
Monoclonal Antibodies as Neurological Therapeutics

 ,
, 
 and
and
Abstract
1. Introduction
2. Nomenclature
3. Basic Categories of Monoclonal Antibodies
3.1. Murine Antibodies
3.2. Chimeric Antibodies
3.3. Humanized Antibodies
3.4. Fully Human Monoclonal Antibodies
4. Mechanism of Action
4.1. Direct Mechanisms
4.2. Indirect or Immune-Mediated Actions
4.3. Conjugated mAbs
4.4. Bispecific Monoclonal Antibodies
5. Doses, Routes of Administration and Pharmacokinetics
6. Indications in Neurology
6.1. Multiple Sclerosis
6.2. Migraine
6.3. Neuromyelitis Optica Spectrum Disorder (NMOSD)
6.4. Idiopathic Inflammatory Myopathies (IIM)
6.5. Myasthenia Gravis (MG)
6.6. Immune-Mediated Peripheral Neuropathies
6.7. Neurooncology
6.8. Alzheimer’s Disease (AD)
6.9. Parkinson’s Disease (PD)
6.10. Duchene’s Muscular Dystrophy (DMD)
7. Safety Considerations of mAbs
7.1. Infusion-Related Reactions (IRRs)
7.2. Anaphylactic Reactions
7.3. Cytokine Release Syndrome (CRS)
7.4. MAb Immunogenicity and Neutralization
7.5. Opportunistic Infections
7.6. Malignancies
7.7. Secondary Autoimmunity
7.8. Summary of Safety
8. Concluding Comments
Author Contributions
Funding
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Kung, P.; Goldstein, G.; Reinherz, E.L.; Schlossman, S.F. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979, 206, 347–349. [Google Scholar] [CrossRef]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [PubMed]
- Grillo-López, A.J.; White, C.A.; Dallaire, B.K.; Varns, C.L.; Shen, C.D.; Wei, A.; Leonard, J.E.; McClure, A.; Weaver, R.; Cairelli, S.; et al. Rituximab: The first monoclonal antibody approved for the treatment of lymphoma. Curr. Pharm. Biotechnol. 2000, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Warner, J.L.; Arnason, J.E. Alemtuzumab use in relapsed and refractory chronic lymphocytic leukemia: A history and discussion of future rational use. Ther. Adv. Hematol. 2012, 3, 375–389. [Google Scholar] [CrossRef]
- Karlin, L.; Coiffier, B. Ofatumumab in the treatment of non-Hodgkin’s lymphomas. Expert Opin. Biol. Ther. 2015, 15, 1085–1091. [Google Scholar] [CrossRef]
- Kaneko, A. Tocilizumab in rheumatoid arthritis: Efficacy, safety and its place in therapy. Ther. Adv. Chronic Dis. 2013, 4, 15–21. [Google Scholar] [CrossRef]
- Coles, A.J.; Compston, D.A.S.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Lake, S.L.; Moran, S.; Palmer, J.; Smith, M.S.; et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801. [Google Scholar] [CrossRef]
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomized controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Food and Drug Administration: LEMTRADA (Alemtuzumab). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/103948s5158lbl.pdf (accessed on 20 November 2020).
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C.; Johnston, S. Bevacizumab for recurrent alkylator-refractory anaplastic oligodendroglioma. Cancer 2009, 115, 1734–1743. [Google Scholar] [CrossRef]
- Norden, A.D.; Young, G.S.; Setayesh, K.; Muzikansky, A.; Klufas, R.; Ross, G.L.; Ciampa, A.S.; Ebbeling, L.G.; Levy, B.; Drappatz, J.; et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology 2008, 70, 779–787. [Google Scholar] [CrossRef]
- Affronti, M.L.; Jackman, J.G.; McSherry, F.; Herndon, J.E., 2nd; Massey, E.C., Jr.; Lipp, E.; Desjardins, A.; Friedman, H.S.; Vlahovic, G.; Vredenburgh, J.; et al. Phase II Study to Evaluate the Efficacy and Safety of Rilotumumab and Bevacizumab in Subjects with Recurrent Malignant Glioma. Oncologist 2018, 23, 889-e98. [Google Scholar] [CrossRef]
- Bielekova, B. Daclizumab Therapy for Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a034470. [Google Scholar] [CrossRef]
- Gold, R.; Radue, E.W.; Giovannoni, G.; Selmaj, K.; Havrdova, E.K.; Montalban, X.; Stefoski, D.; Sprenger, T.; Robinson, R.R.; Fam, S.; et al. Long-term safety and efficacy of daclizumab beta in relapsing-remitting multiple sclerosis: 6-year results from the SELECTED open-label extension study. J. Neurol. 2020, 267, 2851–2864. [Google Scholar] [CrossRef]
- Giovannoni, G.; Gold, R.; Selmaj, K.; Havrdova, E.; Montalban, X.; Radue, E.W.; Stefoski, D.; McNeill, M.; Amaravadi, L.; Sweetser, M.; et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): A multicentre, randomised, double-blind extension trial. Lancet Neurol. 2014, 13, 472–481. [Google Scholar] [CrossRef]
- Kappos, L.; Wiendl, H.; Selmaj, K.; Arnold, D.L.; Havrdova, E.; Boyko, A.; Kaufman, M.; Rose, J.; Greenberg, S.; Sweetser, M.; et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2015, 373, 1418–1428. [Google Scholar] [CrossRef]
- Diao, L.; Hang, Y.; Othman, A.A.; Mehta, D.; Amaravadi, L.; Nestorov, I.; Tran, J.Q. Population PK-PD analyses of CD25 occupancy, CD56bright NK cell expansion, and regulatory T cell reduction by daclizumab HYP in subjects with multiple sclerosis. Br. J. Clin. Pharmacol. 2016, 82, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. EMA Urgently Reviewing Multiple Sclerosis Medicine Zinbryta Following Cases of Inflammatory Brain Disorders; Press Release 02/03/2018. Available online: https://www.ema.europa.eu/en/news/ema-urgently-reviewing-multiple-sclerosis-medicine-zinbryta-following-cases-inflammatory-brain (accessed on 20 November 2020).
- Luessi, F.; Engel, S.; Spreer, A.; Bittner, S.; Zipp, F. GFAPalpha IgG-associated encephalitis upon daclizumab treatment of MS. Neurol. NeuroImmunol. Neuroinflamm. 2018, 5, e481. [Google Scholar] [CrossRef] [PubMed]
- Avasarala, J. DRESS Syndrome and Daclizumab Failure-Were Potentially Dangerous Signs Missed in Clinical Trials? Drug Target. Insights 2018, 12, 1177392818785136. [Google Scholar] [CrossRef] [PubMed]
- Cortese, I.; Ohayon, J.; Fenton, K.; Lee, C.C.; Raffeld, M.; Cowen, E.W.; DiGiovanna, J.J.; Bielekova, B. Cutaneous adverse events in multiple sclerosis patients treated with daclizumab. Neurology 2016, 86, 847–855. [Google Scholar] [CrossRef]
- Cohan, S.L.; Lucassen, E.B.; Romba, M.C.; Linch, S.N. Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis. Biomedicines 2019, 7, 18. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Xue, T.; Yang, Y.; Lu, Q.; Gao, B.; Chen, Z.; Wang, Z. Efficacy and Safety of Monoclonal Antibody Therapy in Neuromyelitis Optica Spectrum Disorders: Evidence from Randomized Controlled Trials. Mult. Scler. Relat. Disord. 2020, 43, 102166. [Google Scholar] [CrossRef]
- Akaishi, T.; Nakashima, I. Efficiency of antibody therapy in demyelinating diseases. Int. Immunol. 2017, 29, 327–335. [Google Scholar] [CrossRef]
- Howard, J.F.; Utsugisawa, K.; Benatar, M.; Murai, H.; Barohn, R.J.; Illa, I.; Jacob, S.; Vissing, J.; Burns, T.M.; Kissel, J.T.; et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): A phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017, 16, 976–986. [Google Scholar] [CrossRef]
- Nishimura, J.; Yamamoto, M.; Hayashi, S.; Ohyashiki, K.; Ando, K.; Brodsky, A.L.; Noji, H.; Kitamura, K.; Eto, T.; Takahashi, T.; et al. Genetic variants in C5 and poor response to eculizumab. N. Engl. J. Med. 2014, 370, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Saper, J.; Cady, R.; Schaeffler, B.A.; Biondi, D.M.; Hirman, J.; Pederson, S.; Allan, B.; Smith, J. Eptinezumab in episodic migraine: A randomized, double-blind, placebo-controlled study (PROMISE-1). Cephalalgia 2020, 40, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Reuter, U.; Hallström, Y.; Broessner, G.; Bonner, J.H.; Zhang, F.; Sapra, S.; Picard, H.; Mikol, D.D.; Lenz, R.A. A Controlled Trial of Erenumab for Episodic Migraine. N. Engl. J. Med. 2017, 377, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Goadsby, P.J.; Reuter, U.; Silberstein, S.; Dodick, D.W.; Xue, F.; Zhang, F.; Paiva da Silva Lima, G.; Cheng, S.; Mikol, D.D. Long-term efficacy and safety of erenumab in migraine prevention: Results from a 5-year, open-label treatment phase of a randomized clinical trial. Eur. J. Neurol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Dodick, D.W.; Ashina, M.; Brandes, J.L.; Kudrow, D.; Lanteri-Minet, M.; Osipova, V.; Palmer, K.; Picard, H.; Mikol, D.D.; Lenz, R.A. ARISE: A phase 3 randomized trial of erenumab for episodic migraine. Cephalalgia 2018, 38, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Tepper, S.; Ashina, M.; Reuter, U.; Brandes, J.L.; Doležil, D.; Silberstein, S.; Winner, P.; Leonardi, D.; Mikol, D.; Lenz, R. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double-blind, placebo–controlled phase 2 trial. Lancet Neurol. 2017, 16, 425–434. [Google Scholar] [CrossRef]
- Ashina, M.; Dodick, D.; Goadsby, P.J.; Reuter, U.; Silberstein, S.; Zhang, F.; Gage, J.R.; Cheng, S.; Mikol, D.D.; Lenz, R.A. Erenumab (AMG 334) in episodic migraine: Interim analysis of an ongoing open-label study. Neurology 2017, 89, 1237–1243. [Google Scholar] [CrossRef]
- Dodick, D.W.; Silberstein, S.D.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Effect of Fremanezumab Compared With Placebo for Prevention of Episodic Migraine: A Randomized Clinical Trial. JAMA 2018, 319, 1999–2008. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Silberstein, S.D.; Yeung, P.P.; Cohen, J.M.; Ning, X.; Yang, R.; Dodick, D.W. Long-term safety, tolerability, and efficacy of fremanezumab in migraine: A randomized study. Neurology 2020, 95, e2487–e2499. [Google Scholar] [CrossRef]
- Stauffer, V.L.; Dodick, D.W.; Zhang, Q.; Carter, J.N.; Ailani, J.; Conley, R.R. Evaluation of Galcanezumab for the Prevention of Episodic Migraine: The EVOLVE-1 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1080–1088. [Google Scholar] [CrossRef]
- Skljarevski, V.; Matharu, M.; Millen, B.A.; Ossipov, M.H.; Kim, B.K.; Yang, J.Y. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. Cephalalgia 2018, 38, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Detke, H.C.; Goadsby, P.J.; Wang, S.; Friedman, D.I.; Selzler, K.J.; Aurora, S.K. Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. Neurology 2018, 91, e2211–e2221. [Google Scholar] [CrossRef] [PubMed]
- Gklinos, P.; Mitsikostas, D.D. Galcanezumab in migraine prevention: A systematic review and meta-analysis of randomized controlled trials. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420918088. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Dodick, D.W.; Leone, M.; Bardos, J.N.; Oakes, T.M.; Millen, B.A.; Zhou, C.; Dowsett, S.A.; Aurora, S.K.; Ahn, A.H.; et al. Trial of galcanezumab in prevention of episodic cluster headache. N. Engl. J. Med. 2019, 381, 132–141. [Google Scholar] [CrossRef]
- Camporeale, A.; Kudrow, D.; Sides, R.; Wang, S.; Van Dycke, A.; Selzler, K.J.; Stauffer, V.L. A phase 3, long-term, open-label safety study of Galcanezumab in patients with migraine. BMC Neurol. 2018, 18, 188. [Google Scholar] [CrossRef]
- Martinez, J.M.; Hindiyeh, N.; Anglin, G.; Kalidas, K.; Hodsdon, M.E.; Kielbasa, W.; Moser, B.A.; Pearlman, E.M.; Garces, S. Assessment of immunogenicity from galcanezumab phase 3 trials in patients with episodic or chronic migraine. Cephalalgia 2020, 40, 978–989. [Google Scholar] [CrossRef]
- Agius, M.A.; Klodowska-Duda, G.; Maciejowski, M.; Potemkowski, A.; Li, J.; Patra, K.; Wesley, J.; Madani, S.; Barron, G.; Katz, E.; et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult. Scler. 2019, 25, 235–245. [Google Scholar] [CrossRef]
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010, 133, 349–361. [Google Scholar] [CrossRef]
- Kuroda, R.; Suzuki, J.; Muramatsu, M.; Tasaki, A.; Yano, M.; Imai, N.; Serizawa, M.; Kobari, M. Efficacy of infliximab in neuro-Behçet’s disease presenting with isolated longitudinally extensive transverse myelitis. J. Neurol. 2013, 260, 3167–3170. [Google Scholar] [CrossRef] [PubMed]
- Fritz, D.; Timmermans, W.M.C.; van Laar, J.A.M.; van Hagen, P.M.; Siepman, T.A.M.; van de Beek, D.; Brouwer, M.C. Infliximab treatment in pathology-confirmed neurosarcoidosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e847. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Schürpf, T.; Springer, T.A. How natalizumab binds and antagonizes α4 integrins. J. Biol. Chem. 2013, 288, 32314–32325. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef]
- Miller, D.H.; Soon, D.; Fernando, K.T.; MacManus, D.G.; Barker, G.J.; Yousry, T.A.; Fisher, E.; O’Connor, P.W.; Phillips, J.T.; Polman, C.H.; et al. AFFIRM Investigators. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology 2007, 68, 1390–1401. [Google Scholar] [CrossRef]
- Butzkueven, H.; Kappos, L.; Wiendl, H.; Trojano, M.; Spelman, T.; Chang, I.; Kasliwal, R.; Jaitly, S.; Campbell, N.; Ho, P.R.; et al. Tysabri Observational Program (TOP) Investigators. Long-term safety and effectiveness of natalizumab treatment in clinical practice: 10 years of real-world data from the Tysabri Observational Program (TOP). J. Neurol. Neurosurg. Psychiatry 2020, 91, 660–668. [Google Scholar] [CrossRef]
- Defer, G.; Mariotte, D.; Derache, N.; Toutirais, O.; Legros, H.; Cauquelin, B.; Le Mauff, B. CD49d expression as a promising biomarker to monitor natalizumab efficacy. J. Neurol. Sci. 2012, 314, 138–142. [Google Scholar] [CrossRef]
- Vennegoor, A.; Rispens, T.; Strijbis, E.M.; Seewann, A.; Uitdehaag, B.M.; Balk, L.J.; Barkhof, F.; Polman, C.H.; Wolbink, G.; Killestein, J. Clinical relevance of serum natalizumab concentration and anti-natalizumab antibodies in multiple sclerosis. Mult. Scler. 2013, 19, 593–600. [Google Scholar] [CrossRef]
- Jensen, P.E.; Koch-Henriksen, N.; Sellebjerg, F.; Sorensen, P.S. Prediction of antibody persistency from antibody titres to natalizumab. Mult. Scler. 2012, 18, 1493–1499. [Google Scholar] [CrossRef]
- Lundkvist, M.; Engdahl, E.; Holmen, C.; Moverare, R.; Olsson, T.; Hillert, J.; Fogdell-Hahn, A. Characterization of anti-natalizumab antibodies in multiple sclerosis patients. Mult. Scler. 2013, 19, 757–764. [Google Scholar] [CrossRef]
- Chisari, C.G.; Grimaldi, L.M.; Salemi, G.; Ragonese, P.; Iaffaldano, P.; Bonavita, S.; Sparaco, M.; Rovaris, M.; D’Arma, A.; Lugaresi, A.; et al. Clinical effectiveness of different natalizumab interval dosing schedules in a large Italian population of patients with multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.M.; Pula, J.H.; Garwacki, D.; Cotter, J.; Kattah, J.C. Cryptococcal meningitis in a multiple sclerosis patient taking natalizumab. J. Neurol. Sci. 2014, 340, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Dahdaleh, D.; Altmann, D.M.; Malik, O.; Nicholas, R.S. Breathlessness, night sweats, and weight loss on natalizumab. Lancet 2012, 380, 726–727. [Google Scholar] [CrossRef]
- Rudick, R.A.; Stuart, W.H.; Calabresi, P.A.; Confavreux, C.; Galetta, S.L.; Radue, E.W.; Lublin, F.D.; Weinstock-Guttman, B.; Wynn, D.R.; Lynn, F.; et al. SENTINEL Investigators. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 911–923. [Google Scholar] [CrossRef]
- Antezana, A.; Sigal, S.; Herbert, J.; Kister, I. Natalizumab-induced hepatic injury: A case report and review of literature. Mult. Scler. Relat. Disord. 2015, 4, 495–498. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Sul, J.; Patel, A.; Gordon, M.L.; Steinklein, J.; Sanguinetti, S.; Pramanik, B.; Orban, Z.; Koralnik, I.; Harel, A. Progressive Multifocal Leukoencephalopathy in a Patient on Ocrelizumab Monotherapy. 62nd Annual meeting of the American Academy of Neurology (AAN), Toronto, Canada, Abstract S29.001. Neurology 2020, 94 (Suppl. 15), 4875. [Google Scholar]
- Ocrelizumab EMA Summary of Project Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/ocrevus-epar-product-information_en.pdf (accessed on 20 November 2020).
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef]
- Golay, J.; Semenzato, G.; Rambaldi, A.; Foà, R.; Gaidano, G.; Gamba, E.; Pane, F.; Pinto, A.; Specchia, G.; Zaja, F.; et al. Lessons for the clinic from rituximab pharmacokinetics and pharmacodynamics. MAbs 2013, 5, 826–837. [Google Scholar] [CrossRef]
- Komori, M.; Lin, Y.C.; Cortese, I.; Blake, A.; Ohayon, J.; Cherup, J.; Maric, D.; Kosa, P.T.; Wu, T.; Bielekova, B. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann. Clin. Transl. Neurol. 2016, 3, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Waubant, E.; Arnold, D.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, S.G.; Kristoffersson, A.; Boström, I.; Feresiadou, A.; Burman, J.; Landtblom, A.M. Rapidly increasing off-label use of rituximab in multiple sclerosis in Sweden—Outlier or predecessor? Acta Neurol. Scand. 2018, 138, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Pellkofer, H.L.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Gerdes, L.A.; Havla, J.; Bittner, R.; Canis, M.; Meinl, E.; Hohlfeld, R.; et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011, 76, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Lamb, S.; Morgan, K.; Chen, A.; Waubant, E.; Genain, C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005, 64, 1270–1272. [Google Scholar] [CrossRef]
- Jacob, A.; Weinshenker, B.G.; Violich, I.; McLinskey, N.; Krupp, L.; Fox, R.J.; Wingerchuk, D.M.; Boggild, M.; Constantinescu, C.S.; Miller, A.; et al. Treatment of neuromyelitis optica with rituximab: Retrospective analysis of 25 patients. Arch Neurol. 2008, 65, 1443–1448. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kim, W.; Li, X.F.; Jung, I.-J.; Kim, H.J. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011, 68, 1412–1420. [Google Scholar] [CrossRef]
- Bedi, G.S.; Brown, A.D.; Delgado, S.R.; Usmani, N.; Lam, B.L.; Sheremata, W.A. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult. Scler. 2011, 17, 1225–1230. [Google Scholar] [CrossRef]
- Kim, S.H.; Huh, S.Y.; Lee, S.J.; Joung, A.; Kim, H.J. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013, 70, 1110–1117. [Google Scholar] [CrossRef]
- Levine, T.D. Rituximab in the treatment of dermatomyositis. Arthritis Rheum. 2005, 52, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, M.; Dalakas, M. Practical considerations on the use of rituximab in autoimmune neurological disorders. Therap. Adv. Neurol. Disord. 2010, 3, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Oddis, C.V.; Reed, A.M.; Aggarwal, R.; Rider, L.G.; Ascherman, D.P.; Levesque, M.C.; Barohn, R.J.; Feldman, B.M.; Harris-Love, M.O.; Koontz, D.C.; et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheum 2013, 65, 314e24. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Bandos, A.; Reed, A.M.; Ascherman, D.P.; Barohn, R.J.; Feldman, B.M.; Miller, F.W.; Rider, L.G.; Harris-Love, M.O.; Levesque, M.C.; et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 2014, 66, 740e9. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Loganathan, P.; Koontz, D.; Qi, Z.; Reed, A.M.; Oddis, C.V. Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatol 2017, 56, 247e54. [Google Scholar] [CrossRef] [PubMed]
- Brauner, S.; Eriksson-Dufva, A.; Albert Hietala, M.; Frisell, T.; Press, R.; Piehl, F. Comparison between rituximab treatment for new-onset generalized myasthenia gravis and refractory generalized myasthenia gravis. JAMA Neurol. 2020, e200851. [Google Scholar] [CrossRef] [PubMed]
- Beecher, G.; Anderson, D.; Siddiqi, Z.A. Rituximab in refractory myasthenia gravis: Extended prospective study results. Muscle Nerve. 2018, 58, 452–455. [Google Scholar] [CrossRef]
- Illa, I.; Diaz-Manera, J.; Rojas-Garcia, R.; Pradas, J.; Rey, A.; Blesa, R.; Juarez, C.; Gallardo, E. Sustained response to rituximab in anti-AchR and anti-MuSKpositive myasthenia gravis patients. J. Neuroimmun. 2008, 201–202, 90–94. [Google Scholar] [CrossRef]
- Iorio, R.; Damato, V.; Alboini, P.E.; Evoli, A. Efficacy and safety of rituximab for myasthenia gravis: A systematic review and meta-analysis. J. Neurol. 2015, 262, 1115–1119. [Google Scholar] [CrossRef]
- Tandan, R.; Hehir, M.K.; Waheed, W.; Howard, D.B. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve 2017, 56, 185–196. [Google Scholar] [CrossRef]
- Hehir, M.K.; Hobson-Webb, L.D.; Benatar, M. Rituximab as treatment for anti-MuSK myasthenia gravis. Neurology 2017, 89, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Stieglbauer, K.; Pihler, R.; Topakian, R. 10-year-outcomes after rituximab for myasthenia gravis: Efficacy, safety, costs of in hospital care, and impact on childbearing potential. J. Neurol. Sci. 2017, 375, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, A.; Noury, J.-B.; Genetest, S.; Nadaj-Pakleza, A.; Cassereau, J.; Baron, C.; Videt, D.; Michel, L.; Pereon, Y.; Wiertlewki, S.; et al. Efficacy and safety of Rituximab in myasthenia gravis: A French multicentre real-life study. Eur. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dıaz-Manera, J.; Martınez-Hernandez, E.; Querol, L.; Klooster, R.; Rojas-García, R.; Suárez-Calvet, X.; Muñoz-Blanco, J.L.; Mazia, C.; Straasheijm, K.R.; Gallardo, E.; et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012, 78, 189–193. [Google Scholar] [CrossRef]
- Marino, M.; Basile, U.; Spagni, G. long-lasting rituximab-induced reduction of specific—But not total—IgG4 in MuSK-positive myasthenia gravis. Front. Immunol. 2020, 11, 613. [Google Scholar] [CrossRef]
- Lebrun, C.; Bourg, V.; Bresch, S.; Cohen, M.; Rosenthal-Allieri, M.A.; Desnuelle, C. Ticchioni, Therapeutic target of memory B cells depletion helps to tailor administration frequency of rituximab in myasthenia gravis. J. Neuroimmunol. 2016, 298, 79–81. [Google Scholar] [CrossRef]
- Ruegg, S.; Fuhr, P.; Steck, A. Rituximab stabilizes multifocal motor neuropathy increasingly less responsive to IVIg. Neurology 2004, 63, 2178–2179. [Google Scholar] [CrossRef]
- Gorson, K.C.; Natarajan, N.; Ropper, A.H.; Weinstein, R. Rituximab treatment in patients with IVIg-dependent immune polyneuropathy: A prospective pilot trial. Muscle Nerve 2007, 35, 66–69. [Google Scholar] [CrossRef]
- Dalakas, M.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.; Hahn, A.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293. [Google Scholar] [CrossRef]
- Léger, J.M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. RIMAG Study Group (France and Switzerland). Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy. Neurology 2013, 80, 2217–2225. [Google Scholar] [CrossRef]
- Gazzola, S.; Delmont, E.; Franques, J.; Boucraut, J.; Salort-Campana, E.; Verschueren, A.; Sagui, E.; Hubert, A.M.; Pouget, J.; Attarian, S. Predictive factors of efficacy of rituximab in patients with anti-MAG neuropathy. J. Neurol. Sci. 2017, 377, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Muley, S.A.; Jacobsen, B.; Parry, G.; Usman, U.; Ortega, E.; Walk, D.; Allen, J.; Pasnoor, M.; Varon, M.; Dimachkie, M.M. Rituximab in refractory chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2020, 61, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Van Vollenhoven, R.F.; Emery, P.; Bingham, C.O., 3rd; Keystone, E.C.; Fleischmann, R.M.; Furst, D.E.; Tyson, N.; Collinson, N.; Lehane, P.B. Long-term safety of rituximab in rheumatoid arthritis: 9.5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann. Rheum. Dis. 2013, 72, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: A randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet. Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef]
- Araki, M.; Aranami, T.; Matsuoka, T.; Nakamura, M.; Miyake, S.; Yamamura, T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod. Rheumatol. 2013, 23, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Kieseier, B.C.; Stüve, O.; Dehmel, T.; Goebels, N.; Leussink, V.I.; Mausberg, A.K.; Ringelstein, M.; Turowski, B.; Aktas, O.; Anoch, G.; et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: Implication for cellular immune responses. JAMA Neurol. 2012, 70, 390–393. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Tocilizumab in the Treatment of Refractory Polymyositis and Dermatomyositis (TIM). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02043548,NCT02043548 (accessed on 20 November 2010).
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56, Update in: Nature 2017, 546, 564. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. 2020.221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease (EMERGE). Available online: https://clinicaltrials.gov/ct2/show/NCT02484547,NCT02484547 (accessed on 20 November 2020).
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen claims aducanumab turnaround. Nat. Rev. Neurol. 2020, 16, 63–64. [Google Scholar] [CrossRef]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Zhang, H.; Saadoun, S.; Phuan, P.W.; Lam, C.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann. Neurol. 2012, 71, 314–322. [Google Scholar] [CrossRef]
- Duan, T.; Tradtrantip, L.; Phuan, P.W.; Bennett, J.L.; Verkman, A.S. Affinity-matured ‘aquaporumab’ anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology 2020, 162, 107827. [Google Scholar] [CrossRef] [PubMed]
- Clinical trials.gov. A Study to Evaluate the Efficacy, Safety and PD and PK of HBM9161 in MG Patients. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04346888?term=HBM9161&draw=2&rank=2 (accessed on 20 November 2020).
- Kwon, S.; Iba, M.; Kim, C.; Masliah, E. Immunotherapies for Aging-Related Neurodegenerative Diseases-Emerging Perspectives and New Targets. Neurotherapeutics 2020, 17, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Kuchimanchi, M.; Monine, M.; Kandadi Muralidharan, K.; Woodward, C.; Penner, N. Phase II Dose Selection for Alpha Synuclein-Targeting Antibody Cinpanemab (BIIB054) Based on Target Protein Binding Levels in the Brain CPT. Pharmacomet. Syst. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401—A clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- EISAI 2020 News Release. Initiation of New Phase III Clinical Study (ahead 3-45) of Ban2401 Preclinical (Asymptomatic) ALZHEIMER’S Disease. Available online: https://www.eisai.com/news/2020/news202042.html (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study to Evaluate Safety, Tolerability, and Efficacy of Lecanemab in Subjects with Early Alzheimer’s Disease. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT01767311,NCT01767311 (accessed on 20 November 2020).
- Howard, J.F., Jr.; Bril, V.; Burns, T.M.; Mantegazza, R.; Bilinska, M.; Szczudlik, A.; Beydoun, S.; Garrido, F.J.R.R.; Piehl, F.; Rottoli, M.; et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 2019, 92, e2661–e2673. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A Safety and Tolerability Study of ARGX-113 in Patients with Myasthenia Gravis Who Have Generalized Muscle Weakness. (ADAPT+). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03770403,NCT03770403 (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study to Assess the Safety and Efficacy of a Subcutaneous Formulation of Efgartigimod in Adults with Chronic Inflammatory Demyelinating Polyneuropathy (CIDP, an Autoimmune Disorder That Affects the Peripheral Nerves) (ADHERE). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04281472,NCT04281472 (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study of Gantenerumab in Participants with Mild Alzheimer Disease. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02051608?term=gantenerumab&draw=2&rank=9 (accessed on 20 November 2020).
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. SCarlet RoAD Investigators. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Biogen. Biogen Reports Top-Line Results from Phase 2 Study in Progressive Supranuclear Palsy. 2019. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-reports-top-line-results-phase-2-study-progressive (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study to Evaluate the Safety, Tolerability, Efficacy, Pharmacokinetics and Pharmacodynamics of M281 Administered to Adults With Generalized Myasthenia Gravis. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03772587,NCT03772587 (accessed on 20 November 2020).
- Mi, S.; Hu, B.; Hahm, K.; Luo, Y.; Kam Hui, E.S.; Yuan, Q.; Wong, W.M.; Wang, L.; Su, H.; Chu, T.H.; et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat. Med. 2007, 13, 1228–1233. [Google Scholar] [CrossRef]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Frederiksen, J.; Skeen, M.; Jaffe, G.J.; Butzkueven, H.; et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): A randomised, placebo controlled, phase 2 trial. Lancet Neurol. 2017, 16, 189–199. [Google Scholar] [CrossRef]
- Cadavid, D.; Mellion, M.; Hupperts, M.; Edwards, K.R.; Calabresi, P.A.; Drulović, J.; Giovannoni, G.; Hartung, H.P.; Arnold, D.L.; Fisher, E.; et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2019, 18, 845–856. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. An Efficacy and Safety Study of Ravulizumab in Adult Participants with NMOSD. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04201262,NCT04201262 (accessed on 20 November 2020).
- ClinicalTrials.Gov. Safety and Efficacy Study of Ravulizumab in Adults with Generalized Myasthenia Gravis. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03920293,NCT03920293 (accessed on 20 November 2020).
- Wen, P.Y.; Schiff, D.; Cloughesy, T.F.; Raizer, J.J.; Laterra, J.; Smitt, M.; Wolf, M.; Oliner, K.S.; Anderson, A.; Zhu, M.; et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro-Oncology 2011, 13, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Bril, V.; Benatar, M.; Andersen, H.; Vissing, J.; Brock, M.; Greve, B.; Kiessling, P.; Woltering, F.; Griffin, L.; Van den Bergh, P. MG0002 Investigators. Efficacy and safety of rozanolixizumab in moderate-to-severe generalised myasthenia gravis: A phase 2 RCT. Neurology 2020, 20. [Google Scholar] [CrossRef]
- World Health Organization. International Nonproprietary Names (INN) for Biological and Biotechnological Substances (A Review). Available online: http://www.who.int/medicines/services/inn/BioRev2014.pdf (accessed on 25 July 2017).
- World Health Organization. Revised Monoclonal Antibody (mAb) Nomenclature Scheme. Geneva, 26 May 2017. Available online: http://www.who.int/medicines/services/inn/Revised_mAb_nomenclature_scheme.pdf?ua=1 (accessed on 25 July 2017).
- Stevenson, J.G.; Green, L. Biologics, Pharmacovigilance, and Patient Safety: It’s all in the Name. J. Manag. Care Spec. Pharm. 2016, 22, 927–930. [Google Scholar] [CrossRef]
- Weiner, L.M.; Adams, G.P.; Von Mehren, M. Therapeutic monoclonal antibodies: General principles. In Cancer Principles and Practice and Oncology, 6th ed.; DeVita, V.T., Hellman, H., Rosenberg, S.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 495–508. [Google Scholar]
- Chirmule, N.; Jawa, V.; Meibohm, B. Immunogenicity to therapeutic proteins: Impact on PK/PD and efficacy. AAPS J. 2012, 14, 296–302. [Google Scholar] [CrossRef]
- Van den Bemt, B.J.; Wolbink, G.J.; Hekster, Y.A.; van Riel, P.L.C.M.; Benraad, B.; van den Hoogen, F.H.J. Anti-infliximab antibodies are already detectable in most patients with rheumatoid arthritis halfway through an infusion cycle: An open-label pharmacokinetic cohort study. BMC Musculoskelet. Disord. 2011, 12, 12. [Google Scholar] [CrossRef]
- Boulianne, G.L.; Hozumi, N.; Shulman, M.J. Production of functional chimaeric mouse/human antibody. Nature 1984, 312, 643–646. [Google Scholar] [CrossRef]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Steinitz, M. Three decades of human monoclonal antibodies: Past, present and future developments. Human. Antibodies 2009, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jespers, L.S.; Roberts, A.; Mahler, S.M.; Winter, G.; Hoogenboom, H.R. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology (NY) 1994, 12, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R.; Strohl, L.M. 8-Monoclonal antibody targets and mechanisms of action. In Therapeutic Antibody Engineering; Series in Biomedicine; Woodhead Publishing Limited: Cambridge, UK, 2012; pp. 163–595. ISBN 9781907568374. [Google Scholar] [CrossRef]
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Zafir-Lavie, I.; Michaeli, Y.; Reiter, Y. Novel antibodies as anticancer agents. Oncogene 2007, 26, 3714–3733. [Google Scholar] [CrossRef]
- Russell, F.A.; King, R.; Smillie, S.-J.; Kodji, X.; Brain, S.D. Calcitonin Gene-Related Peptide: Physiology and Pathophysiology. Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef]
- Mitsikostas, D.D.; Rapoport, A.M. New players in the preventive treatment of migraine. BMC Med. 2015, 13, 279. [Google Scholar] [CrossRef]
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011, 19, 101–113. [Google Scholar] [CrossRef]
- Dobson, C.L.; Main, S.; Newton, P.; Chodorge, M.; Cadwallander, K.; Humphreys, R.; Albert, V.; Vaughan, T.J.; Minter, R.R.; Edwards, B.M. Human monomeric antibody fragments to TRAIL-R1 and TRAIL-R2 that display potent in vitro agonism. mAbs 2009, 1, 552–562. [Google Scholar] [CrossRef]
- Suzuki, M.; Kato, C.; Kato, A. Therapeutic antibodies: Their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol. 2015, 28, 133–139. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439. [Google Scholar] [CrossRef]
- Yu, J.; Song, Y.; Tian, W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45. [Google Scholar] [CrossRef]
- Teicher, B.A.; Chari, R.V. Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. 90 Y-ibritumomab tiuxetan: A nearly forgotten opportunity. Oncotarget 2016, 7, 7597–7609. [Google Scholar] [CrossRef]
- Turshudzhyan, A. The role of ado-trastuzumab emtansine in current clinical practice. J. Oncol. Pharm. Pract. 2021, 27, 150–155. [Google Scholar] [CrossRef]
- Husain, B.; Ellerman, D. Expanding the Boundaries of Biotherapeutics with Bispecific Antibodies. BioDrugs 2018, 32, 441–464. [Google Scholar] [CrossRef]
- Weber, F.; Bohrmann, B.; Niewoehner, J.; Fischer, J.A.A.; Rueger, P.; Tiefenthaler, G.; Moelleken, J.; Bujotzek, A.; Brady, L.; Singer, T.; et al. Brain shuttle antibody for Alzheimer’s disease with attenuated peripheral effector function due to an inverted binding mode. Cell Rep. 2018, 22, 149–162. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug. Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C.; et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N. Engl. J. Med. 2017, 377, 809–818. [Google Scholar] [CrossRef]
- He, P.; Xin, W.; Schulz, P.; Sierks, M.R. Bispecific Antibody Fragment Targeting APP and Inducing α-Site Cleavage Restores Neuronal Health in an Alzheimer’s Mouse Model. Mol. Neurobiol. 2019, 56, 7420–7432. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Z.; Lv, L.; Qiao, H.; Chen, X.; Zou, C. A bispecific antibody (ScBsAbAgn-2/TSPO) target for Ang-2 and TSPO resulted in therapeutic effects against glioblastomas. Biochem. Biophys. Res. Commun. 2016, 472, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/ VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef]
- Newsome, B.W.; Ernstoff, M.S. The clinical pharmacology of therapeutic monoclonal antibodies in the treatment of malignancy; have the magic bullets arrived? Br. J. Clin. Pharmacol. 2008, 66, 6. [Google Scholar] [CrossRef]
- Loffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef]
- Goebl, N.A.; Babbey, C.M.; Datta-Mannan, A.; Witcher, D.R.; Wroblewski, V.J.; Dunn, K.W. Neonatal Fc receptor mediates internalization of Fc in transfected human endothelial cells. Mol. Biol. Cell. 2008, 19, 5490–5505. [Google Scholar] [CrossRef]
- Goel, N.; Stephens, S. Certolizumab pegol. mAbs 2010, 2, 137–147. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Bolz, J.; Peschke, M.; Duscha, A.; Hellwig, K.; Lee, D.H.; Linker, R.A.; Gold, R.; Haghikia, A. Peripheral CD19(+) B-cell counts and infusion intervals as a surrogate for long-term B-cell depleting therapy in multiple sclerosis and neuromyelitis optica/neuromyelitis optica spectrum disorders. J. Neurol. 2019, 266, 57–67. [Google Scholar] [CrossRef]
- Oller-Salvia, B.; Sanchez-Navarro, M.; Giralt, E.; Teixido, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef]
- Sharma, G.; Lakkadwala, S.; Modgil, A.; Singh, J. The role of cell-penetrating peptide and transferrin on enhanced delivery of drug to brain. Int. J. Mol. Sci. 2016, 17, 806. [Google Scholar] [CrossRef]
- Thom, G.; Hatcher, J.; Hearn, A.; Paterson, J.; Rodrigo, N.; Beljean, A.; Gurrell, I.; Webster, C. Isolation of blood-brain barrier-crossing antibodies from a phage display library by competitive elution and their ability to penetrate the central nervous system. MAbs 2018, 10, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Razpotnik, R.; Novak, N.; Curin, Šerbec, V.; Rajcevic, U. Targeting Malignant Brain Tumors with Antibodies. Front. Immunol. 2017, 8, 1181. [Google Scholar] [CrossRef] [PubMed]
- The Lenercept Multiple Sclerosis Study Group; The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457–465. [Google Scholar] [CrossRef]
- Vollmer, T.L.; Wynn, D.R.; Alam, M.S.; Valdes, J. A phase 2, 24-week, randomized, placebo-controlled, double-blind study examining the efficacy and safety of an anti-interleukin-12 and -23 monoclonal antibody in patients with relapsing-remitting or secondary progressive multiple sclerosis. Mult. Scler. 2011, 17, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.M.; Constantinescu, C.S.; Raychaudhuri, A.; Kim, L.; Fidelus-Gort, R.; Kasper, L.H. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: A phase II, double-blind, placebo-controlled, randomised, dose ranging study. Lancet Neurol. 2008, 7, 796–804. [Google Scholar] [CrossRef]
- European Medicines Agency. LEMTRADA Product Information. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lemtrada (accessed on 20 November 2020).
- Hartung, H.-P.; Aktas, O. Bleak prospects for primary progressive multiple sclerosis therapy: Downs and downs, but a glimmer of hope. Ann. Neurol. 2009, 66, 429–432. [Google Scholar] [CrossRef]
- Vogel, A.C.; Schmidt, H.; Loud, S.; McBurney, R.; Mateen, F.J. Impact of the COVID-19 pandemic on the health care of >1000 People living with multiple sclerosis: A cross-sectional study. Mult. Scler. Relat. Disord. 2020, 46, 102512. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Joyner, C.J.; Sanz, I.; Lee, F.E.-H. Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front. Immunol. 2019, 10, 2138. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Kokoti, L.; Drellia, K.; Papadopoulos, D.; Mitsikostas, D.D. Placebo and nocebo phenomena in anti-CGRP monoclonal antibody trials for migraine prevention: A meta-analysis. J. Neurol. 2020, 267, 1158–1170. [Google Scholar] [CrossRef]
- Drellia, K.; Kokoti, L.; Dilligianni, C.; Papadopoulos, D.; Mitsikostas, D.D. Anti-CGRP Monoclonal Antibodies For Migraine Prevention: A Systematic Review And Likelihood To Help Or Harm Analysis. Cephalalgia 2021, in press. [Google Scholar]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kümpfel, T. Neuromyelitis Optica Study Group (NEMOS). Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed]
- Ayzenberg, I.; Kleiter, I.; Schröder, A.; Hellwig, K.; Chan, A.; Yamamura, T.; Gold, R. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013, 70, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.; Paik, J.J. Inflammatory muscle disease-An update. Best. Pract. Res. Clin. Rheumatol. 2020, 34, 101484. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.E.; Dastmalchi, M. Possible pathogenic mechanisms in inflammatory myopathies. Rheum. Dis. Clin. N. Am. 2002, 28, 799–822. [Google Scholar] [CrossRef]
- Schiffenbauer, A.; Garg, M.; Castro, C.; Pokrovnichka, A.; Joe, G.; Shrader, J.; Cabalar, I.V.; Faghihi-Kashani, S.; Harris-Love, M.O.; Plotz, P.H.; et al. A randomized, double-blind, placebo-controlled trial of infliximab in refractory polymyositis and dermatomyositis. Semin. Arthritis Rheum. 2018, 47, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G.; Badrising, U.A.; Benveniste, O.; Lloyd, T.E.; Needham, M.; Chinoy, H.; Aoki, M.; Machado, P.M.; Liang, C.; Reardon, K.A.; et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): A randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. 2019, 18, 834–844. [Google Scholar] [CrossRef]
- Dalakas, M.; Rakocevic, G.; Schmidt, J.; Salajegheh, M.; McElroy, B.; Harris-Love, M.O.; Shrader, J.A.; Levy, E.W.; Dambrosia, J.; Kampen, R.L.; et al. Effect of alemtuzumab (CAMPATH-1H) in patients with inclusion-body myositis. Brain 2009, 132, 1536–1544. [Google Scholar] [CrossRef]
- Dalakas, M.C. Immunotherapy in myasthenia gravis in the era of biologics. Nat. Rev. Neurol. 2019, 15, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Espié, P.; He, Y.; Koo, P.; Sickert, D.; Dupuy, C.; Chokoté, E.; Schuler, R.; Mergentaler, H.; Ristov, J.; Milojevic, J.; et al. First-in-human clinical trial to assess pharmacokinetics, pharmacodynamics, safety, and tolerability of iscalimab, an anti-CD40 monoclonal antibody. Am. J. Transplant. 2020, 20, 463–473. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Safety, Tolerability, Pharmacokinetics and Efficacy of CFZ533 in Moderate to Severe Myasthenia Gravis. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT02565576,NCT02565576 (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study of RVT-1401 in Myasthenia Gravis (MG) Patients. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03863080,NCT03863080 (accessed on 20 November 2020).
- Dalakas, M.C. Advances in the diagnosis, immunopathogenesis and therapies of IgM-anti-MAG antibody-mediated neuropathies. Ther. Adv. Neurol. Disord. 2018, 11, 1756285617746640. [Google Scholar] [CrossRef] [PubMed]
- Köller, H.; Kieseier, B.C.; Jander, S.; Hartung, H.P. Chronic inflammatory demyelinating polyneuropathy. N. Engl. J. Med. 2005, 352, 1343–1356. [Google Scholar] [CrossRef]
- Misawa, S.; Kuwabara, S.; Sato, Y.; Yamaguchi, N.; Nagashima, K.; Katayama, K.; Sekiguchi, Y.; Iwai, Y.; Amino, H.; Suichi, T.; et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018, 17, 519–529. [Google Scholar] [CrossRef]
- Lampson, L.A. Monoclonal antibodies in neuro-oncology. mAbs 2011, 3, 153–160. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, T.D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. daclizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Gedeon, P.C.; Streicker, M.A.; Schaller, T.H.; Archer, G.E.; Jokinen, M.P.; Sampson, J.H. GLP toxicology study of a fully-human T cell redirecting CD3:EGFRvIII binding immunotherapeutic bispecific antibody. PLoS ONE 2020, 15, e0236374. [Google Scholar] [CrossRef]
- Schaller, T.H.; Snyder, D.J.; Spasojevic, I.; Gedeon, P.C.; Sanchez-Perez, L.; Sampson, J.H. First in human dose calculation of a single-chain bispecific antibody targeting glioma using the MABEL approach. J. Immunother. Cancer 2020, 8, e000213. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Landen, J.W.; Cohen, S.; Billing, C.B., Jr.; Cronenberger, C.; Styren, S.; Burstein, A.H.; Sattler, C.; Lee, J.H.; Jack, C.R., Jr.; Kantarci, K.; et al. Multiple-dose ponezumab for mild-to-moderate Alzheimer’s disease: Safety and efficacy. Alzheimers Dement (NY) 2017, 3, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Farlow, M.; Aisen, P.S. Alzheimer’s Disease Cooperative Study Data Analysis and Publication Committee. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 1460. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab 3000 and 3001 Clinical Study Investigators. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef] [PubMed]
- Godyń, J.; Jończyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef]
- Oxford, A.E.; Stewart, E.S.; Rohn, T.T. Clinical Trials in Alzheimer’s Disease: A Hurdle in the Path of Remedy. Int. J. Alzheimers Dis. 2020, 5380346. [Google Scholar] [CrossRef]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Lee, J.H.; Kim, Y.J.; Lee, H.M.; Lyoo, C.H.; Ryu, Y.H.; Lee, M.S. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016, 87, 375–383. [Google Scholar] [CrossRef]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J.; et al. Tau and Ab imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Penke, B.; Szucs, M.; Bogar, F. Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: Their role in Alzheimer’s pathogenesis. Molecules 2020, 25, 1659. [Google Scholar] [CrossRef]
- Panza, F.; Solfrizzi, V.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Santamato, A.; Tortelli, R.; Galizia, I.; Prete, C.; Daniele, A.; et al. Tau-based therapeutics for Alzheimer’s disease: Active and passive immunotherapy. Immunotherapy 2016, 8, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Schofield, D.J.; Irving, L.; Calo, L.; Bogstedt, A.; Rees, G.; Nuccitelli, A.; Narwal, R.; Petrone, M.; Roberts, J.; Brown, L.; et al. Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. [Google Scholar] [CrossRef] [PubMed]
- St Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet Muscle. 2017, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Abdel-Hamid, H.Z.; Mah, J.K.; Campbell, C.; Guglieri, M.; Muntoni, F.; Takeshima, Y.; McDonald, C.M.; Kostera-Pruszczyk, A.; Karachunski, P.; et al. Randomized phase 2 trial and open-label extension of domagrozumab in Duchenne muscular dystrophy. Neuromuscul. Disord. 2020, 30, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Demlova, R.; Valík, D.; Obermannova, R.; ZdraŽilová-Dubská, L. The safety of therapeutic monoclonal antibodies: Implications for cancer therapy including immuno-checkpoint inhibitors. Physiol. Res. 2016, 65, S455. [Google Scholar] [CrossRef]
- Calogiuri, G.; Ventura, M.T.; Mason, L.; Valacca, A.; Buquicchio, R.; Cassano, N.; Vena, G. A Hypersensitivity reactions to last generation chimeric, humanized and human recombinant monoclonal antibodies for therapeutic use. Curr. Pharm. Des. 2008, 14, 2883–2891. [Google Scholar] [CrossRef]
- Cáceres, M.C.; Guerrero-Martín, J.; Pérez-Civantos, D.; Palomo-López, P.; Delgado-Mingorance, J.I.; Durán-Gómez, N. The importance of early identification of infusion-related reactions to monoclonal antibodies. Ther. Clin. Risk Manag. 2019, 15, 965–977. [Google Scholar] [CrossRef]
- Kang, S.P.; Saif, M.W. Infusion-related and hypersensitivity reactions of monoclonal antibodies used to treat colorectal cancer –identification, prevention, and management. J. Support. Oncol. 2007, 5, 451–457. [Google Scholar]
- Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: An updated practice parameter. Ann. Allergy Asthma Immunol. 2010, 105, 259–273. [Google Scholar] [CrossRef]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-α-1, 3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.G.; Hourihane, J.O.; Bousquet, J.; Bruijnzeel-Koomen, C.; Dreborg, S.; Haahtela, T.; Kowalski, M.L.; Mygind, N.; Ring, J.; van Cauwenberge, P.; et al. A revised nomenclature for allergy. An EAACI position statement from the EAACI nomenclature task force. Allergy 2001, 56, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Doessegger, L.; Banholzer, M.L. Clinical development methodology for infusion-related reactions with monoclonal antibodies. Clin. Transl. Immunol. 2015, 4, e39. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunol. Ther. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef]
- Bugelski, P.J.; Achuthanandam, R.; Capocasale, R.J.; Treacy, G.; Bouman-Thio, E. Monoclonal antibody-induced cytokine-release syndrome. Expert Rev. Clin. Immunol. 2009, 5, 499–521. [Google Scholar] [CrossRef]
- Quistrebert, J.; Hässler, S.; Bachelet, D.; Mbogning, C.; Musters, A.; Tak, P.P.; Wijbrandts, C.A.; Herenius, M.; Bergstra, S.A.; Akdemir, G.; et al. Incidence and risk factors for adalimumab and infliximab anti-drug antibodies in rheumatoid arthritis: A European retrospective multicohort analysis. Semin. Arthritis Rheum. 2019, 48, 967–975. [Google Scholar] [CrossRef]
- Linker, R.; Kieseier, B. Innovative monoclonal antibody therapies in multiple sclerosis. Ther. Adv. Neurol. Disord. 2008, 1, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Epstein, D.J.; Dunn, J.; Deresinski, S. Infectious Complications of Multiple Sclerosis Therapies: Implications for Screening, Prophylaxis, and Management. Open. Forum. Infect Dis. 2018, 5, ofy174. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Tuccori, M.; Maggi, F. Progressive multifocal leukoencephalopathy and anti-CD20 monoclonal antibodies: What do we know after 20 years of rituximab. Rev. Med. Virol. 2019, 29, e2077. [Google Scholar] [CrossRef] [PubMed]
- Rau, D.; Lang, M.; Harth, A.; Naumann, M.; Weber, F.; Tumani, H.; Bayas, A. Listeria meningitis complicating alemtuzumab treatment in multiple sclerosis—report of two cases. Int. J. Mol. Sci. 2015, 16, 14669–14676. [Google Scholar] [CrossRef] [PubMed]
- Penkert, H.; Delbridge, C.; Wantia, N.; Wiestler, B.; Korn, T. Fulminant Central Nervous System Nocardiosis in a Patient Treated With Alemtuzumab for Relapsing-Remitting Multiple Sclerosis. JAMA Neurol. 2016, 73, 757–759. [Google Scholar] [CrossRef]
- Clerico, M.; De Mercanti, S.; Artusi, C.A.; Durelli, L.; Naismith, R.T. Active CMV infection in two patients with multiple sclerosis treated with alemtuzumab. Mult. Scler. 2017, 23, 874–876. [Google Scholar] [CrossRef]
- Yann, K.; Jackson, F.; Sharaf, N.; Mihalova, T.; Talbot, P.; Rog, D.; Pace, A. Acute respiratory distress syndrome following alemtuzumab therapy for relapsing multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 14, 1–3. [Google Scholar] [CrossRef]
- Brownlee, W.J.; Chataway, J. Opportunistic infections after alemtuzumab: New cases of norcardial infection and cytomegalovirus syndrome. Mult. Scler. 2017, 23, 876–877. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Hafler, D.A. Regulatory T cells in autoimmune neuroinflammation. Immunol. Rev. 2014, 259, 231–244. [Google Scholar] [CrossRef]
- Tuohy, O.; Costelloe, L.; Hill-Cawthorne, G.; Bjornson, I.; Harding, K.; Robertson, N.; May, K.; Button, T.; Azzopardi, L.; Kousin-Ezewu, O. Alemtuzumab treatment of multiple sclerosis: Long-term safety and efficacy. J. Neurol. Neurosurg. Psychiatry 2015, 86, 208–215. [Google Scholar] [CrossRef]
- Coles, A.J.; Habek, M.; Bass, A.; Brinar, V.; Vladic, A.; Margolin, D.; Lu, M.; Fox, E. Durable efficacy of alemtuzumab over 10 years: Long-term follow-up of patients with RRMS from the CAMMS223 study. Neurology 2016, 86, P3.053. [Google Scholar]
- Menge, T.; Stüve, O.; Kieseier, B.C.; Hartung, H.-P. Alemtuzumab: The advantages and challenges of a novel therapy in MS. Neurology 2014, 83, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Bass, A.D.; Nadj, C.; Agius, M.A.; Steingo, B.; Selmaj, K.W.; Thoits, T.; Guerreiro, A.; Van Wijmeersch, B.; Ziemssen, T.; et al. Immune thrombocytopenia in alemtuzumab-treated MS patients: Incidence, detection, and management. Mult. Scler. 2020, 26, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, M.R.; Wallin, E.F.; Jayne, D.R. Anti-glomerular basement membrane disease after alemtuzumab. N. Engl. J. Med. 2008, 359, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Cox, A.; Le Page, E.; Jones, J.; Trip, S.A.; Deans, J.; Seaman, S.; Miller, D.H.; Hale, G.; Waldmann, H.; et al. The window of therapeutic opportunity in multiple sclerosis: Evidence from monoclonal antibody therapy. J. Neurol. 2006, 253, 98–108. [Google Scholar] [CrossRef]
- Richter, S.; Wagner, B.; Celius, E.G. Two cases of diabetes mellitus type 1 after alemtuzumab treatment for multiple sclerosis: Another probable secondary autoimmune disease. J. Neurol. 2019, 266, 1270–1271. [Google Scholar] [CrossRef]
- Krämer, J.; Krömer-Olbrisch, T.; Lakomek, H.J.; Schellinger, P.D.; Foell, D.; Meuth, S.G.; Straeten, V. Case Report: Adult Still’s Disease in an Alemtuzumab-Treated Multiple Sclerosis Patient. Front. Immunol. 2020, 11, 2099. [Google Scholar] [CrossRef]
- Aouad, P.; Yiannikas, C.; Fernando, S.L.; Parratt, J. A case of autoimmune myositis after treatment with alemtuzumab for multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2018, 4, 2055217318819012. [Google Scholar] [CrossRef]
- Alcalá, C.; Pzére-Miralles, F.; Gascón, F.; Evole, M.; Estutia, M.; Gil-Perotín, S.; Casanova, B. Recurrent and universal alopecia areata following alemtuzumab treatment in multiple sclerosis: A secondary autoimmune disease. Mult. Scler. Relat. Disord. 2019, 27, 406–408. [Google Scholar] [CrossRef]
- Ruck, T.; Pfeuffer, S.; Schulte-Mecklenbeck, A.; Gross, C.C.; Lindner, M.; Metze, D.; Ehrchen, J.; Sondermann, W.; Pul, R.; Kleischnitz, C.; et al. Vitiligo after alemtuzumab treatment: Secondary autoimmunity is not all about B cells. Neurology 2018. [Google Scholar] [CrossRef]
- Jones, J.L.; Phuah, C.L.; Cox, A.L.; Thompson, S.A.; Ban, M.; Shawcross, J.; Walton, A.; Sawcer, S.J.; Compston, A.; Coles, A.J. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J. Clin. Investig. 2009, 119, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Kopp, T.I.; Delcoigne, B.; Arkema, E.V.; Jacobsen, R.K.; Magyari, M.; Ibfelt, E.H.; Locht, H.; Sellebjerg, F.; Cordtz, R.L.; Jensen, D.V.; et al. Risk of neuroinflammatory events in arthritis patients treated with tumour necrosis factor alpha inhibitors: A collaborative population-based cohort study from Denmark and Sweden. Ann. Rheum. Dis. 2020, 79, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Richez, C.; Sordet, C.; Rist, S.; Gossec, L.; Direz, G.; Houvenagel, E.; Berthelot, J.M.; Pagnoux, C.; Dernis, E.; et al. Club Rhumatismes et Inflammation Section of the SFR Pattern of demyelination occurring during anti-TNF-α therapy: A French national survey. Rheumatology (Oxford) 2013, 52, 868–874. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
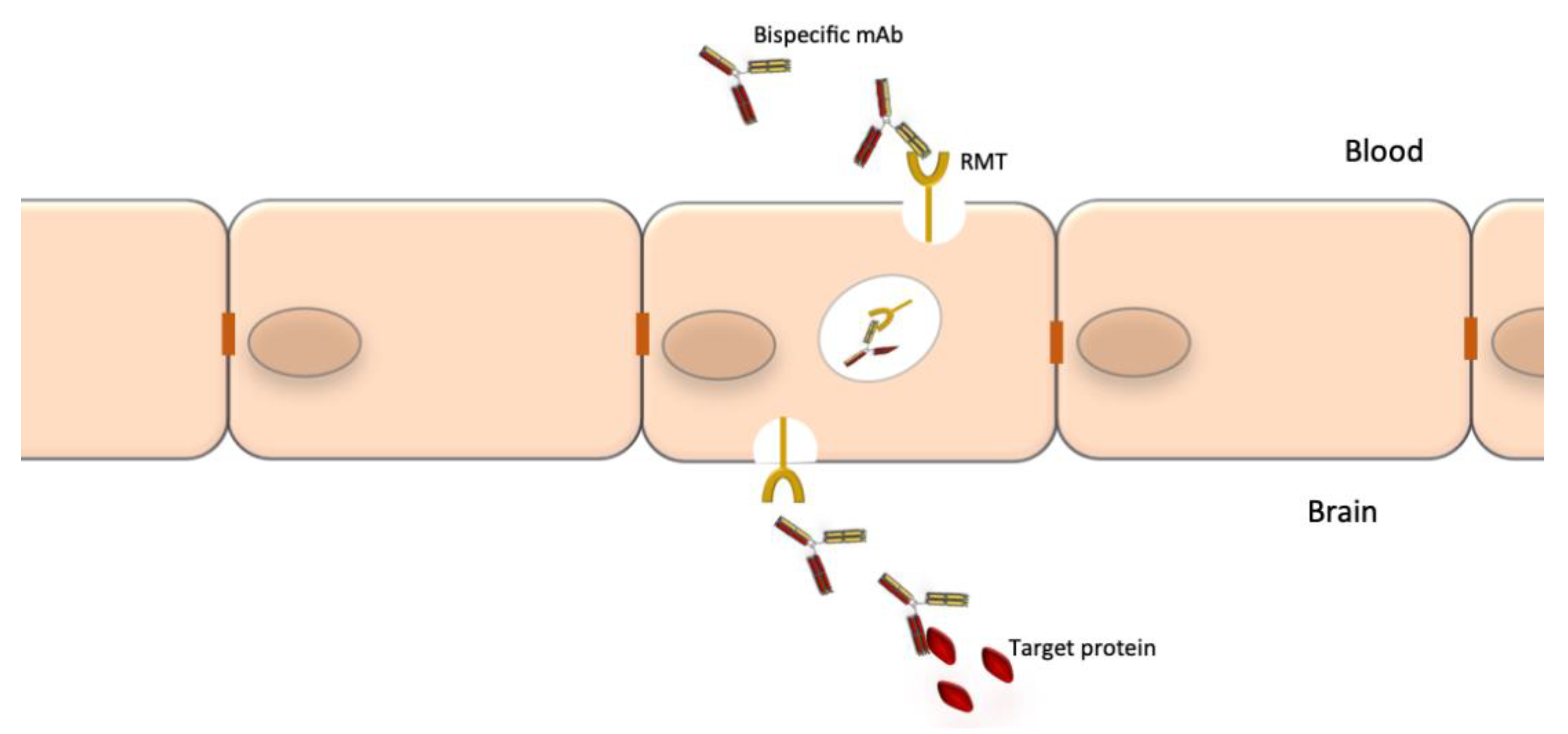{kind=link}
| Name | Type | Target | Action | Route | Neurological Indication | Adverse Effects of Special Interest | References |
|---|---|---|---|---|---|---|---|
| Alemtuzumab | humanized IgG1 | CD52 | Depletes CD52+ T and B cells | IV | RR-MS * | Infusion reactions Secondary autoimmunity Cerebrovascular accidents | [7,10,11,12,13] |
| Bevacizumab | humanized IgG1 | VEGF | Inhibition of angiogenesis | IV | Glioblastoma * | hypertension, gastrointestinal perforation, bleeding, PRES | [14,15,16,17] |
| Daclizumab | humanized IgG1 | IL2R-α (CD25) | Blocks the high affinity IL-2 receptor containing the α subunit | SC | RR-MS * | Autoimmune encephalitis, hepatitis and rashes | [18,19,20,21,22,23,24,25,26,27] |
| Eculizumab | humanized IgG2/4 | C5 complement protein | Inhibition of the terminal C5 complement pathway | IV | Anti-AChR Ab+ MG * AQP-4+ NMOSD * | Meningococcal infections | [28,29,30,31,32,33] |
| Eptinezumab | humanized IgG1 | CGRP ligand | Selectively bind to isoforms a and b of CGRP | IV | EM* and CM * | Nasopharyngitis Hypersensitivity reactions | [34] |
| Erenumab | fully human IgG2 | CGRP receptor | Competitively and reversibly binds the CGRP receptor | SC | EM * and CM * | Constipation Injection site reactions | [35,36,37,38,39] |
| Fremanezumab | humanized IgG2 | CGRP ligand | Selectively bind to isoforms a and b of CGRP | SC | EM * and CM * | Injection site reactions | [40,41] |
| Galcanezumab | humanized IgG4 | CGRP ligand | Binds CGRP and prevents its biological activity | SC | EM * and CM * Cluster headache | Injection site reactions | [42,43,44,45,46,47,48] |
| Inebilizumab | humanized IgG1 | CD19 | Depletes B cells and some short-lived plasmablasts and plasma cells | IV | AQP-4+ NMOSD | Infusion reactions, infections | [49,50] |
| Infliximab | chimeric IgG1 | TNF-α blockade | TNF-α signaling blockade | IV | DM/PM Behcet disease Neurosarcoidosis | Infusion reactions CNS demyelination | [29,51,52,53,54] |
| Natalizumab | humanized IgG4 | α4β1 integrin (CD49d) | Inhibits the entry of lymphocytes into the brain parenchyma | IV | RR-MS * | PML, hepatotoxicity | [55,56,57,58,59,60,61,62,63,64,65,66,67] |
| Ocrelizumab | humanized IgG1 | CD20 | Depletes B cells | IV | RR-MS * PP-MS * | Infusion reactions, infections | [68,69,70,71] |
| Ofatumumab | fully human IgG1 | CD20 | Depletes B cells | SC | RR-MS * | Injections site reactions, infections, neutropenia | [8,72] |
| Rituximab | chimeric IgG1 | CD20 | Depletes B cells | IV | RR-MS NMOSD; MG; CIDP; MMN, anti-MAG neuropathy PM/DM | Infusion reactions PML | [4,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106] |
| Satralizumab | humanized IgG2 | IL-6 receptor | IL-6 receptor signaling blockade | SC | Anti-AQP4 Ab+ NMOSD * | Infections, neutropenia, elevated liver enzymes | [107,108] |
| Tocilizumab | humanized IgG1 | IL-6 receptor | IL-6 receptor signaling blockade | IV | NMOSD CRS | Infusion reactions, Infections | [9,109,110,111] |
| Name | Type | Target | Action | Stage of Development | Neurological Indication | References |
|---|---|---|---|---|---|---|
| Aducanumab (BIIB037) | fully human IgG1 | Aβ | Binding of the aggregated Aβ forms | In phase IΙΙ | Prodromal to mild AD | [112,113,114,115,116] |
| Aquaporumab | fully human (mutated Fc) | AQP-4 | Competitively inhibits binding of anti-AQP-4 auto-Abs | not yet in clinical trials | NMOSD | [117,118] |
| Batoclimab (HBM9161) | fully human IgG1 | FcRn | Reduction of auto-antibody levels | In phase II | MG | [119] |
| Cinpanemab (BIIB054) | humanized IgG1 | α-synuclein | Prevention of accumulation and aggregation of α-synuclein | In phase II | PD | [120,121] |
| Donanemab (N3pG) | humanized IgG1 | Aβ | Binding aggregated Aβ forms | In phase II | Mild AD | [122,123,124] |
| Efgartigimod | Antibody fragment | FcRn | Reduction of auto-antibody levels | In phase II for CIDP completed phase III for MG | MG CIDP | [125,126,127] |
| Gantenerumab (RG1450) | fully human IgG1 | Aβ | Binding aggregated Aβ forms | In two phase III trials | Prodromal and mild AD | [128,129] |
| Gosuranemab (BIIB092) | humanized IgG4 | tau | Targeting abnormal forms of tau protein or soluble oligomers | In phase II | Prodromal to mild AD | [130,131] |
| Nipocalimab (M 281) | fully human IgG1 | FcRn | Reduction of auto-antibody levels | Completed phase II trial | MG | [132] |
| Opicinumab (BIIB033) | fully human IgG1 | LINGO-1 | Promotion of remyelination | In phase II | MS | [133,134,135] |
| Ravulizumab (ALXN1210) | humanized IgG2/4 | C5 | Inhibition of the C5 terminal complement pathway | In phase III | AQP-4+ NMOSD, MG | [136,137] |
| Rilotumumab (AMG102) | fully human IgG2 | HGF | Prevents activation of the c-Met receptor and tumor cell growth | In phase II | Glioblastoma | [17,138] |
| Rozanolixizumab (UCB 7665) | humanized IgG4 | FcRn | Reduction of auto-antibody levels | Completed a phase II study | MG | [139] |
| Semorinemab (RG6100) | humanized IgG4 | tau | Targeting all isoforms of tau protein | In phase II | Prodromal to mild AD | [130] |
| Tilavonemab (ABBV 8E12) | humanized IgG4 | tau | Targeting abnormal extracellular forms of tau protein | In phase II | Prodromal to mild AD | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gklinos, P.; Papadopoulou, M.; Stanulovic, V.; Mitsikostas, D.D.; Papadopoulos, D. Monoclonal Antibodies as Neurological Therapeutics. Pharmaceuticals 2021, 14, 92. https://doi.org/10.3390/ph14020092
Gklinos P, Papadopoulou M, Stanulovic V, Mitsikostas DD, Papadopoulos D. Monoclonal Antibodies as Neurological Therapeutics. Pharmaceuticals. 2021; 14(2):92. https://doi.org/10.3390/ph14020092
Chicago/Turabian StyleGklinos, Panagiotis, Miranta Papadopoulou, Vid Stanulovic, Dimos D. Mitsikostas, and Dimitrios Papadopoulos. 2021. "Monoclonal Antibodies as Neurological Therapeutics" Pharmaceuticals 14, no. 2: 92. https://doi.org/10.3390/ph14020092
APA StyleGklinos, P., Papadopoulou, M., Stanulovic, V., Mitsikostas, D. D., & Papadopoulos, D. (2021). Monoclonal Antibodies as Neurological Therapeutics. Pharmaceuticals, 14(2), 92. https://doi.org/10.3390/ph14020092