces ‡ | - | - | - | 71.54 | (6.53) | |||
| Tissues | Time after Injection | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 h | 6 h | 1 h | 6 h | |||||
| [77Br]9 | [125I]ICO1686 | |||||||
| Blood | 5.64 | (0.19) | 5.35 | (0.99) | 0.50 | (0.20) | 0.17 | (0.03) |
| Liver | 15.89 | (0.72) | 6.76 | (1.78) | 30.32 | (2.78) | 10.16 | (3.50) |
| Kidney | 7.75 | (0.85) | 4.91 | (0.80) | 2.64 | (1.45) | 1.05 | (0.41) |
| Small intestine | 64.52 | (3.35) | 12.46 | (5.93) | 86.29 | (2.99) | 11.72 | (8.01) |
| Large intestine | 24.33 | (17.81) | 59.40 | (18.22) | 27.65 | (21.75) | 95.22 | (32.67) |
| Spleen | 9.19 | (2.12) | 2.04 | (2.36) | 1.80 | (0.75) | 0.25 | (0.07) |
| Pancreas | 12.95 | (5.14) | 4.28 | (0.91) | 2.71 | (0.87) | 0.36 | (0.22) |
| Lung | 11.42 | (1.52) | 6.29 | (0.52) | 1.62 | (0.67) | 0.37 | (0.15) |
| Heart | 5.36 | (1.07) | 2.99 | (1.05) | 0.61 | (0.20) | 0.53 | (0.86) |
| Stomach ‡ | 1.31 | (0.39) | 1.27 | (0.89) | 1.49 | (0.52) | 1.17 | (1.68) |
| Bone | 1.93 | (1.51) | 1.95 | (2.50) | 0.42 | (0.16) | 0.22 | (0.15) |
| Muscle | 3.42 | (1.19) | 1.79 | (0.30) | 0.36 | (0.18) | 0.10 | (0.05) |
| Brain | 1.49 | (0.16) | 1.39 | (0.17) | 0.04 | (0.01) | 0.02 | (0.03) |
| H1975 | 4.51 | (0.17) | 3.48 | (0.50) | 0.68 | (0.11) | 0.10 | (0.02) |
| H441 | 3.71 | (0.13) | 3.30 | (0.54) | 0.44 | (0.06) | 0.19 | (0.06) |
| Tissues | Time after Injection | |||||||
|---|---|---|---|---|---|---|---|---|
| 15 min | 1 h | 4 h | 24 h | |||||
| Blood | 6.96 | (0.58) | 6.98 | (0.45) | 6.42 | (0.64) | 3.48 | (0.36) |
| Liver | 2.45 | (0.29) | 2.61 | (0.17) | 2.35 | (0.30) | 1.18 | (0.12) |
| Kidney | 5.10 | (0.83) | 4.58 | (0.21) | 4.51 | (0.58) | 2.62 | (0.34) |
| Small intestine | 3.94 | (0.24) | 3.92 | (0.16) | 3.18 | (0.29) | 1.99 | (0.22) |
| Large intestine | 3.12 | (0.44) | 2.78 | (0.22) | 2.37 | (0.30) | 1.55 | (0.16) |
| Spleen | 4.74 | (0.31) | 4.30 | (0.26) | 3.75 | (0.73) | 2.25 | (0.36) |
| Pancreas | 5.09 | (0.78) | 4.38 | (0.27) | 3.80 | (0.37) | 2.22 | (0.32) |
| Lung | 8.02 | (0.95) | 7.12 | (0.48) | 5.81 | (0.59) | 3.62 | (0.32) |
| Heart | 3.72 | (0.47) | 3.23 | (0.09) | 2.41 | (0.20) | 1.50 | (0.19) |
| Stomach ‡ | 3.94 | (0.54) | 4.59 | (1.18) | 5.44 | (1.23) | 4.18 | (1.00) |
| Bone | 3.72 | (0.44) | 3.25 | (0.24) | 3.00 | (0.38) | 1.78 | (0.30) |
| Muscle | 2.28 | (0.18) | 1.92 | (0.11) | 1.63 | (0.14) | 0.98 | (0.04) |
| Brain | 1.08 | (0.13) | 1.49 | (0.07) | 1.45 | (0.14) | 0.84 | (0.09) |
| Neck ‡ | 2.45 | (0.50) | 3.02 | (0.35) | 2.46 | (0.42) | 1.27 | (0.28) |
| Urine ‡ | - | - | - | 20.74 | (4.69) | |||
| Feces ‡ | - | - | - | 1.33 | (0.49) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fawwaz, M.; Mishiro, K.; Nishii, R.; Makino, A.; Kiyono, Y.; Shiba, K.; Kinuya, S.; Ogawa, K. A Radiobrominated Tyrosine Kinase Inhibitor for EGFR with L858R/T790M Mutations in Lung Carcinoma. Pharmaceuticals 2021, 14, 256. https://doi.org/10.3390/ph14030256
Fawwaz M, Mishiro K, Nishii R, Makino A, Kiyono Y, Shiba K, Kinuya S, Ogawa K. A Radiobrominated Tyrosine Kinase Inhibitor for EGFR with L858R/T790M Mutations in Lung Carcinoma. Pharmaceuticals. 2021; 14(3):256. https://doi.org/10.3390/ph14030256
Chicago/Turabian StyleFawwaz, Muammar, Kenji Mishiro, Ryuichi Nishii, Akira Makino, Yasushi Kiyono, Kazuhiro Shiba, Seigo Kinuya, and Kazuma Ogawa. 2021. "A Radiobrominated Tyrosine Kinase Inhibitor for EGFR with L858R/T790M Mutations in Lung Carcinoma" Pharmaceuticals 14, no. 3: 256. https://doi.org/10.3390/ph14030256
APA StyleFawwaz, M., Mishiro, K., Nishii, R., Makino, A., Kiyono, Y., Shiba, K., Kinuya, S., & Ogawa, K. (2021). A Radiobrominated Tyrosine Kinase Inhibitor for EGFR with L858R/T790M Mutations in Lung Carcinoma. Pharmaceuticals, 14(3), 256. https://doi.org/10.3390/ph14030256