
MicroRNA-Based Therapeutics for Drug-Resistant Colorectal Cancer


Abstract
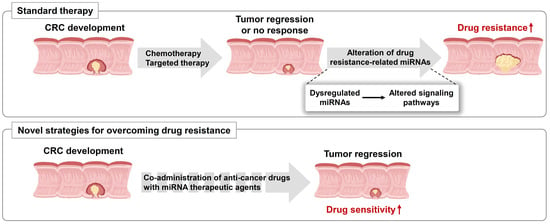
1. Introduction
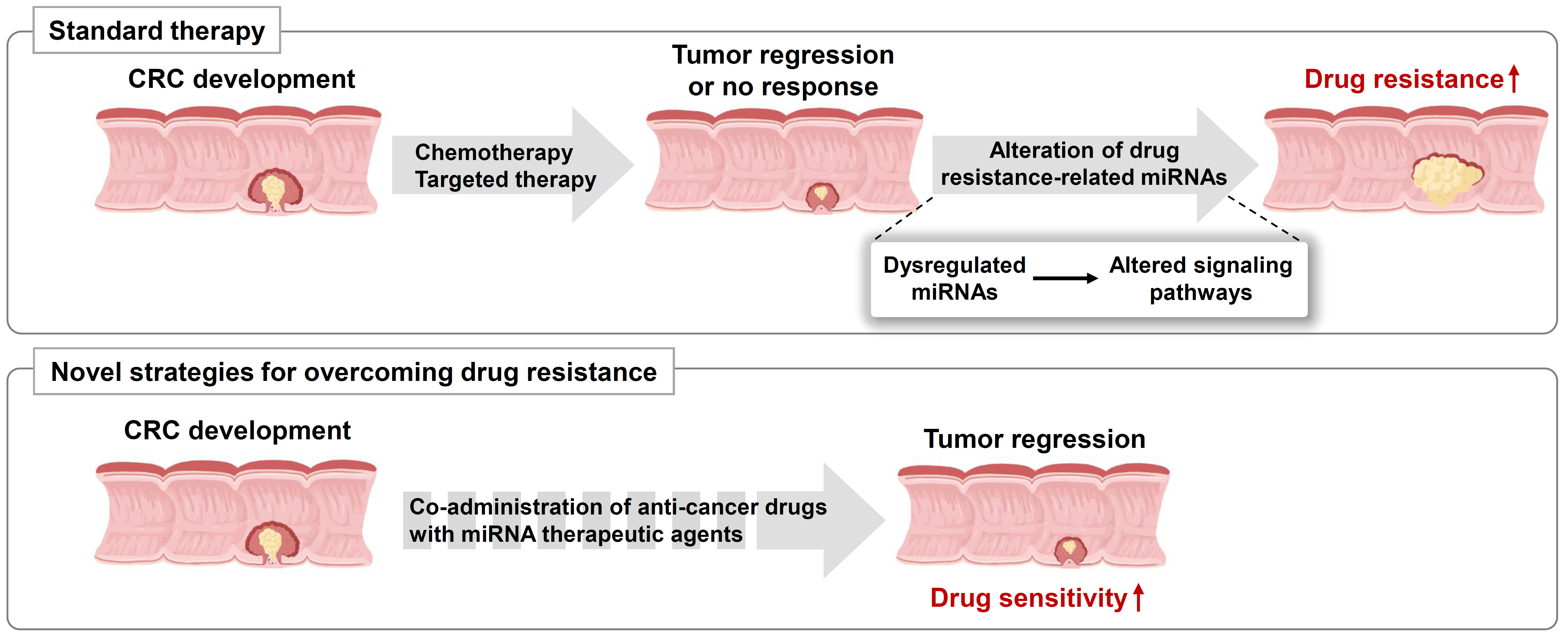
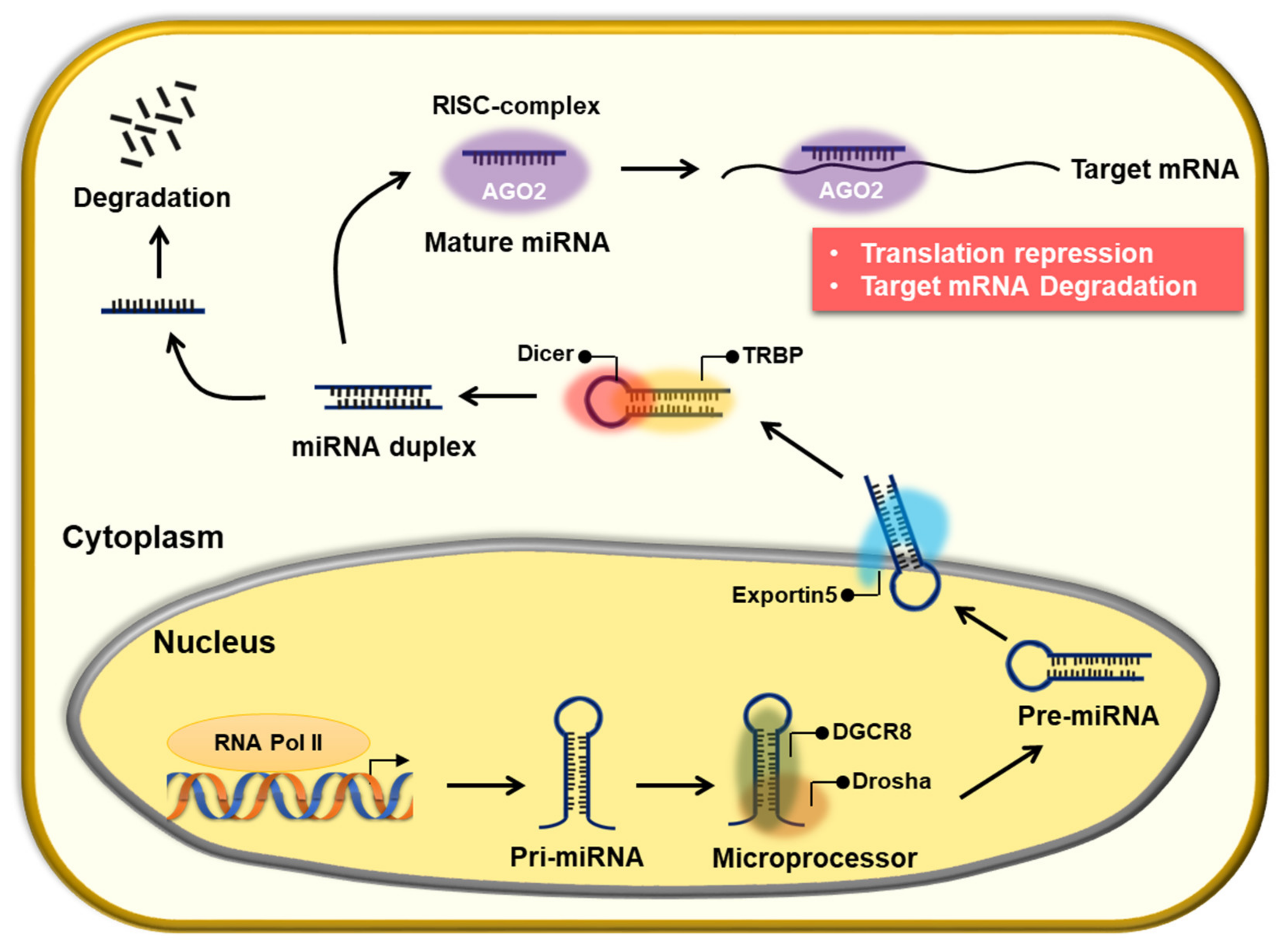
2. The Role of miRNAs in Chemoresistance
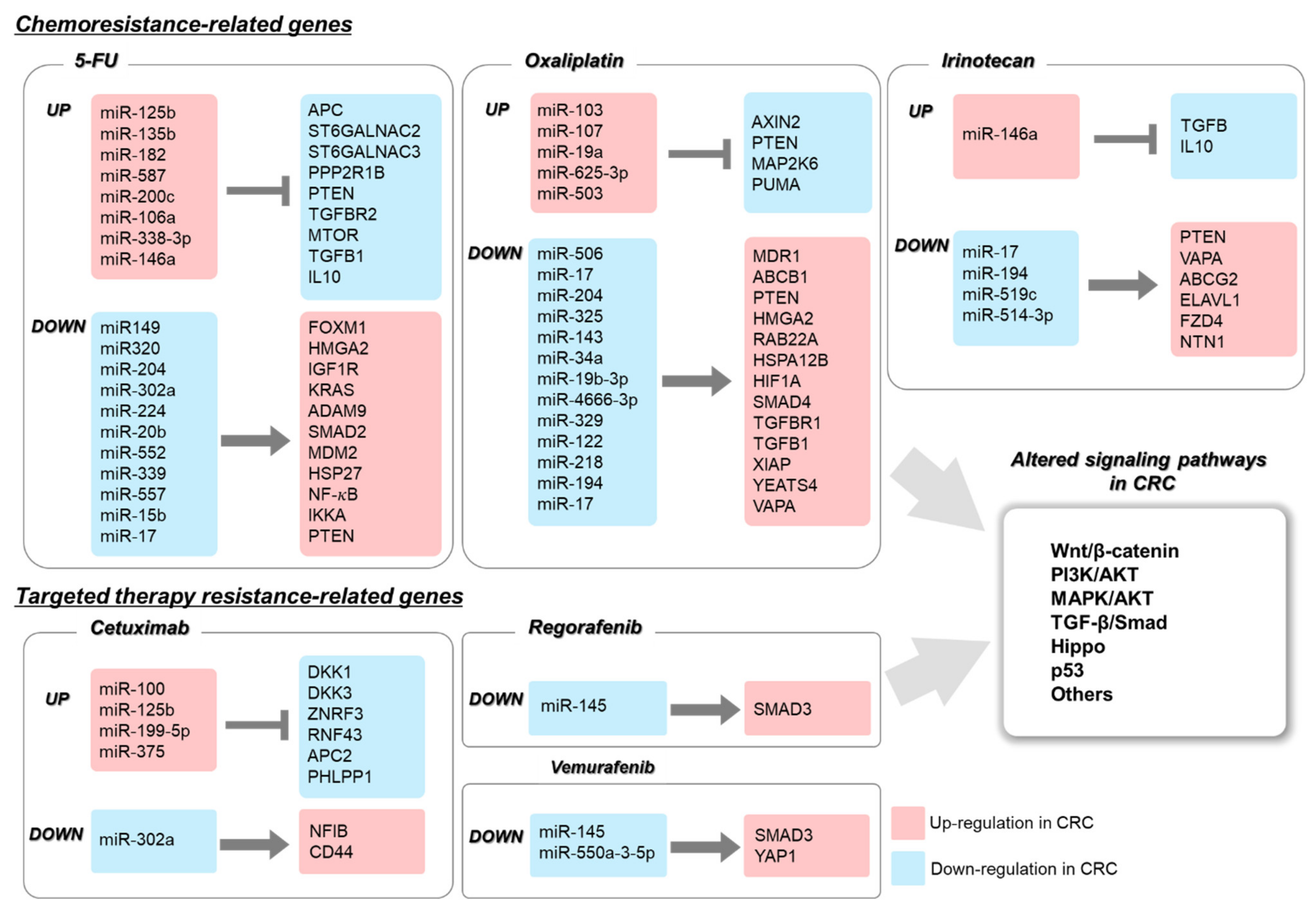
2.1. The Role of miRNA in 5-FU-Resistant CRC
2.2. The Role of miRNA in Oxaliplatin-Resistant CRC
2.3. The Role of miRNA in Irinotecan-Resistant CRC

{kind=link}
{kind=link}
{kind=link}
| miRNA | Drug | Target Gene | Signaling Pathway | Level | Reference |
|---|---|---|---|---|---|
| miR-125b | 5-FU | APC | Wnt/β-catenin | Up | [28] |
| miR149 | 5-FU | FOXM1 | Wnt/β-catenin | Down | [29] |
| miR320 | 5-FU | FOXM1 | Wnt/β-catenin | Down | [29] |
| miR-135b | 5-FU | ST6GALNAC2 | PI3K/AKT | Up | [31] |
| miR-182 | 5-FU | ST6GALNAC3 | PI3K/AKT | Up | [31] |
| miR-204 | 5-FU | HMGA2 | PI3K/AKT | Down | [32] |
| miR-587 | 5-FU | PPP2R1B | PI3K/AKT | Up | [33] |
| miR-200c | 5-FU | PTEN | PI3K/AKT | Up | [34] |
| miR-302a | 5-FU | IGF-1R | PI3K/AKT | Down | [35] |
| miR-224 | 5-FU | KRAS | PI3K/AKT | Down | [36] |
| miR-20b | 5-FU | ADAM9 | PI3K/AKT | Down | [37] |
| miR-552 | 5-FU | Smad2 | TGF-β | Down | [38] |
| miR-106a | 5-FU | TGF-βR2 | TGF-β | Up | [39] |
| miR-338-3p | 5-FU | mTOR | p53 | Up | [40] |
| miR-339 | 5-FU | MDM2 | p53 | Down | [41] |
| miR-557 | 5-FU | HSP27 | Other | Down | [42] |
| miR-15b | 5-FU | NF-𝜅B, IKKα | Other | Down | [43] |
| miR-103 | oxaliplatin | Axin2 | Wnt/β-catenin | Up | [45] |
| miR-107 | oxaliplatin | Axin2 | Wnt/β-catenin | Up | [45] |
| miR-506 | oxaliplatin | MDR1/P-gp | Wnt/β-catenin | Down | [46] |
| miR-17 | 5-FU, oxaliplatin, irinotecan | PTEN | PI3K/AKT | Down | [47] |
| miR-19a | oxaliplatin | PTEN | PI3K/AKT | Up | [48] |
| miR-204 | oxaliplatin | HMGA2, RAB22A | PI3K/AKT | Down | [32,49] |
| miR-325 | oxaliplatin | HSPA12B | PI3K/AKT | Down | [50] |
| miR-143 | oxaliplatin | HIF-1α | PI3K/AKT | Down | [51] |
| miR-34a | oxaliplatin | Smad4 | TGF-β | Down | [52] |
| miR-19b-3p | oxaliplatin | Smad4 | TGF-β | Down | [53] |
| miR-4666-3p | oxaliplatin | TGF-βR1 | TGF-β | Down | [54] |
| miR-329 | oxaliplatin | TGF-β1 | TGF-β | Down | [54] |
| miR-625-3p | oxaliplatin | MAP2K6 | Other | Up | [55] |
| miR-122 | oxaliplatin | XIAP | Other | Down | [56] |
| miR-503 | oxaliplatin | PUMA | Other | Up | [57] |
| miR-218 | oxaliplatin | YEATS4 | Other | Down | [58] |
| miR-194 | oxaliplatin, irinotecan | VAPA | PI3K/AKT | Down | [60] |
| miR-146a | 5-FU, irinotecan | TGF-β, IL-10 | TGF-β | Up | [61] |
| miR-519c | irinotecan | ABCG2, HuR | Other | Down | [62] |
| miR-514-3p | irinotecan | FZD4, NTN1 | Other | Down | [63] |
3. Molecular Targeted Therapeutic Drugs and Resistance
3.1. VEGF/VEGFR Targeted Therapy
3.2. EGFR-Targeted Therapy
3.3. BRAF-Targeted Therapy
3.4. Roles of miRNAs in Targeted Therapy Resistance
| miRNA | Drug | Target Gene | Signaling Pathway | Level | Reference |
|---|---|---|---|---|---|
| miR-100 | Cetuximab | DKK1, DKK3, ZNRF3, RNF43, APC2 | Wnt/β-catenin | Up | [93] |
| miR-125b | Cetuximab | DKK1, DKK3, ZNRF3, RNF43, APC2 | Wnt/β-catenin | Up | [93] |
| miR-302a | Cetuximab | NFIB, CD44 | MAPK/AKT | Down | [94] |
| miR-199-5p | Cetuximab | PHLPP1 | PI3K/AKT | Up | [95] |
| miR-375 | Cetuximab | PHLPP1 | PI3K/AKT | Up | [95] |
| miR-145 | Regorafenib | SMAD3 | TGF-β/SMAD | Down | [96] |
| miR-145 | Vemurafenib | SMAD3 | TGF-β/SMAD | Down | [97] |
| miR-550a-3-5p | Vemurafenib | YAP1 | Hippo | Down | [98] |
3.5. Roles of miRNAs in Immunotherapy
4. Clinical Application of miRNAs as Anti-Cancer Drug Resistance Biomarkers and Therapeutic Targets
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Center, M.M.; Jemal, A.; Smith, R.A.; Ward, E. Worldwide Variations in Colorectal Cancer. CA Cancer J. Clin. 2009, 59, 366–378. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.; Barzi, A.; Jemal, A.J. Colorectal Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-De-Diego, C.; Dieste, A.P.; Cerrada, E.; Yoldi, M.J.R. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Lee, P.Y.; Fletcher, W.S.; Sullivan, E.S.; Vetto, J.T. Colorectal cancer in young patients: Characteristics and outcome. Am. Surg. 1994, 60, 607–612. [Google Scholar] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z. The effect of immune microenvironment on the progression and prognosis of colorectal cancer. J. Med. Oncol. 2014, 31, 82. [Google Scholar] [CrossRef]
- American Cancer Society. Global Cancer Facts & Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 2019, 110, 473–481. [Google Scholar] [CrossRef]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. WNT Signaling and Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Spitzner, M.; Reineke, S.; Möller, J.; Auslander, N.; Kramer, F.; Hu, Y.; Beissbarth, T.; Wolff, H.A.; Rave-Fränk, M.J.M.C.R. Chemoradiotherapy resistance in colorectal cancer cells is mediated by Wnt/β-catenin signaling. J. Med. Case Rep. 2017, 15, 1481–1490. [Google Scholar] [CrossRef]
- Che, J.; Pan, L.; Yang, X.; Liu, Z.; Huang, L.; Wen, C.; Lin, A.; Liu, H.J.M. Thymidine phosphorylase expression and prognosis in colorectal cancer treated with 5-fluorouracil-based chemotherapy: A meta-analysis. Oncology 2017, 7, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, F.M. Monoclonal Antibodies as Magic Bullets. Pharm. Res. 1988, 5, 1–9. [Google Scholar] [CrossRef]
- Dosset, M.; Vargas, T.R.; Lagrange, A.; Boidot, R.; Végran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Marie-Joseph, E.L.; et al. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. OncoImmunology 2018, 7, e1433981. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Marliot, F.; Ou, F.-S.; Bifulco, C.B.; Lugli, A.; Zlobec, I.; Rau, T.T.; Hartmann, A.; Masucci, G.V.; et al. Validation of the Immunoscore (IM) as a prognostic marker in stage I/II/III colon cancer: Results of a worldwide consortium-based analysis of 1,336 patients. J. Clin. Oncol. 2016, 34, 3500. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.J.B.C. MicroRNAs are important regulators of drug resistance in colorectal cancer. J. Biol. Chem. 2017, 398, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Huang, M.; Wang, Y.; Wang, L.; Chen, C.; Chu, X. MicroRNAs as Regulators, Biomarkers and Therapeutic Targets in the Drug Resistance of Colorectal Cancer. Cell. Physiol. Biochem. 2016, 40, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.W.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nat. Cell Biol. 2010, 465, 584–589. [Google Scholar] [CrossRef]
- Michlewski, G.; Cáceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jazbutyte, V.; Thum, T. MicroRNA-21: From cancer to cardiovascular disease. J. Cardiovasc. Diagn. Ther. 2010, 11, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.-Y. MicroRNAs in Human Diseases: From Cancer to Cardiovascular Disease. Immune Netw. 2011, 11, 135–154. [Google Scholar] [CrossRef]
- Diosdado, B.; Van De Wiel, M.A.; Droste, J.S.T.S.; Mongera, S.; Postma, C.; Meijerink, W.J.H.J.; Carvalho, B.; Meijer, G.A. MiR-17-92 cluster is associated with 13q gain and c-myc expression during colorectal adenoma to adenocarcinoma progression. Br. J. Cancer 2009, 101, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Chirshev, E.; Oberg, K.C.; Ioffe, Y.J.; Unternaehrer, J.J. Let-7 as biomarker, prognostic indicator, and therapy for precision medicine in cancer. Clin. Transl. Med. 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.D.; Damas, N.D.; Lees, M.P.; Jacobsen, A.B.; Lund, A.H. miR-339-5p regulates the p53 tumor-suppressor pathway by targeting MDM2. Oncogene 2014, 34, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Zhou, J.; Chen, Z.-R.; Chng, W.-J. p53mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.; Hu, L. The regulatory effects of metformin on the [SNAIL/miR-34]:[ZEB/miR-200] system in the epithelial-mesenchymal transition(EMT) for colorectal cancer(CRC). Eur. J. Pharmacol. 2018, 834, 45–53. [Google Scholar] [CrossRef]
- Yeh, M.; Oh, C.S.; Yoo, J.Y.; Kaur, B.; Lee, T.J. Pivotal role of microRNA-138 in human cancer. Am. J. Cancer Res. 2019, 9, 1118–1126. [Google Scholar]
- Yu, X.; Shi, W.; Zhang, Y.; Wang, X.; Sun, S.; Song, Z.; Liu, M.; Zeng, Q.; Cui, S.; Qu, X. CXCL12/CXCR4 axis induced miR-125b promotes invasion and confers 5-fluorouracil resistance through enhancing autophagy in colorectal cancer. Sci. Rep. 2017, 7, srep42226. [Google Scholar] [CrossRef]
- Liu, X.; Xie, T.; Mao, X.; Xue, L.; Chu, X.; Chen, L. MicroRNA-149 Increases the Sensitivity of Colorectal Cancer Cells to 5-Fluorouracil by Targeting Forkhead Box Transcription Factor FOXM1. Cell. Physiol. Biochem. 2016, 39, 617–629. [Google Scholar] [CrossRef]
- Wan, L.-Y.; Deng, J.; Xiang, X.-J.; Zhang, L.; Yu, F.; Chen, J.; Sun, Z.; Feng, M.; Xiong, J.-P. miR-320 enhances the sensitivity of human colon cancer cells to chemoradiotherapy in vitro by targeting FOXM1. Biochem. Biophys. Res. Commun. 2015, 457, 125–132. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Y.; Zhao, L.; Pan, Y.; Shan, Y.; Lifen, Z.; Jia, L. Upregulation of microRNA-135b and microRNA-182 promotes chemoresistance of colorectal cancer by targeting ST6GALNAC2 via PI3K/AKT pathway. Mol. Carcinog. 2017, 56, 2669–2680. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liangfang, S.; Shen, L.; Shen, L. MicroRNA-204 modulates colorectal cancer cell sensitivity in response to 5-fluorouracil-based treatment by targeting high mobility group protein A2. Biol. Open 2016, 5, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; A Talmon, G.; Wang, J. MicroRNA-587 antagonizes 5-FU-induced apoptosis and confers drug resistance by regulating PPP2R1B expression in colorectal cancer. Cell Death Dis. 2015, 6, e1845. [Google Scholar] [CrossRef] [PubMed]
- Heydari, K.; Saidijam, M.; Sharifi, M.R.; Dermani, F.K.; Asl, S.S.; Shabab, N.; Najafi, R. The Effect of miR-200c Inhibition on Chemosensitivity (5- FluoroUracil) in Colorectal Cancer. Pathol. Oncol. Res. 2018, 24, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, J.; Zhao, Z.; Han, J.; Jiang, T.; Chen, Y.; Hou, N.; Huang, C. MicroRNA-302a enhances 5-fluorouracil-induced cell death in human colon cancer cells. Oncol. Rep. 2017, 37, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Amankwatia, E.B.; Chakravarty, P.; Carey, F.A.; Weidlich, S.; Steele, R.J.C.; Munro, A.J.; Wolf, C.R.; Smith, G. MicroRNA-224 is associated with colorectal cancer progression and response to 5-fluorouracil-based chemotherapy by KRAS-dependent and -independent mechanisms. Br. J. Cancer 2015, 112, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Cheng, J.; Zhang, J.; Zhang, Y.; Chen, X.; Luo, S.; Xie, J. miR-20b reduces 5-FU resistance by suppressing the ADAM9/EGFR signaling pathway in colon cancer. Oncol. Rep. 2017, 37, 123–130. [Google Scholar] [CrossRef]
- Zhao, P.; Ma, Y.-G.; Zhao, Y.; Liu, D.; Dai, Z.-J.; Yan, C.-Y.; Guan, H.-T. MicroRNA-552 deficiency mediates 5-fluorouracil resistance by targeting SMAD2 signaling in DNA-mismatch-repair-deficient colorectal cancer. Cancer Chemother. Pharmacol. 2019, 84, 427–439. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Wang, H.; Wu, D. MiR-106a-5p promotes 5-FU resistance and the metastasis of colorectal cancer by targeting TGFβR2. Int. J. Clin. Exp. Pathol. 2018, 11, 5622–5634. [Google Scholar] [PubMed]
- Han, J.; Li, J.; Tang, K.; Zhang, H.; Guo, B.; Hou, N.; Huang, C. miR-338-3p confers 5-fluorouracil resistance in p53 mutant colon cancer cells by targeting the mammalian target of rapamycin. Exp. Cell Res. 2017, 360, 328–336. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Wang, X.; Wu, R.; Lin, M.; Laddha, S.V.; Yang, Q.; Chan, C.S.; Feng, Z. MicroRNA-339-5p inhibits colorectal tumorigenesis through regulation of the MDM2/p53 signaling. Oncotarget 2014, 5, 9106–9117. [Google Scholar] [CrossRef]
- Jiang, H.; Ju, H.; Zhang, L.; Lu, H.; Jie, K. microRNA-577 suppresses tumor growth and enhances chemosensitivity in colorectal cancer. J. Biochem. Mol. Toxicol. 2017, 31, e21888. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, Q.; Zhang, C.; Wang, G.; Yao, Y.; Huang, X.; Zhan, F.; Zhu, Y.; Shi, J.; Chen, J.; et al. miR-15b-5p resensitizes colon cancer cells to 5-fluorouracil by promoting apoptosis via the NF-κB/XIAP axis. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, R.K. Review of cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncol. Rev. 2014, 8, 256. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Lang, Y.-D.; Lin, H.-N.; Liu, Y.-R.; Liao, C.-C.; Nana, A.W.; Yen, Y.; Chen, R.-H. miR-103/107 prolong Wnt/β-catenin signaling and colorectal cancer stemness by targeting Axin2. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin, C.; Zhang, Y.; Zhang, X.; Zhang, C.; Zhang, P.; Xie, X.; Ren, Z. miR-506 enhances the sensitivity of human colorectal cancer cells to oxaliplatin by suppressing MDR1/P-gp expression. Cell Prolif. 2017, 50, e12341. [Google Scholar] [CrossRef]
- Fang, L.; Li, H.; Wang, L.; Hu, J.; Jin, T.; Wang, J.; Yang, B.B. MicroRNA-17-5p promotes chemotherapeutic drug resistance and tumour metastasis of colorectal cancer by repressing PTEN expression. Oncotarget 2014, 5, 2974–2987. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Zhang, J.; Xu, Y.; Shao, J.; Hu, Y.; Shu, P.; Cheng, H. Inhibition of miR-19a partially reversed the resistance of colorectal cancer to oxaliplatin via PTEN/PI3K/AKT pathway. Aging 2020, 12, 5640–5650. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhang, B.; Wang, W.; Fei, B.; Quan, C.; Zhang, J.; Song, M.; Bian, Z.; Wang, Q.; Ni, S.; et al. miR-204-5p Inhibits Proliferation and Invasion and Enhances Chemotherapeutic Sensitivity of Colorectal Cancer Cells by Downregulating RAB22A. Clin. Cancer Res. 2014, 20, 6187–6199. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, H.; Song, Y.; Gu, Q.; Zhang, L.; Xie, Q.; Xu, J.; Zhang, M. MiR-325 Promotes Oxaliplatin-Induced Cytotoxicity Against Colorectal Cancer Through the HSPA12B/PI3K/AKT/Bcl-2 Pathway. Dig. Dis. Sci. 2020, 1–10. [Google Scholar] [CrossRef]
- Qian, X.; Yu, J.; Yin, Y.; He, J.; Wang, L.; Li, Q.; Zhang, L.-Q.; Li, C.; Shi, Z.-M.; Xu, Q.; et al. MicroRNA-143 inhibits tumor growth and angiogenesis and sensitizes chemosensitivity to oxaliplatin in colorectal cancers. Cell Cycle 2013, 12, 1385–1394. [Google Scholar] [CrossRef]
- Sun, C.; Wang, F.-J.; Zhang, H.-G.; Xu, X.-Z.; Jia, R.-C.; Yao, L.; Qiao, P.-F. miR-34a mediates oxaliplatin resistance of colorectal cancer cells by inhibiting macroautophagy via transforming growth factor-β/Smad4 pathway. World J. Gastroenterol. 2017, 23, 1816–1827. [Google Scholar] [CrossRef]
- Jiang, T.; Ye, L.; Han, Z.; Liu, Y.; Yang, Y.; Peng, Z.; Fan, J. miR-19b-3p promotes colon cancer proliferation and oxaliplatin-based chemoresistance by targeting SMAD4: Validation by bioinformatics and experimental analyses. J. Exp. Clin. Cancer Res. 2017, 36, 1–14. [Google Scholar] [CrossRef]
- Ye, J.; Lei, J.; Fang, Q.; Shen, Y.; Xia, W.; Hu, X.; Xu, Q.; Yuan, H.; Huang, J.; Ni, C. miR-4666-3p and miR-329 Synergistically Suppress the Stemness of Colorectal Cancer Cells via Targeting TGF-β/Smad Pathway. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.H.; Lyskjær, I.; Jersie-Christensen, R.R.; Tarpgaard, L.S.; Primdal-Bengtson, B.; Nielsen, M.M.; Pedersen, J.S.; Hansen, T.P.; Hansen, F.; Olsen, J.V.; et al. miR-625-3p regulates oxaliplatin resistance by targeting MAP2K6-p38 signalling in human colorectal adenocarcinoma cells. Nat. Commun. 2016, 7, 12436. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Zhu, Y.; Zhang, J.; Zhu, Z.; Ning, Z.; Chen, H.; Liu, L.; Chen, Z.; Meng, Z. miR-122 Targets X-Linked Inhibitor of Apoptosis Protein to Sensitize Oxaliplatin-Resistant Colorectal Cancer Cells to Oxaliplatin-Mediated Cytotoxicity. Cell. Physiol. Biochem. 2018, 51, 2148–2159. [Google Scholar] [CrossRef]
- Xu, K.; Chen, G.; Qiu, Y.; Yuan, Z.; Li, H.; Yuan, X.; Sun, J.; Xu, J.; Liang, X.; Yin, P. miR-503-5p confers drug resistance by targeting PUMA in colorectal carcinoma. Oncotarget 2017, 8, 21719–21732. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Cheng, J.; Zhang, J.; Zhang, Y.; Chen, X.; Xie, J.; Luo, S. Downregulation of YEATS4 by miR-218 sensitizes colorectal cancer cells to L-OHP-induced cell apoptosis by inhibiting cytoprotective autophagy. Oncol. Rep. 2016, 36, 3682–3690. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Mitchell, E.P.; Hoff, P.M. Irinotecan in the treatment of colorectal cancer. Cancer Treat. Rev. 2006, 32, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-Y.; Ye, S.-P.; Pan, S.-L.; Kuo, T.-T.; Liu, B.C.; Chen, Y.-L.; Huang, T.-C. Overexpression of miR-194 Reverses HMGA2-driven Signatures in Colorectal Cancer. Theranostics 2017, 7, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Khorrami, S.; Hosseini, A.Z.; Mowla, S.J.; Soleimani, M.; Rakhshani, N.; Malekzadeh, R. MicroRNA-146a induces immune suppression and drug-resistant colorectal cancer cells. Tumor Biol. 2017, 39, 1010428317698365. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Leung, W.; Ng, S.S.-M. Exploiting a novel miR-519c–HuR–ABCG2 regulatory pathway to overcome chemoresistance in colorectal cancer. Exp. Cell Res. 2015, 338, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.-L.; Yan, T.-T.; Shen, C.-Q.; Tang, J.-Y.; Kong, X.; Wang, Y.-C.; Chen, J.; Liu, Q.; He, J.; Zhong, M.; et al. The distinct role of strand-specific miR-514b-3p and miR-514b-5p in colorectal cancer metastasis. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mésange, P.; Poindessous, V.; Sabbah, M.; Escargueil, A.E.; De Gramont, A.; Larsen, A.K. Intrinsic bevacizumab resistance is associated with prolonged activation of autocrine VEGF signaling and hypoxia tolerance in colorectal cancer cells and can be overcome by nintedanib, a small molecule angiokinase inhibitor. Oncotarget 2014, 5, 4709–4721. [Google Scholar] [CrossRef]
- Norguet, E.; Dahan, L.; Gaudart, J.; Gasmi, M.; Ouafik, L.; Seitz, J.-F. Cetuximab after bevacizumab in metastatic colorectal cancer: Is it the best sequence? Dig. Liver Dis. 2011, 43, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lambrechts, D.; Prenen, H.; Jain, R.K.; Carmeliet, P. Lessons From the Adjuvant Bevacizumab Trial on Colon Cancer: What Next? J. Clin. Oncol. 2011, 29, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xiao, Z.; Hong, Z.; Jiao, H.; Zhu, S.; Zhao, Y.; Bi, J.; Qiu, J.; Zhang, D.; Yan, J.; et al. FOXF1 promotes angiogenesis and accelerates bevacizumab resistance in colorectal cancer by transcriptionally activating VEGFA. Cancer Lett. 2018, 439, 78–90. [Google Scholar] [CrossRef]
- Hsu, H.-C.; Lapke, N.; Chen, S.-J.; Lu, Y.-J.; Jhou, R.-S.; Yeh, C.; Tsai, W.-S.; Hung, H.-Y.; Hsieh, C.-H.; Yang, T.-S.; et al. PTPRT and PTPRD Deleterious Mutations and Deletion Predict Bevacizumab Resistance in Metastatic Colorectal Cancer Patients. Cancers 2018, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Sobrero, A.F.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; Adenis, A.; et al. Phase III CORRECT trial of regorafenib in metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2012, 30, 3502. [Google Scholar] [CrossRef]
- Cai, M.-H.; Xu, X.-G.; Yan, S.-L.; Sun, Z.; Ying, Y.; Wang, B.; Tu, Y.-X. Regorafenib suppresses colon tumorigenesis and the generation of drug resistant cancer stem-like cells via modulation of miR-34a associated signaling. J. Exp. Clin. Cancer Res. 2018, 37, 151. [Google Scholar] [CrossRef]
- Mirone, G.; Perna, S.; Shukla, A.; Marfé, G. Involvement of Notch-1 in Resistance to Regorafenib in Colon Cancer Cells. J. Cell. Physiol. 2015, 231, 1097–1105. [Google Scholar] [CrossRef]
- Ou, B.; Cheng, X.; Xu, Z.; Chen, C.; Shen, X.; Zhao, J.; Lu, A. A positive feedback loop of β-catenin/CCR2 axis promotes regorafenib resistance in colorectal cancer. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Andre, T.; Chibaudel, B. Aflibercept (zaltrap (®)) approved in metastatic colorectal cancer. Bull. Cancer 2013, 100, 1023–1025. [Google Scholar]
- Bouygues, A.; Mesange, P.; Ayadi, M.; Chiron, M.; Dochy, E.; André, T.; De Gramont, A.; Larsen, A. P-221 Acquired 5-FU resistance in CRC models is accompanied by upregulation of VEGF-VEGFR1 signaling, increased migration and invasion that can be attenuated by aflibercept. Ann. Oncol. 2015, 26, iv64. [Google Scholar] [CrossRef][Green Version]
- Stanel, S.C.; Sjöberg, J.; Salmonson, T.; Foggi, P.; Caleno, M.; Melchiorri, D.; Gravanis, I.; Tzogani, K.; Pignatti, F. European Medicines Agency approval summary: Zaltrap for the treatment of patients with oxaliplatin-resistant metastatic colorectal cancer. ESMO Open 2017, 2, e000190. [Google Scholar] [CrossRef]
- Giordano, G.; Remo, A.; Porras, A.; Pancione, M. Remo Immune Resistance and EGFR Antagonists in Colorectal Cancer. Cancers 2019, 11, 1089. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, W.A.; Ahnen, D.J. Targeting EGFR in colorectal cancer. N. Engl. J. Med. 2008, 359, 1834. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Takegawa, N.; Yonesaka, K.; Sakai, K.; Ueda, H.; Watanabe, S.; Nonagase, Y.; Okuno, T.; Takeda, M.; Maenishi, O.; Tsurutani, J.; et al. HER2 genomic amplification in circulating tumor DNA from patients with cetuximab-resistant colorectal cancer. Oncotarget 2016, 7, 3453–3460. [Google Scholar] [CrossRef]
- Yang, Y.-N.; Zhang, R.; Du, J.-w.; Yuan, H.-H.; Li, Y.-J.; Wei, X.-L.; Du, X.-X.; Jiang, S.-L.; Han, Y. Predictive role of UCA1-containing exosomes in cetuximab-resistant colorectal cancer. J. Catherization Cardiovasc. Interv. 2018, 18, 164. [Google Scholar] [CrossRef]
- Bendell, J.C.; Atreya, C.E.; André, T.; Tabernero, J.; Gordon, M.S.; Bernards, R.; Van Cutsem, E.; Tejpar, S.; Sidhu, R.; Go, W.Y.; et al. Efficacy and tolerability in an open-label phase I/II study of MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in combination in patients (pts) with BRAF V600E mutated colorectal cancer (CRC). J. Clin. Oncol. 2014, 32, 3515. [Google Scholar] [CrossRef]
- Barry, G.S.; Cheang, M.C.; Chang, H.L.; Kennecke, H. Genomic markers of panitumumab resistance including ERBB2/HER2 in a phase II study of KRAS wild-type (wt) metastatic colorectal cancer (mCRC). Oncotarget 2016, 7, 18953–18964. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Clin. Oncol. 2015, 6, 660. [Google Scholar]
- Phipps, A.I.; Buchanan, D.D.; Makar, K.W.; Burnett-Hartman, A.N.; Coghill, A.E.; Passarelli, M.N.; Baron, J.A.; Ahnen, D.J.; Win, A.K.; Potter, J.D.; et al. BRAF Mutation Status and Survival after Colorectal Cancer Diagnosis According to Patient and Tumor Characteristics. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1792–1798. [Google Scholar] [CrossRef]
- Kalady, M.F.; DeJulius, K.L.; Sanchez, J.A.; Jarrar, A.; Liu, X.; Manilich, E.; Skacel, M.; Church, J.M. BRAF Mutations in Colorectal Cancer Are Associated with Distinct Clinical Characteristics and Worse Prognosis. Dis. Colon Rectum 2012, 55, 128–133. [Google Scholar] [CrossRef]
- Hazar-Rethinam, M.; Kleyman, M.; Han, G.C.; Liu, D.; Ahronian, L.G.; Shahzade, H.A.; Chen, L.; Parikh, A.R.; Allen, J.N.; Clark, J.W.; et al. Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAFV600E Colorectal Cancer. Cancer Discov. 2018, 8, 417–427. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Capone, E.; Barcaroli, D.; D’Agostino, D.; Volpe, S.; Benfante, A.; Van Hooff, S.; Iacobelli, V.; Rossi, C.; Iacobelli, S.; et al. ErbB-3 activation by NRG-1β sustains growth and promotes vemurafenib resistance in BRAF-V600E colon cancer stem cells (CSCs). Oncotarget 2015, 6, 16902–16911. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.M.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients with Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef]
- Tabernero, J.; Chan, E.; Baselga, J.; Blay, J.-Y.; Segelov, E.; Hyman, D.M.; Raje, N.S.; Wolf, J.; Sirzen, F.; Veronese, M.L.; et al. VE-BASKET, a Simon 2-stage adaptive design, phase II, histology-independent study in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m): Activity of vemurafenib (VEM) with or without cetuximab (CTX) in colorectal cancer (CRC). J. Clin. Oncol. 2014, 32, 3518. [Google Scholar] [CrossRef]
- Tabernero, J.; Van Geel, R.; Guren, T.K.; Yaeger, R.D.; Spreafico, A.; Faris, J.E.; Yoshino, T.; Yamada, Y.; Kim, T.; Bendell, J.C.; et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J. Clin. Oncol. 2016, 34, 3544. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, X.; Liu, Q.; Mingli, Y.; Graves-Deal, R.; Cao, Z.; Singh, B.; Franklin, J.L.; Wang, J.; Bhuminder, S.; et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/β-catenin signaling. Nat. Med. 2017, 23, 1331–1341. [Google Scholar] [CrossRef]
- Sun, L.; Fang, Y.; Wang, X.; Han, Y.; Du, F.; Li, C.; Hu, H.; Liu, H.; Liu, Q.; Wang, J.; et al. miR-302a Inhibits Metastasis and Cetuximab Resistance in Colorectal Cancer by Targeting NFIB and CD44. Theranostics 2019, 9, 8409–8425. [Google Scholar] [CrossRef]
- Mussnich, P.; Rosa, R.; Bianco, R.; Fusco, A.; D’Angelo, D. MiR-199a-5p and miR-375 affect colon cancer cell sensitivity to cetuximab by targeting PHLPP1. Expert Opin. Ther. Targets 2015, 19, 1017–1026. [Google Scholar] [CrossRef]
- Wei, F.; Wang, M.; Li, Z.; Wang, Y.; Zhou, Y. Long Non-Coding RNA MIR570MG Causes Regorafenib Resistance in Colon Cancer by Repressing miR-145/SMAD3 Signaling. Front. Oncol. 2020, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Hu, J.; Zhu, X.-D.; Liu, X.; Wang, C.-C.; Li, W.-H.; Chen, Z.-Y. Overexpression of miR-145 increases the sensitivity of vemurafenib in drug-resistant colo205 cell line. Tumor Biol. 2013, 35, 2983–2988. [Google Scholar] [CrossRef]
- Choe, M.H.; Yoon, Y.; Kim, J.; Hwang, S.-G.; Han, Y.-H.; Kim, J.-S. miR-550a-3-5p acts as a tumor suppressor and reverses BRAF inhibitor resistance through the direct targeting of YAP. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Ashizawa, M.; Okayama, H.; Ishigame, T.; Min, A.K.T.; Saito, K.; Ujiie, D.; Murakami, Y.; Kikuchi, T.; Nakayama, Y.; Noda, M.; et al. microRNA-148a-3p regulates immunosuppression in DNA mismatch repair-deficient colorectal cancer by targeting PD-L1. Mol. Cancer Res. 2019, 17, 1403–1413. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, Y.; Zhang, Y.; Wang, Y.; Alotaibi, F.; Qiu, L.; Wang, H.; Peng, S.; Liu, Y.; Li, Q.; et al. miRNA-5119 regulates immune checkpoints in dendritic cells to enhance breast cancer immunotherapy. Cancer Immunol. Immunother. 2020, 69, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Ma, S.; Liu, M.; Xu, Z.; Li, Y.; Guo, H.; Ge, Y.; Liu, Y.; Zheng, D.; Shi, J. A double feedback loop mediated by microRNA-23a/27a/24-2 regulates M1 versus M2 macrophage polarization and thus regulates cancer progression. Oncotarget 2016, 7, 13502–13519. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wei, J.; Wang, F.; Kong, L.-Y.; Ling, X.-Y.; Nduom, E.; Gabrusiewicz, K.; Doucette, T.; Yang, Y.; Yaghi, N.K.; et al. Effect of miR-142-3p on the M2 Macrophage and Therapeutic Efficacy Against Murine Glioblastoma. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Lin, R.; Chen, L.; Chen, G.; Hu, C.; Jiang, S.; Sevilla, J.; Wan, Y.; Sampson, J.H.; Zhu, B.; Li, Q.-J. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. J. Clin. Investig. 2014, 124, 5352–5367. [Google Scholar] [CrossRef]
- Wei, J.; Nduom, E.K.; Kong, L.-Y.; Hashimoto, Y.; Xu, S.; Gabrusiewicz, K.; Ling, X.; Huang, N.; Qiao, W.; Zhou, S.; et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016, 18, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Doellgast, G.J. Quantitation of peroxidase-antibody binding to membrane fragments using column chromatography. Anal. Biochem. 1979, 98, 53–59. [Google Scholar] [CrossRef]
- Taylor, D.D.; Homesley, H.D.; Doellgast, G.J. Binding of specific peroxidase-labeled antibody to placental-type phosphatase on tumor-derived membrane fragments. Cancer Res. 1980, 40, 4064–4069. [Google Scholar]
- Koga, K.; Matsumoto, K.; Akiyoshi, T.; Kubo, M.; Yamanaka, N.; Tasaki, A.; Nakashima, H.; Nakamura, M.; Kuroki, S.; Tanaka, M.; et al. Purification, characterization and biological significance of tumor-derived exosomes. Anticancer Res. 2005, 25, 3703–3707. [Google Scholar] [PubMed]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef]
- Xue, X.-Y.; Liu, Y.-X.; Wang, C.; Gu, X.-J.; Xue, Z.-Q.; Zang, X.-L.; Ma, X.-D.; Deng, H.; Liu, R.; Pan, L.; et al. Identification of exosomal miRNAs as diagnostic biomarkers for cholangiocarcinoma and gallbladder carcinoma. Signal Transduct. Target. Ther. 2020, 5, 77. [Google Scholar] [CrossRef]
- Vautrot, V.; Chanteloup, G.; Elmallah, M.I.Y.; Cordonnier, M.; Aubin, F.; Garrido, C.; Gobbo, J. Exosomal miRNA: Small Molecules, Big Impact in Colorectal Cancer. J. Oncol. 2019, 2019, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhong, J.; Zhong, B.; Huang, J.; Jiang, L.; Jiang, Y.; Yuan, J.; Sun, J.; Dai, L.; Yang, C.; et al. Exosomes as potential sources of biomarkers in colorectal cancer. Cancer Lett. 2020, 476, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Chabot, S.; Orio, J.; Castanier, R.; Bellard, E.; Nielsen, S.J.; Golzio, M.; Teissié, J. LNA-Based Oligonucleotide Electrotransfer for miRNA Inhibition. Mol. Ther. 2012, 20, 1590–1598. [Google Scholar] [CrossRef]
- Ørom, U.A.V.; Kauppinen, S.; Lund, A.H. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene 2006, 372, 137–141. [Google Scholar] [CrossRef]
- Ebert, M.S.; Neilson, J.R.; A Sharp, P. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Rama, A.R.; Perazzoli, G.; Cabeza, L.; Mesas, C.; Quiñonero, F.; García-Pinel, B.; Vélez, C. Novel MicroRNA Sponges to Specifically Modulate Gene Expression in Colon Cancer Cells. Nucleic Acid Ther. 2020, 30, 325–334. [Google Scholar] [CrossRef]
- Wen, D.; Danquah, M.; Chaudhary, A.K.; Mahato, R.I. Small molecules targeting microRNA for cancer therapy: Promises and obstacles. J. Control. Release 2015, 219, 237–247. [Google Scholar] [CrossRef]
- Monroig-Bosque, P.D.C.; Shah, M.Y.; Fu, X.; Fuentes-Mattei, E.; Ling, H.; Ivan, C.; Nouraee, N.; Huang, B.; Chen, L.; Pileczki, V.; et al. OncomiR-10b hijacks the small molecule inhibitor linifanib in human cancers. Sci. Rep. 2018, 8, 13106. [Google Scholar] [CrossRef]
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef]
- Ramchandani, D.; Lee, S.K.; Yomtoubian, S.; Han, M.S.; Tung, C.-H.; Mittal, V. Nanoparticle Delivery of miR-708 Mimetic Impairs Breast Cancer Metastasis. Mol. Cancer Ther. 2019, 18, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Z.; Xiang, J.; Gu, X. MicroRNA-155 acts as a tumor suppressor in colorectal cancer by targeting CTHRC1 in vitro. Oncol. Lett. 2018, 15, 5561–5568. [Google Scholar] [CrossRef]
- Qian, Z.; Gong, L.; Mou, Y.; Han, Y.; Zheng, S. MicroRNA-203a-3p is a candidate tumor suppressor that targets thrombospondin 2 in colorectal carcinoma. Oncol. Rep. 2019, 42, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Vychytilova-Faltejskova, P.; Merhautova, J.; Machackova, T.; Gutierrez-Garcia, I.; Garcia-Solano, J.; Radova, L.; Brchnelova, D.; Slaba, K.; Svoboda, M.; Halamkova, J.; et al. MiR-215-5p is a tumor suppressor in colorectal cancer targeting EGFR ligand epiregulin and its transcriptional inducer HOXB9. Oncology 2017, 6, 1–14. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Takayama, A.; Arai, T.; Kawauchi, J.; Sudo, H. MicroRNA-8073: Tumor suppressor and potential therapeutic treatment. PLoS ONE 2018, 13, e0209750. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, X.; Zhang, H.; Xiang, Y.; Chen, J.; Yin, Y.; Cai, X.; Wang, K.; Wang, G.; Ba, Y.; et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009, 28, 1385–1392. [Google Scholar] [CrossRef]
- Pekow, J.; Meckel, K.; Dougherty, U.; Butun, F.; Mustafi, R.; Lim, J.; Crofton, C.; Chen, X.; Joseph, L.; Bissonnette, M. Tumor suppressors miR-143 and miR-145 and predicted target proteins API5, ERK5, K-RAS, and IRS-1 are differentially expressed in proximal and distal colon. Am. J. Physiol. Liver Physiol. 2015, 308, G179–G187. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, K.; Wang, X.; Zhang, R.; Li, X. MicroRNA-192 acts as a tumor suppressor in colon cancer and simvastatin activates miR-192 to inhibit cancer cell growth. Mol. Med. Rep. 2019, 19, 1753–1760. [Google Scholar] [CrossRef]
- Wu, N.; Fesler, A.; Liu, H.; Ju, J. Development of novel miR-129 mimics with enhanced efficacy to eliminate chemoresistant colon cancer stem cells. Oncotarget 2017, 9, 8887–8897. [Google Scholar] [CrossRef]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Zhai, H.; Ju, J. miR-129 promotes apoptosis and enhances chemosensitivity to 5-fluorouracil in colorectal cancer. Cell Death Dis. 2013, 4, e659. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Lu, F.-Y.; Shi, R.-H.; Feng, Y.-D.; Zhao, X.-D.; Lu, Z.-P.; Xiao, L.; Zhou, G.-Q.; Qiu, J.-M.; Cheng, C.-E. MiR-26b regulates 5-FU-resistance in human colorectal cancer via down-regulation of Pgp. Am. J. Cancer Res. 2018, 8, 2518–2527. [Google Scholar] [PubMed]
- Xie, Y.; Shao, Y.; Deng, X.; Wang, M.; Chen, Y. MicroRNA-298 Reverses Multidrug Resistance to Antiepileptic Drugs by Suppressing MDR1/P-gp Expression in vitro. Front. Neurosci. 2018, 12, 602. [Google Scholar] [CrossRef] [PubMed]
- Medarova, Z.; Pantazopoulos, P.; Yoo, B. Screening of potential miRNA therapeutics for the prevention of multi-drug resistance in cancer cells. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Chen, Y.; Xianyu, Y.; Jiang, X. Surface Modification of Gold Nanoparticles with Small Molecules for Biochemical Analysis. Acc. Chem. Res. 2017, 50, 310–319. [Google Scholar] [CrossRef]
- Theis, T.; Yoo, M.; Park, C.S.; Chen, J.; Kügler, S.; Gibbs, K.M.; Schachner, M. Lentiviral Delivery of miR-133b Improves Functional Recovery After Spinal Cord Injury in Mice. Mol. Neurobiol. 2016, 54, 4659–4671. [Google Scholar] [CrossRef]
- De Lima, M.C.P.; Simões, S.; Pires, P.; Faneca, H.; Düzgüneş, N. Cationic lipid—DNA complexes in gene delivery: From biophysics to biological applications. Adv. Drug Deliv. Rev. 2001, 47, 277–294. [Google Scholar] [CrossRef]
- Huang, Z.; Shi, T.; Zhou, Q.; Shi, S.; Zhao, R.; Shi, H.; Dong, L.; Zhang, C.; Zeng, K.; Chen, J.; et al. miR-141 Regulates colonic leukocytic trafficking by targeting CXCL12β during murine colitis and human Crohn’s disease. Gut 2013, 63, 1247–1257. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, B.; Zhou, L.; Shi, Y.; Li, Z.; Xia, Y.; Tian, J. Imaging Dendrimer-Grafted Graphene Oxide Mediated Anti-miR-21 Delivery with an Activatable Luciferase Reporter. ACS Appl. Mater. Interfaces 2016, 8, 9014–9021. [Google Scholar] [CrossRef]
- Wen, D.; Peng, Y.; Liu, D.; Weizmann, Y.; Mahato, R.I. Mesenchymal stem cell and derived exosome as small RNA carrier and Immunomodulator to improve islet transplantation. J. Control. Release 2016, 238, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Laffont, B.; Corduan, A.; Plé, H.; Duchez, A.-C.; Cloutier, N.; Boilard, E.; Provost, P. Activated platelets can deliver mRNA regulatory Ago2•microRNA complexes to endothelial cells via microparticles. Blood 2013, 122, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, X.; Du, L.; Wang, Y.-S.; Liu, X.; Tian, H.; Wang, L.; Li, P.; Zhao, Y.; Duan, W.; et al. Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, E.; Choi, J.; Kim, J.-S.; Han, T.-S. MicroRNA-Based Therapeutics for Drug-Resistant Colorectal Cancer. Pharmaceuticals 2021, 14, 136. https://doi.org/10.3390/ph14020136
Jung E, Choi J, Kim J-S, Han T-S. MicroRNA-Based Therapeutics for Drug-Resistant Colorectal Cancer. Pharmaceuticals. 2021; 14(2):136. https://doi.org/10.3390/ph14020136
Chicago/Turabian StyleJung, Eunsun, Jinhyeon Choi, Jang-Seong Kim, and Tae-Su Han. 2021. "MicroRNA-Based Therapeutics for Drug-Resistant Colorectal Cancer" Pharmaceuticals 14, no. 2: 136. https://doi.org/10.3390/ph14020136
APA StyleJung, E., Choi, J., Kim, J.-S., & Han, T.-S. (2021). MicroRNA-Based Therapeutics for Drug-Resistant Colorectal Cancer. Pharmaceuticals, 14(2), 136. https://doi.org/10.3390/ph14020136