
Pharmacogenetics of Carbamazepine and Valproate: Focus on Polymorphisms of Drug Metabolizing Enzymes and Transporters


 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. CBZ and VPA Mechanisms of Action
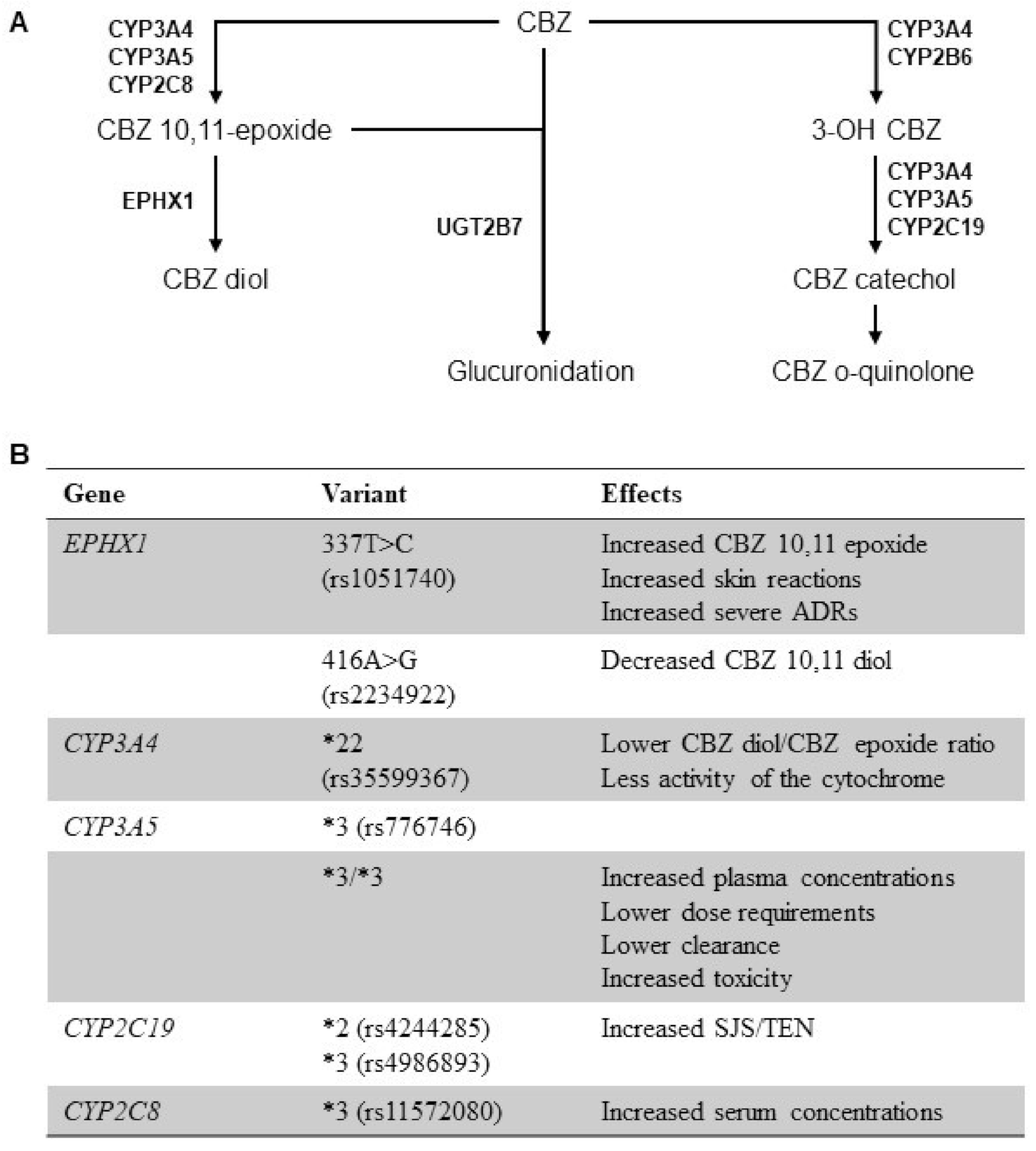
3. Genetic Polymorphisms of Drug Metabolizing Enzymes and CBZ PK
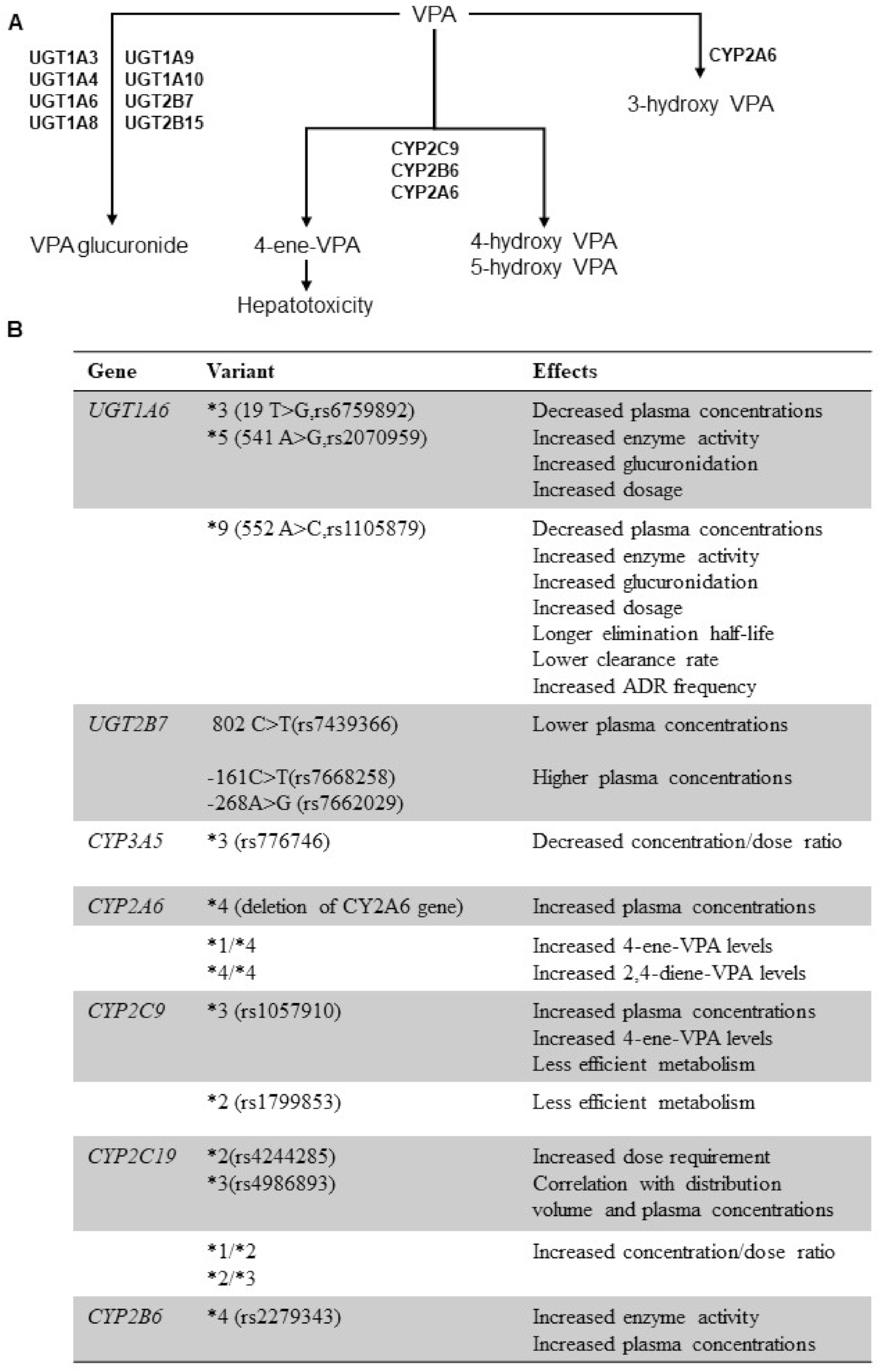
4. Genetic Polymorphisms of Drug Metabolizing Enzymes and VPA PK
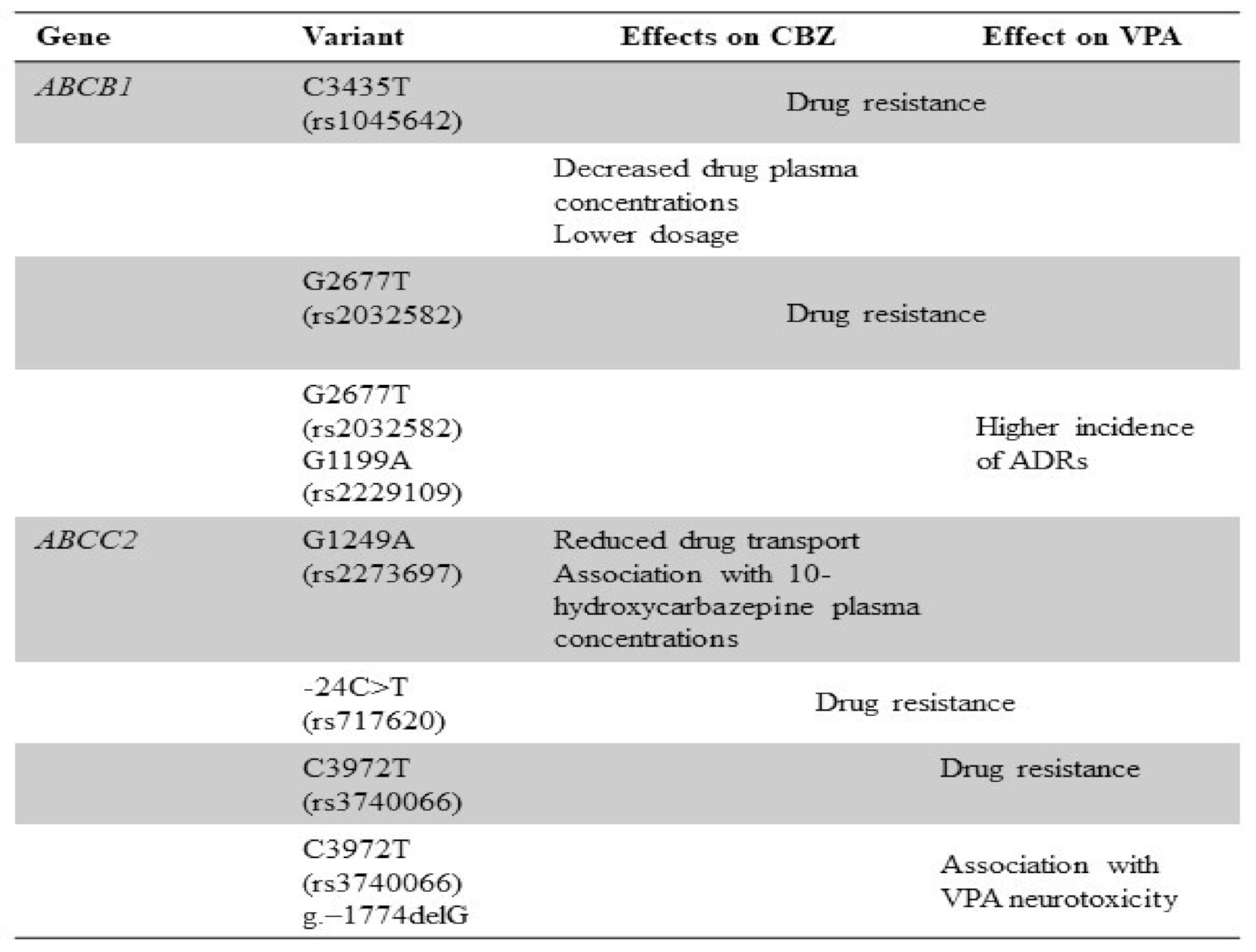
5. Genetic Polymorphisms of Drug Transporters and CBZ/VPA Response
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shim, I.H.; Woo, Y.S.; Kim, M.D.; Bahk, W.M. Antidepressants and Mood Stabilizers: Novel Research Avenues and Clinical Insights for Bipolar Depression. Int. J. Mol. Sci. 2017, 18, 2406. [Google Scholar] [CrossRef]
- Ferrari, A.J.; Stockings, E.; Khoo, J.-P.; Erskine, H.E.; Degenhardt, L.; Vos, T.; Whiteford, H.A. The Prevalence and Burden of Bipolar Disorder: Findings from the Global Burden of Disease Study 2013. Bipolar. Disord. 2016, 18, 440–450. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Calabrese, J.R. Bipolar depression: The clinical characteristics and unmet needs of a complex disorder. Curr. Med. Res. Opin. 2019, 35, 1993–2005. [Google Scholar] [CrossRef]
- Bailly, D. Pharmacological treatment of bipolar disorder in children and adolescents. Encephale 2017, 43, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Corponi, F.; Fabbri, C.; Serretti, A. Pharmacogenetics in Psychiatry. Adv. Pharm. 2018, 83, 297–331. [Google Scholar] [CrossRef]
- Bousman, C.; Maruf, A.A.; Müller, D.J. Towards the integration of pharmacogenetics in psychiatry: A minimum, evidence-based genetic testing panel. Curr. Opin. Psychiatry. 2019, 32, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug-Resistant Epilepsy. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef]
- Cuéllar-Barboza, A.B.; McElroy, S.L.; Veldic, M.; Singh, B.; Kung, S.; Romo-Nava, F.; Nunez, N.A.; Cabello-Arreola, A.; Coombes, B.J.; Prieto, M.; et al. Potential Pharmacogenomic Targets in Bipolar Disorder: Considerations for Current Testing and the Development of Decision Support Tools to Individualize Treatment Selection. Int. J. Bipolar Disord. 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.H. Bipolar Disorder. Prim. Care 2016, 43, 269–284. [Google Scholar] [CrossRef]
- McCormick, U.; Murray, B.; McNew, B. Diagnosis and Treatment of Patients with Bipolar Disorder: A Review for Advanced Practice Nurses. J. Am. Assoc. Nurse Pract. 2015, 27, 530–542. [Google Scholar] [CrossRef]
- Tremain, H.; Fletcher, K.; Murray, G. Number of Episodes in Bipolar Disorder: The Case for More Thoughtful Conceptualization and Measurement. Bipolar Disord. 2020, 22, 231–244. [Google Scholar] [CrossRef]
- Joshi, A.; Bow, A.; Agius, M. Pharmacological Therapies in Bipolar Disorder: A Review of Current Treatment Options. Psychiatr. Danub. 2019, 31 (Suppl. 3), 595–603. [Google Scholar] [PubMed]
- Hui Poon, S.; Sim, K.; Baldessarini, R.J. Pharmacological Approaches for Treatment-resistant Bipolar Disorder. Curr. Neuropharmacol. 2015, 13, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Grunze, H.C. Anticonvulsants in bipolar disorder. J Ment Health. 2010, 19, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanova, R.; Stefanova, I. Basic Mechanisms of Action of the Antiepileptic Drugs. Acta Medica Bulgarica. 2017, 44, 2. [Google Scholar] [CrossRef][Green Version]
- Friedman, S.D.; Dager, S.R.; Parow, A.; Hirashima, F.; Demopulos, C.; Stoll, A.L.; Lyoo, I.K.; Dunner, D.L.; Renshaw, P.F. Lithium and valproic acid treatment effects on brain chemistry in bipolar disorder. Biol. Psychiatry 2004, 56, 340–348. [Google Scholar] [CrossRef]
- Chiu, C.T.; Wang, Z.; Hunsberger, J.G.; Chuang, D.M. Therapeutic potential of mood stabilizers lithium and valproic acid: Beyond bipolar disorder. Pharm. Rev. 2013, 65, 105–142. [Google Scholar] [CrossRef]
- Ayano, G. Bipolar Disorders and Carbamazepine: Pharmacokinetics, Pharmacodynamics, Therapeutic Effects and Indications of Carbamazepine: Review of Articles. J. Neuropsychopharmacol. Ment. Health 2016, 1. [Google Scholar] [CrossRef]
- Rapoport, S.I.; Basselin, M.; Kim, H.W.; Rao, J.S. Bipolar disorder and mechanisms of action of mood stabilizers. Brain Res. Rev. 2009, 61, 185–209. [Google Scholar] [CrossRef]
- Rapoport, S.I. Lithium and the other mood stabilizers effective in bipolar disorder target the rat brain arachidonic acid cascade. ACS Chem. Neurosci. 2014, 5, 459–467. [Google Scholar] [CrossRef]
- Basselin, M.; Chang, L.; Chen, M.; Bell, J.M.; & Rapoport, S.I. Chronic carbamazepine administration attenuates dopamine D2-like receptor-initiated signaling via arachidonic acid in rat brain. Neurochem. Res. 2008, 33, 1373–1383. [Google Scholar] [CrossRef]
- Ghasemi, M.; Phillips, C.; Trillo, L.; De Miguel, Z.; Das, D.; Salehi, A. The role of NMDA receptors in the pathophysiology and treatment of mood disorders. Neurosci. Biobehav. Rev. 2014, 47, 336–358. [Google Scholar] [CrossRef]
- D’Onofrio, S.; Mahaffey, S.; Garcia-Rill, E. Role of Calcium Channels in Bipolar Disorder. Curr. Psychopharmacol. 2017, 6, 122–135. [Google Scholar] [CrossRef][Green Version]
- Chiu Tikhonov, D.B.; Zhorov, B.S. Mechanism of sodium channel block by local anesthetics, antiarrhythmics, and anticonvulsants. J. Gen. Physiol. 2017, 149, 465–481. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Hanna, M.G. The Role of Calcium Channels in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 4, a022723. [Google Scholar] [CrossRef]
- Cain, S.M.; Tyson, J.R.; Jones, K.L.; Snutch, T.P. Thalamocortical neurons display suppressed burst-firing due to an enhanced Ih current in a genetic model of absence epilepsy. Pflug. Arch. Eur. J. Physiol. 2015, 467, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Baggiani, G.; Ambrosiani, L.; Trincas, P.; Burrai, C.; Bocchetta, A. Psychotropic Medication of Acute Episodes of Mood Disorders: Current Prescription Attitude in Two Psychiatric Wards in Cagliari, Italy. Clin. Pract. Epidemiol. Ment. Health 2018, 14, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Shoja Shafti, S.; Kaviani, H. Extended-release carbamazepine versus lithium in management of acute mania in male inpatients with bipolar I disorder. Psychiatry Clin. Psychopharmacol. 2018, 28, 371–377. [Google Scholar] [CrossRef]
- Weisler, R.H.; Hirschfeld, R.; Cutler, A.J.; Gazda, T.; Ketter, T.A.; Keck, P.E.; Swann, A.; Kalali, A. Extended-Release Carbamazepine Capsules as Monotherapy in Bipolar Disorder: Pooled Results from Two Randomised, Double-Blind, Placebo-Controlled Trials. CNS Drugs 2006, 20, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Keck, P.E.J.; McElroy, S.L. Carbamazepine and Valproate in the Maintenance Treatment of Bipolar Disorder. J. Clin. Psychiatry 2002, 63 (Suppl. 10), 13–17. [Google Scholar]
- Chen, C.-H.; Lin, S.-K. Carbamazepine Treatment of Bipolar Disorder: A Retrospective Evaluation of Naturalistic Long-Term Outcomes. BMC Psychiatry 2012, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Bahji, A.; Ermacora, D.; Stephenson, C.; Hawken, E.R.; Vazquez, G. Comparative efficacy and tolerability of pharmacological treatments for the treatment of acute bipolar depression: A systematic review and network meta-analysis. J. Affect. Disord. 2020, 269, 154–184. [Google Scholar] [CrossRef]
- Hartong, E.G.T.M.; Moleman, P.; Hoogduin, C.A.L.; Broekman, T.G.; Nolen, W.A. Prophylactic Efficacy of Lithium versus Carbamazepine in Treatment-Naive Bipolar Patients. J. Clin. Psychiatry 2003, 64, 144–151. [Google Scholar] [CrossRef]
- Janszky, J.; Tényi, D.; Bóné, B. Valproate in the treatment of epilepsy and status epilepticus. Ideggyogy. Szle. 2017, 70, 258–264. [Google Scholar] [CrossRef]
- Johannessen, C.U.; Johannessen, S.I. Valproate: Past, Present, and Future. Cns Drug Rev. 2003, 9, 199–216. [Google Scholar] [CrossRef]
- Jochim, J.; Rifkin-Zybutz, R.P.; Geddes, J.; Cipriani, A. Valproate for Acute Mania. Cochrane Database Syst. Rev. 2019, 10, CD004052. [Google Scholar] [CrossRef]
- Vieta, E.; Sanchez-Moreno, J. Acute and Long-Term Treatment of Mania. Dialogues Clin. Neurosci. 2008, 10, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Reid, K.; Young, A.H.; Macritchie, K.; Geddes, J. Valproic Acid, Valproate and Divalproex in the Maintenance Treatment of Bipolar Disorder. Cochrane Database Syst. Rev. 2013, 2013, CD003196. [Google Scholar] [CrossRef]
- Kang, M.G.; Qian, H.; Keramatian, K.; Chakrabarty, T.; Saraf, G.; Lam, R.W.; Wong, H.; Yatham, L.N. Lithium vs Valproate in the Maintenance Treatment of Bipolar I Disorder: A Post-Hoc Analysis of a Randomized Double-Blind Placebo-Controlled Trial. Aust. N. Z. J. Psychiatry 2020, 54, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Musetti, L.; Tundo, A.; Benedetti, A.; Massimetti, G.; Cambiali, E.; Pergentini, I.; Del Grande, C.; Dell’Osso, L. Lithium, Valproate, and Carbamazepine Prescribing Patterns for Long-Term Treatment of Bipolar I and II Disorders: A Prospective Study. Hum. Psychopharmacol. 2018, 33, e2676. [Google Scholar] [CrossRef]
- Wingård, L.; Brandt, L.; Bodén, R.; Kieler, H.; Andersen, M.; Reutfors, J. Monotherapy vs. combination therapy for post mania maintenance treatment: A population-based cohort study. Eur. Neuropsychopharmacol. 2019, 29, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Fornaro, M.; De Berardis, D.; Koshy, A.S. Prevalence and clinical features associated with bipolar disorder polypharmacy: A systematic review. Neuropsychiatr. Dis. Treat. 2016, 12, 719–735. [Google Scholar] [CrossRef] [PubMed]
- Missio, G.; Moreno, D.H.; Demetrio, F.N.; Soeiro-de-Souza, M.G.; Dos Santos Fernandes, F.; Barros, V.B.; Moreno, R.A. A Randomized Controlled Trial Comparing Lithium plus Valproic Acid versus Lithium plus Carbamazepine in Young Patients with Type 1 Bipolar Disorder: The LICAVAL Study. Trials 2019, 20, 608. [Google Scholar] [CrossRef] [PubMed]
- Rakitin, A.; Kõks, S.; Haldre, S. Metabolic syndrome and anticonvulsants: A comparative study of valproic acid and carbamazepine. Seizure. 2016, 38, 11–16. [Google Scholar] [CrossRef]
- Djordjevic, N.; Jankovic, S.M.; Milovanovic, J.R. Pharmacokinetics and Pharmacogenetics of Carbamazepine in Children. Eur. J. Drug Metab. Pharm. 2017, 42, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Gierbolini, J.; Giarratano, M.; Benbadis, S.R. Carbamazepine-Related Antiepileptic Drugs for the Treatment of Epilepsy—A Comparative Review. Expert Opin. Pharmacother. 2016, 885–888. [Google Scholar] [CrossRef]
- Franco, V.; Perucca, E. The Pharmacogenomics of Epilepsy. Expert Rev. Neurother. 2015, 15, 1161–1170. [Google Scholar] [CrossRef]
- Makmor-Bakry, M.; Sills, G.J.; Hitiris, N.; Butler, E.; Wilson, E.A.; Brodie, M.J. Genetic Variants in Microsomal Epoxide Hydrolase Influence Carbamazepine Dosing. Clin. Neuropharmacol. 2009, 32, 205–212. [Google Scholar] [CrossRef]
- Daci, A.; Beretta, G.; Vllasaliu, D.; Shala, A.; Govori, V.; Norata, G.D.; Krasniqi, S. Polymorphic Variants of SCN1A and EPHX1 Influence Plasma Carbamazepine Concentration, Metabolism and Pharmacoresistance in a Population of Kosovar Albanian Epileptic Patients. PLoS ONE 2015, 10, e0142408. [Google Scholar] [CrossRef]
- Zhao, G.X.; Shen, M.L.; Zhang, Z.; Wang, P.; Xie, C.X.; He, G.H. Association between EPHX1 polymorphisms and carbamazepine metabolism in epilepsy: A meta-analysis. Int. J. Clin. Pharm. 2019, 41, 1414–1428. [Google Scholar] [CrossRef] [PubMed]
- He, X.-J.; Jian, L.-Y.; He, X.-L.; Tang, M.; Wu, Y.; Xu, Y.-Y.; Sun, X.-J.; Zhao, L.-M. Association of ABCB1, CYP3A4, EPHX1, FAS, SCN1A, MICA, and BAG6 Polymorphisms with the Risk of Carbamazepine-Induced Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis in Chinese Han Patients with Epilepsy. Epilepsia 2014, 55, 1301–1306. [Google Scholar] [CrossRef]
- Chbili, C.; Fathallah, N.; Laouani, A.; Nouira, M.; Hassine, A.; Ben Amor, S.; Ben Ammou, S.; Ben Salem, C.; Saguem, S. Effects of EPHX1 and CYP3A4*22 Genetic Polymorphisms on Carbamazepine Metabolism and Drug Response among Tunisian Epileptic Patients. J. Neurogenet. 2016, 30, 16–21. [Google Scholar] [CrossRef]
- Caruso, A.; Bellia, C.; Pivetti, A.; Agnello, L.; Bazza, F.; Scazzone, C.; Bivona, G.; Lo Sasso, B.; Ciaccio, M. Effects of EPHX1 and CYP3A4 polymorphisms on carbamazepine metabolism in epileptic patients. Pharm. Pers. Med. 2014, 7, 117–120. [Google Scholar]
- Saiz-Rodríguez, M.; Almenara, S.; Navares-Gómez, M.; Ochoa, D.; Román, M.; Zubiaur, P.; Koller, D.; Santos, M.; Mejía, G.; Borobia, A.M.; et al. Effect of the Most Relevant CYP3A4 and CYP3A5 Polymorphisms on the Pharmacokinetic Parameters of 10 CYP3A Substrates. Biomedicines 2020, 8, 94. [Google Scholar] [CrossRef]
- Park, P.W.; Seo, Y.H.; Ahn, J.Y.; Kim, K.A.; Park, J.Y. Effect of CYP3A5*3 genotype on serum carbamazepine concentrations at steady state in Korean epileptic patients. J. Clin. Pharm. Ther. 2009, 34, 569–574. [Google Scholar] [CrossRef]
- Ganesapandian, M.; Ramasamy, K.; Adithan, S.; Narayan, S.K. Influence of Cytochrome P450 3A5 (CYP3A5) Genetic Polymorphism on Dose-Adjusted Plasma Levels of Carbamazepine in Epileptic Patients in South Indian Population. Indian J. Pharmacol. 2019, 51, 384–388. [Google Scholar] [CrossRef]
- Panomvana, D.; Traiyawong, T.; Towanabut, S. Effect of CYP3A5 genotypes on the pharmacokinetics of carbamazepine when used as monotherapy or co-administered with phenytoin, phenobarbital or valproic acid in Thai patients. J. Pharm. Pharm. Sci. Publ. Can. Soc. Pharm. Sci. Soc. Can. Sci. Pharm. 2013, 16, 502–510. [Google Scholar] [CrossRef]
- Wang, P.; Yin, T.; Ma, H.-Y.; Liu, D.-Q.; Sheng, Y.-H.; Wang, C.; Zhou, B.-T. Effects of CYP3A4/5 and ABCB1 Genetic Polymorphisms on Carbamazepine Metabolism and Transport in Chinese Patients with Epilepsy Treated with Carbamazepine in Monotherapy and Bitherapy. Epilepsy Res. 2015, 117, 52–57. [Google Scholar] [CrossRef]
- Al-Gahtany, M.; Karunakaran, G.; Munisamy, M. Pharmacogenetics of CYP3A5 on Carbamazepine Pharmacokinetics in Epileptic Patients Developing Toxicity. BMC Genom. 2014, P2. [Google Scholar] [CrossRef]
- Hong, T.P.; Huynh Hieu, T.M.; Vo, T.; Manh, H.T. Effect of CYP3A5 genotypes on serum carbamazepine concentrations at steady state in Vietnamese epileptic patients. Res. J. Pharm. Technol. 2020, 13, 2802. [Google Scholar] [CrossRef]
- Milovanovic, D.D.; Radosavljevic, I.; Radovanovic, M.; Milovanovic, J.R.; Obradovic, S.; Jankovic, S.; Milovanovic, D.; Djordjevic, N. CYP3A5 polymorphism in Serbian paediatric epileptic patientson carbamazepine treatment. Ser. J. Exp. Clin. Res. 2015, 16, 93–99. [Google Scholar] [CrossRef]
- Puranik, Y.G.; Birnbaum, A.K.; Marino, S.E.; Ahmed, G.; Cloyd, J.C.; Remmel, R.P.; Leppik, I.E.; Lamba, J.K. Association of carbamazepine major metabolism and transport pathway gene polymorphisms and pharmacokinetics in patients with epilepsy. Pharmacogenomics 2013, 14, 35–45. [Google Scholar] [CrossRef]
- Lu, Q.; Huang, Y.-T.; Shu, Y.; Xu, P.; Xiang, D.-X.; Qu, Q.; Qu, J. Effects of CYP3A5 and UGT2B7 Variants on Steady-State Carbamazepine Concentrations in Chinese Epileptic Patients. Medicine 2018, 97, e11662. [Google Scholar] [CrossRef]
- Hung, C.-C.; Chang, W.-L.; Ho, J.-L.; Tai, J.J.; Hsieh, T.-J.; Huang, H.-C.; Hsieh, Y.-W.; Liou, H.-H. Association of Polymorphisms in EPHX1, UGT2B7, ABCB1, ABCC2, SCN1A and SCN2A Genes with Carbamazepine Therapy Optimization. Pharmacogenomics 2012, 13, 159–169. [Google Scholar] [CrossRef]
- Ma, C.L.; Jiao, Z.; Wu, X.Y. Association between PK/PD-involved gene polymorphisms and carbamazepine-individualized therapy. Pharmacogenomics 2015, 16, 1499–1512. [Google Scholar] [CrossRef]
- Mittag, N.; Meister, S.; Berg, A.M.; Walther, U.I. A Case Report of a Carbamazepine Overdose with Focus on Pharmacokinetic Aspects. Pharmacogenomics 2016, 49, 76–78. [Google Scholar] [CrossRef]
- Djordjevic, N.; Milovanovic, D.D.; Radovanovic, M.; Radosavljevic, I.; Obradovic, S.; Jakovljevic, M.; Milovanovic, D.; Milovanovic, J.R.; Jankovic, S. CYP1A2 Genotype Affects Carbamazepine Pharmacokinetics in Children with Epilepsy. Eur. J. Clin. Pharm. 2016, 72, 439–445. [Google Scholar] [CrossRef]
- Yip, V.L.M.; Pertinez, H.; Meng, X.; Maggs, J.L.; Carr, D.F.; Park, B.K.; Marson, A.G.; Pirmohamed, M. Evaluation of clinical and genetic factors in the population pharmacokinetics of carbamazepine. Br. J. Clin. Pharm. 2020. [Google Scholar] [CrossRef]
- Tanno, L.K.; Kerr, D.S.; dos Santos, B.; Talib, L.L.; Yamaguti, C. The Absence of CYP3A5*3 Is a Protective Factor to Anticonvulsants Hypersensitivity Reactions: A Case-Control Study in Brazilian Subjects. PLoS ONE 2015, 10, e0136141. [Google Scholar] [CrossRef]
- Laska, A.J.; Han, M.J.; Lospinoso, J.A.; Brown, P.J.; Beachkofsky, T.M. CYP2C19*2 Status in Patients with Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis. Pharmgenomics. Pers. Med. 2017, 10, 183–186. [Google Scholar] [CrossRef]
- Milovanovic, D.D.; Milovanovic, J.R.; Radovanovic, M.; Radosavljevic, I.; Obradovic, S.; Jankovic, S.; Milovanovic, D.; Djordjevic, N. The Influence of CYP2C8*3 on Carbamazepine Serum Concentration in Epileptic Pediatric Patients. Balk. J. Med. Genet. 2016, 19, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Budde, M.; Degner, D.; Brockmöller, J.; Schulze, T.G. Pharmacogenomic Aspects of Bipolar Disorder: An Update. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Pisanu, C.; Heilbronner, U.; Squassina, A. The Role of Pharmacogenomics in Bipolar Disorder: Moving Towards Precision Medicine. Mol. Diagn. 2018, 22, 409–420. [Google Scholar] [CrossRef]
- Zhu, M.M.; Li, H.L.; Shi, L.H.; Chen, X.P.; Luo, J.; Zhang, Z.L. The pharmacogenomics of valproic acid. J. Hum. Genet. 2017, 62, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Feng, W.; Zhu, L.; Yu, Y.; Yang, W.; Gao, B.; Wu, X.; Zhao, Z.; Fang, F. Genetic polymorphisms and valproic acid plasma concentration in children with epilepsy on valproic acid monotherapy. Seizure 2017, 51, 22–26. [Google Scholar] [CrossRef]
- Fortinguerra, S.; Sorrenti, V.; Giusti, P.; Zusso, M.; Buriani, A. Pharmacogenomic Characterization in Bipolar Spectrum Disorders. Pharmaceutics 2019, 12, 13. [Google Scholar] [CrossRef]
- Sun, Y.P.; Tan, L.; Wang, Y.; Song, J.H. Effect of UGT1A6 genetic polymorphisms on the metabolism of sodium valproate. Zhonghua Yi Xue Za Zhi. 2007, 87, 2033–2035. [Google Scholar]
- Guo, Y.; Hu, C.; He, X.; Qiu, F.; Zhao, L. Effects of UGT1A6, UGT2B7, and CYP2C9 genotypes on plasma concentrations of valproic acid in Chinese children with epilepsy. Drug Metab. Pharm. 2012, 27, 536–542. [Google Scholar] [CrossRef]
- Munisamy, M.; Tripathi, M.; Behari, M.; Raghavan, S.; Jain, D.C.; Ramanujam, B.; Arumugam, K.; Rajakannan, T.; Mallayasamy, S.R.; Behari, V. The Effect of Uridine Diphosphate Glucuronosyltransferase (UGT)1A6 Genetic Polymorphism on Valproic Acid Pharmacokinetics in Indian Patients with Epilepsy: A Pharmacogenetic Approach. Mol. Diagn. 2013, 17, 319–326. [Google Scholar] [CrossRef]
- Goey, A.K.; Sissung, T.M.; Peer, C.J.; Figg, W.D. Pharmacogenomics and Histone Deacetylase Inhibitors. Pharmacogenomics 2016, 17, 1807–1815. [Google Scholar] [CrossRef]
- Chatzistefanidis, D.; Lazaros, L.; Giaka, K.; Nakou, I.; Tzoufi, M.; Georgiou, I.; Kyritsis, A.; Markoula, S. UGT1A6- and UGT2B7-Related Valproic Acid Pharmacogenomics According to Age Groups and Total Drug Concentration Levels. Pharmacogenomics 2016, 17, 827–835. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, S.; Li, X.; Wang, X.; Zeng, S. Genetic variants of human UGT1A3: Functional characterization and frequency distribution in a Chinese Han population. Drug Metab. Dispos 2006, 34, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-M.; Zhang, L.-F.; Wang, G.-J.; Zhang, S.-N.; Zhou, J.-H.; Hao, H.-P. Influence of UDP-Glucuronosyltransferase Polymorphisms on Valproic Acid Pharmacokinetics in Chinese Epilepsy Patients. Eur. J. Clin. Pharm. 2012, 68, 1395–1401. [Google Scholar] [CrossRef]
- Kim, S.C.; Kim, M.G. A meta-analysis of the influence of UGT1A6 genetic polymorphisms on valproic acid pharmacokinetics. Int. J. Clin. Pharmacol. Ther. 2019, 57, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Shastri, S.; Gulati, S.; Kaleekal, T.; Kabra, M.; Gupta, N.; Gupta, Y.K.; Pandey, R.M. Prevalence of UGT1A6 polymorphisms in children with epilepsy on valproate monotherapy. Neurol. India 2015, 63, 35–39. [Google Scholar] [CrossRef]
- Ma, H.; Zhang, T.; Gong, Z.; Zhou, B.; Zou, M.; Xiao, S.; Zhu, W. Effect of UGT2B7 genetic variants on serum valproic acid concentration. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2013, 38, 766–772. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Li, Y.; Yan, J.; Zhang, J.; Wang, B. Correlations between UGT2B7∗2 Gene Polymorphisms and Plasma Concentrations of Carbamazepine and Valproic Acid in Epilepsy Patients. Brain Dev. 2018, 40, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Zhuo, W.Y.; Lin, H.; Peng, Z.K.; Wang, H.M.; Huang, H.W.; Luo, Y.H.; Tang, F.Q. The influence of UGT2B7 genotype on valproic acid pharmacokinetics in Chinese epilepsy patients. Epilepsy Res. 2015, 114, 78–80. [Google Scholar] [CrossRef]
- Du, Z.; Jiao, Y.; Shi, L. Association of UGT2B7 and UGT1A4 Polymorphisms with Serum Concentration of Antiepileptic Drugs in Children. Med. Sci. Monit. 2016, 22, 4107–4113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, P.; Lin, X.Q.; Cai, W.K.; Xu, G.L.; Zhou, M.D.; Yang, M.; He, G.H. Effect of UGT2B7 genotypes on plasma concentration of valproic acid: A meta-analysis. Eur. J. Clin. Pharm. 2018, 74, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.T.; Sun, Y.P.; Ou, J.R.; Song, J.H.; Yu, Y. The influence of cytochrome oxidase CYP2A6, CYP2B6, and CYP2C9 polymorphisms on the plasma concentrations of valproic acid in epileptic patients. Clin. Neurol. Neurosurg. 2010, 112, 320–323. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Ahn, M.H.; Yee, J.; Lee, N.; Han, J.M.; Gwak, H.S. Influence of CYP2C9 and CYP2A6 on plasma concentrations of valproic acid: A meta-analysis. Eur. J. Clin. Pharm. 2020, 76, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Abbott, F.S.; Zanger, U.M.; Chang, T.K. Influence of CYP2C9 genotypes on the formation of a hepatotoxic metabolite of valproic acid in human liver microsomes. Pharm. J. 2003, 3, 335–342. [Google Scholar] [CrossRef]
- Amini-Shirazi, N.; Ghahremani, M.H.; Ahmadkhaniha, R.; Mandegary, A.; Dadgar, A.; Abdollahi, M.; Shadnia, S.; Pakdaman, H.; Kebriaeezadeh, A. Influence of CYP2C9 polymorphism on metabolism of valproate and its hepatotoxin metabolite in Iranian patients. Toxicol. Mech. Methods 2010, 20, 452–457. [Google Scholar] [CrossRef]
- Bűdi, T.; Tóth, K.; Nagy, A.; Szever, Z.; Kiss, Á.; Temesvári, M.; Háfra, E.; Garami, M.; Tapodi, A.; Monostory, K. Clinical significance of CYP2C9-status guided valproic acid therapy in children. Epilepsia 2015, 56, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Gielgens, R.C.; Bok, L.A. Commentary on clinical significance of CYP2C9-status-guided valproic acid therapy in children. Epilepsia 2016, 57, 1338–1339. [Google Scholar] [CrossRef]
- Monostory, K.; Bűdi, T.; Tóth, K.; Nagy, A.; Szever, Z.; Kiss, Á.; Temesvári, M.; Háfra, E.; Tapodi, A.; Garami, M. In response: Commentary on clinical significance of CYP2C9-status-guided valproic acid therapy in children. Epilepsia 2016, 57, 1339–1340. [Google Scholar] [CrossRef]
- Monostory, K.; Nagy, A.; Tóth, K.; Büdi, T.; Kiss, Á; Déri, M.; Csukly, G. Relevance of CYP2C9 Function in Valproate Therapy. Curr. Neuropharmacol. 2019, 17, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, T.; Li, G.; Qiu, F.; Sun, Y.; Zhao, L. Associations of CYP2C9 and CYP2A6 Polymorphisms with the Concentrations of Valproate and its Hepatotoxin Metabolites and Valproate-Induced Hepatotoxicity. Basic Clin. Pharmacol. Toxicol. 2017, 121, 138–143. [Google Scholar] [CrossRef]
- Wang, C.; Wang, P.; Yang, L.P.; Pan, J.; Yang, X.; Ma, H.Y. Association of CYP2C9, CYP2A6, ACSM2A, and CPT1A gene polymorphisms with adverse effects of valproic acid in Chinese patients with epilepsy. Epilepsy Res. 2017, 132, 64–69. [Google Scholar] [CrossRef]
- Stewart, J.D.; Horvath, R.; Baruffini, E.; Ferrero, I.; Bulst, S.; Watkins, P.B.; Fontana, R.J.; Day, C.P.; Chinnery, P.F. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology 2010, 52, 1791–1796. [Google Scholar] [CrossRef]
- Bousman, C.A.; Jaksa, P.; Pantelis, C. Systematic evaluation of commercial pharmacogenetic testing in psychiatry: A focus on CYP2D6 and CYP2C19 allele coverage and results reporting. Pharm. Genom. 2017, 27, 387–393. [Google Scholar] [CrossRef]
- Mei, S.; Feng, W.; Zhu, L.; Li, X.; Yu, Y.; Yang, W.; Gao, B.; Wu, X.; Fang, F.; Zhao, Z. Effect of CYP2C19, UGT1A8, and UGT2B7 on valproic acid clearance in children with epilepsy: A population pharmacokinetic model. Eur. J. Clin. Pharm. 2018, 74, 1029–1036. [Google Scholar] [CrossRef]
- Song, C.; Li, X.; Mao, P.; Song, W.; Liu, L.; Zhang, Y. Impact of CYP2C19 and CYP2C9 gene polymorphisms on sodium valproate plasma concentration in patients with epilepsy. Eur. J. Hosp. Pharm. 2020. [Google Scholar] [CrossRef]
- Smith, R.L.; Haslemo, T.; Refsum, H.; Molden, E. Impact of age, gender and CYP2C9/2C19 genotypes on dose-adjusted steady-state serum concentrations of valproic acid-a large-scale study based on naturalistic therapeutic drug monitoring data. Eur. J. Clin. Pharm. 2016, 72, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, M.; Yan, P.; Ju, X.; Liu, J.; Wang, C.; Shi, J. Effect of genetic polymorphisms of CYP2C19 on the steady-state serum concentrations of valproic acid in chinese han patients with schizophrenia. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Noai, M.; Soraoka, H.; Kajiwara, A.; Tanamachi, Y.; Oniki, K.; Nakagawa, K.; Ishitsu, T.; Saruwatari, J. Cytochrome P450 2C19 Polymorphisms and Valproic Acid-Induced Weight Gain. Acta Neurol. Scand. 2016, 133, 216–223. [Google Scholar] [CrossRef]
- Drokov, A.P.; Lipatova, L.V.; Shnayder, N.A.; Nasyrova, R.F. Pharmacogenetic Markers for Metabolic Impairments in Treatment with Valproic Acid. Neurosci. Behav. Physi. 2020, 50, 13–19. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z. CYP3A5*3 Polymorphism May Influence the Concentration of Valproic Acid. Int. J. Pharmacol. 2017, 13, 495–500. [Google Scholar] [CrossRef]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of ABC transporters in drug resistance, metabolism, and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. J. Am. Soc. Exp. Neurother. 2005, 2, 86–98. [Google Scholar] [CrossRef]
- Bruhn, O.; Lindsay, M.; Wiebel, F.; Kaehler, M.; Nagel, I.; Bohm, R.; Roder, C.; Cascorbi, I. Alternative polyadenylation of ABC-transporters of the C-family (ABCC1, ABCC2, ABCC3) and implications on post-transcriptional micro-RNA regulation. Mol. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Istikharah, R.; Hartienah, S.D.; Vitriyani, S.; Ningrum, V.D.A. Allele Frequency of Carbamazepine Major Efflux Transporter Encoding Gene ABCB1 C3435T among Javanese-Indonesian Population. Open Access Maced. J. Med. Sci. 2020, 8, 406–413. [Google Scholar] [CrossRef]
- Malek, C.; Hedia, K.; Ilhem Ben-Youssef, T.; Lamia, H. ABCB1 Polymorphisms and Drug-Resistant Epilepsy in a Tunisian Population. Dis. Markers 2019, 16. [Google Scholar] [CrossRef]
- Soranzo, N.; Cavalleri, G.L.; Weale, M.E.; Wood, N.W.; Depondt, C.; Marguerie, R.; Sisodiya, S.M.; Goldstein, D.B. Identifying candidate causal variants responsible for altered activity of the ABCB1 multidrug resistance gene. Genome Res. 2004, 14, 1333–1344. [Google Scholar] [CrossRef]
- Meng, H.; Guo, G.; Ren, J.; Zhou, H.; Ge, Y.; Guo, Y. Effects of ABCB1 polymorphisms on plasma carbamazepine concentrations and pharmacoresistance in Chinese patients with epilepsy. Epilepsy Behav. 2011, 21, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Cheng, J. Effects of MDR1 (C3435T) Polymorphism on Resistance, Uptake, and Efflux to Antiepileptic Drugs. DNA Cell Biol. 2019, 38, 250–255. [Google Scholar] [CrossRef]
- Li, S.X.; Liu, Y.Y.; Wang, Q.B. ABCB1 gene C3435T polymorphism and drug resistance in epilepsy: Evidence based on 8604 subjects. Med. Sci. Monit. 2015, 21, 861–868. [Google Scholar] [CrossRef]
- Haerian, B.S.; Roslan, H.; Raymond, A.A.; Tan, C.T.; Lim, K.S.; Zulkifli, S.Z.; Mohamed, E.H.; Tan, H.J.; Mohamed, Z. ABCB1 C3435T polymorphism and the risk of resistance to antiepileptic drugs in epilepsy: A systematic review and meta-analysis. Seizure 2010, 19, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Chouchi, M.; Kaabachi, W.; Klaa, H.; Tizaoui, K.; Turki, I.B.; Hila, L. Relationship between ABCB1 3435TT genotype and antiepileptic drugs resistance in Epilepsy: Updated systematic review and meta-analysis. BMC Neurol. 2017, 17, 32. [Google Scholar] [CrossRef]
- Hosseini, M.; Ebrahimi, A.; Houshmand, M.; Zainali, S.; Tonekaboni, S.H.; Moghaddasi, M. SCN1A and ABCB1 Polymorphisms in Epilepsy. Arch. Neurosci. 2018, 5, e59383. [Google Scholar] [CrossRef]
- Salih, K.S.; Hamdan, F.B.; Al-Mayah, Q.S. Association of ABCB1 gene polymorphism (C1236T and C3435T) with refractory epilepsy in Iraqi patients. Mol. Biol. Rep. 2020, 47, 4245–4254. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Fang, S.; Yan, Y.L.; Zeng, S.S.; Xu, Z.J.; Gong, Z.C. The ABCC2 c.-24C>T polymorphism increases the risk of resistance to antiepileptic drugs: A meta-analysis. J. Clin. Neurosci. 2017, 37, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Orsini, A.; Esposito, M.; Perna, D.; Bonuccelli, A.; Peroni, D.; Striano, P. Personalized medicine in epilepsy patients. J. Transl. Genet Genom. 2018, 2, 16. [Google Scholar] [CrossRef]
- Balestrini, S.; Sisodiya, S.M. Pharmacogenomics in epilepsy. Neurosci. Lett. 2018, 667, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Lee, J.H.; Yi, J.; Cho, Y.J.; Heo, K.; Lee, S.H.; Kim, S.W.; Kim, M.K.; Kim, K.H.; In Lee, B.; et al. A nonsynonymous variation in MRP2/ABCC2 is associated with neurological adverse drug reactions of carbamazepine in patients with epilepsy. Pharm. Genom. 2010, 20, 249–256. [Google Scholar] [CrossRef]
- Riquelme-Alcázar, J.; González-Vargas, R.; Moya, P.R. ABC transporters and drug resistance in epilepsy: Biological plausibility, pharmacogenetics and precision medicine. Rev. Neurol. 2020, 70, 23–32. [Google Scholar] [CrossRef]
- Yang, X.; Yan, Y.; Fang, S.; Zeng, S.; Ma, H.; Qian, L.; Chen, X.; Wei, J.; Gong, Z.; Xu, Z. Comparison of oxcarbazepine efficacy and MHD concentrations relative to age and BMI: Associations among ABCB1, ABCC2, UGT2B7, and SCN2A polymorphisms. Medicine 2019, 98, e14908. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, B.; Liu, Z.; Tang, Y.; Zhang, Y.; Wang, S.; Guo, Y.; Ding, Y.; Wang, S.; Ding, M. Effects of ABCB1, ABCC2, UGT2B7 and HNF4α genetic polymorphisms on oxcarbazepine concentrations and therapeutic efficacy in patients with epilepsy. Seizure 2017, 51, 102–106. [Google Scholar] [CrossRef]
- Al-Eitan, L.N.; Al-Dalalah, I.M.; Mustafa, M.M. Effects of MTHFR and ABCC2 gene polymorphisms on antiepileptic drug responsiveness in Jordanian epileptic patients. Pharmgenomics Pers. Med. 2019, 12, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Luo, X.; Yang, K.; Sun, X.; Li, X.; Zhang, C.; Ma, S.; Liu, Y.; Yin, J. Neural overexpression of multidrug resistance-associated protein 1 and refractory epilepsy: A meta-analysis of nine studies. Int. J. Neurosci. 2016, 126, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Sabin, O.; Bocsan, I.C.; Trifa, A.; Major, Z.Z. Correlation between ABCB1 gene polymorphisms, antiepileptic drug concentrations and treatment response. Rev. Romana Med. Laborator. 2018, 26, 479–487. [Google Scholar] [CrossRef]
- Sabin, O.; Pop, R.; Trifa, A.; Buzoianu, A.D. The influence of CYP2C9, CYP2C19 and ABCB1 polymorphisms on the plasma concentrations of valproic acid in epileptic patients. HVM Bioflux. 2016, 8, 29–33. [Google Scholar]
- Ajmi, M.; Boujaafar, S.; Zouari, N.; Amor, D.; Nasr, A.; Rejeb, N.B.; Amor, S.B.; Omezzine, A.; Benammou, S.; Bouslama, A. Association between ABCB1 polymorphisms and response to first-generation antiepileptic drugs in a Tunisian epileptic population. Int. J. Neurosci. 2018, 128, 705–714. [Google Scholar] [CrossRef]
- Cárdenas-Rodríguez, N.; Carmona-Aparicio, L.; Pérez-Lozano, D.L.; Ortega-Cuellar, D.; Gómez-Manzo, S.; Ignacio-Mejía, I. Genetic variations associated with pharmacoresistant epilepsy (Review). Mol. Med. Rep. 2020, 21, 1685–1701. [Google Scholar] [CrossRef]
- Grewal, G.K.; Kukal, S.; Kanojia, N.; Madan, K.; Saso, L.; Kukreti, R. In Vitro Assessment of the Effect of Antiepileptic Drugs on Expression and Function of ABC Transporters and Their Interactions with ABCC2. Molecules 2017, 22, 1484. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Su, Q.; Qin, J.; Zhou, Y.; Ruan, H.; Chen, Z.; Chen, Z.; Li, H.; Zhou, Y.; Zhou, S.; et al. Correlation of MCT1 and ABCC2 gene polymorphisms with valproic acid resistance in patients with epilepsy on valproic acid monotherapy. Drug Metab. Pharm. 2019, 34, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Cho, Y.J.; Kim, W.J.; Lee, M.G.; Lee, J.H. Genetic Variations of ABCC2 Gene Associated with Adverse Drug Reactions to Valproic Acid in Korean Epileptic Patients. Genom. Inf. 2013, 11, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Escalante-Santiago, D.; Feria-Romero, I.A.; Ribas-Aparicio, R.M.; Rayo-Mares, D.; Fagiolino, P.; Vázquez, M.; Escamilla-Núñez, C.; Grijalva-Otero, I.; López-García, M.A.; Orozco-Suárez, S. MDR1 and MRP2 Gene Polymorphisms in Mexican Epileptic Pediatric Patients with Complex Partial Seizures. Front. Neurol. 2014, 9, 184. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannaccone, T.; Sellitto, C.; Manzo, V.; Colucci, F.; Giudice, V.; Stefanelli, B.; Iuliano, A.; Corrivetti, G.; Filippelli, A. Pharmacogenetics of Carbamazepine and Valproate: Focus on Polymorphisms of Drug Metabolizing Enzymes and Transporters. Pharmaceuticals 2021, 14, 204. https://doi.org/10.3390/ph14030204
Iannaccone T, Sellitto C, Manzo V, Colucci F, Giudice V, Stefanelli B, Iuliano A, Corrivetti G, Filippelli A. Pharmacogenetics of Carbamazepine and Valproate: Focus on Polymorphisms of Drug Metabolizing Enzymes and Transporters. Pharmaceuticals. 2021; 14(3):204. https://doi.org/10.3390/ph14030204
Chicago/Turabian StyleIannaccone, Teresa, Carmine Sellitto, Valentina Manzo, Francesca Colucci, Valentina Giudice, Berenice Stefanelli, Antonio Iuliano, Giulio Corrivetti, and Amelia Filippelli. 2021. "Pharmacogenetics of Carbamazepine and Valproate: Focus on Polymorphisms of Drug Metabolizing Enzymes and Transporters" Pharmaceuticals 14, no. 3: 204. https://doi.org/10.3390/ph14030204
APA StyleIannaccone, T., Sellitto, C., Manzo, V., Colucci, F., Giudice, V., Stefanelli, B., Iuliano, A., Corrivetti, G., & Filippelli, A. (2021). Pharmacogenetics of Carbamazepine and Valproate: Focus on Polymorphisms of Drug Metabolizing Enzymes and Transporters. Pharmaceuticals, 14(3), 204. https://doi.org/10.3390/ph14030204